

Abstract
Acetone metabolism in the aerobic bacterium Xanthobacter strain Py2 proceeds by a carboxylation reaction forming acetoacetate as the first detectable product. In this study, acetone carboxylase, the enzyme catalyzing this reaction, has been purified to homogeneity and characterized. Acetone carboxylase was comprised of three polypeptides with molecular weights of 85,300, 78,300, and 19,600 arranged in an α2β2γ2 quaternary structure. The carboxylation of acetone was coupled to the hydrolysis of ATP and formation of 1 mol AMP and 2 mol inorganic phosphate per mol acetoacetate formed. ADP was also formed during the course of acetone consumption, but only accumulated at low, substoichiometric levels (≈10% yield) relative to acetoacetate. Inorganic pyrophosphate could not be detected as an intermediate or product of acetone carboxylation. In the absence of CO2, acetone carboxylase catalyzed the acetone-dependent hydrolysis of ATP to form both ADP and AMP, with ADP accumulating to higher levels than AMP during the course of the assays. Acetone carboxylase did not have inorganic pyrophosphatase activity. Acetone carboxylase exhibited a Vmax for acetone carboxylation of 0.225 μmol acetoacetate formed min−1⋅mg−1 at 30°C and pH 7.6 and apparent Km values of 7.80 μM (acetone), 122 μM (ATP), and 4.17 mM (CO2 plus bicarbonate). These studies reveal molecular properties of the first bacterial acetone-metabolizing enzyme to be isolated and suggest a novel mechanism of acetone carboxylation coupled to ATP hydrolysis and AMP and inorganic phosphate formation.
Acetone is a toxic molecule that is produced biologically by the fermentative metabolism of certain anaerobic bacteria and during mammalian starvation (1, 2). Acetone is known to undergo further metabolic transformations in microbes and higher organisms, and a variety of diverse bacteria have been found to grow using acetone as a source of carbon and energy (see refs. 3–5 and references cited therein). Studies of acetone-utilizing bacteria have provided evidence for the existence of two distinct pathways of acetone metabolism. For some aerobic bacteria, acetone metabolism has been proposed to proceed by an O2-dependent, monooxygenase-catalyzed oxidation producing acetol (hydroxyacetone) as the initial product (4, 6, 7). For other bacteria, including all anaerobes, acetone metabolism has been proposed to proceed by a CO2-dependent carboxylation-producing acetoacetate or an acetoacetyl derivative as the initial product (8–10). While in vivo and in vitro studies have provided some evidence supporting these proposed bacterial pathways (6–8, 11–13), the enzymes responsible for initiating acetone catabolism have not been purified to date.
One bacterium capable of using acetone as a source of carbon and energy is Xanthobacter strain Py2, an obligately aerobic, Gram-negative bacterium (14). The metabolism of acetone by Xanthobacter Py2 was recently shown to proceed by a CO2-dependent pathway analogous to that discussed above (3). The carboxylation of acetone to form acetoacetate was reconstituted in cell extracts with the addition of ATP (3). This study provided the first direct evidence for the involvement of an ATP-dependent carboxylase in bacterial acetone metabolism. In this study, acetone carboxylase has been purified to homogeneity. The molecular properties of acetone carboxylase are described, and evidence for a novel mechanism of acetone carboxylation coupled to ATP hydrolysis and AMP and inorganic phosphate formation is presented.
MATERIALS AND METHODS
Growth of Bacteria and Preparation of Cell Extracts.
Xanthobacter strain Py2 was grown with 32 mM isopropanol as the carbon source in a 15-liter capacity Microferm fermentor (New Brunswick Scientific) as described (15, 16). Cells were harvested after reaching an OD600 (measured using a Shimadzu UV160U spectrophotometer) between 2.5 and 4.0 by tangential-flow filtration with a Pellicon system (Millipore) and stored at −80°C. Frozen cell paste (98 g for the protocol described below) was resuspended in 2 vol of buffer A [25 mM 4-morpholinepropanesulfonic acid (Mops), pH 7.6/1 mM DTT/1 mM benzamidine] containing 0.1 mM EDTA, 0.1 mM EGTA, and 0.2 mg/ml lysozyme and DNase I. The cell suspension was passed three times through a French pressure cell at 110,000 kPa and 4°C and clarified by centrifugation (105,000 × g for 1 hr at 4°C).
Purification of Acetone Carboxylase.
Purification procedures were performed at 4°C. The supernatant of the cell extract was applied to a 5 × 25-cm column of DEAE-Sepharose equilibrated in buffer A containing 20% glycerol, 0.1 mM EDTA, and 0.1 mM EGTA at a linear flow rate of 28 cm/hr. The column was washed with 1,250 ml of buffer A containing 20% glycerol, followed by 1,250 ml of buffer A containing 20% glycerol and 90 mM KCl. Bound protein was fractionated with a 3-liter linear gradient from 90 mM KCl to 270 mM KCl. Active fractions were pooled, diluted 4-fold with buffer A containing 20% glycerol and applied to a 2.6 × 10-cm column of Macroprep ceramic hydroxyapatite (Bio-Rad). The column was washed with 160 ml of buffer A containing 10% glycerol at 45 cm/hr. A 380-ml linear gradient from 0 to 45 mM of potassium phosphate in buffer A containing 10% glycerol was applied to the column. Active fractions were pooled and concentrated by ultrafiltration using a YM100 membrane (Amicon). The sample was chromatographed in 250-mg portions on a 2.6 × 64-cm Sephacryl S-300 gel filtration column equilibrated with buffer A containing 10% glycerol and 0.2 M KCl at a linear flow rate of 8.5 cm/hr. Active fractions from the five S-300 chromatography procedures were pooled, diluted 4-fold with buffer A containing 20% glycerol, and applied to a 2.6 × 11-cm HiLoad Q-Sepharose column. The column was washed with 130 ml of buffer A containing 20% glycerol and 120 mM KCl at a flow rate of 45 cm/hr. Acetone carboxylase was eluted with an 800-ml linear gradient from 120 to 270 mM KCl. Appropriate fractions were pooled, concentrated by ultrafiltration, and frozen in liquid nitrogen.
Assay of Acetone Consumption and Acetoacetate Formation.
Acetone consumption assays were performed in serum vials (9 ml) containing ATP (0–25 mM), MgCl2 (1 mM in excess of ATP concentration), potassium acetate (80 mM), Mops (100 mM), and a source of enzyme (cell extract, column fractions, or purified enzyme) in a total volume of 1 ml at pH 7.6. Potassium bicarbonate and CO2 gas were added to appropriate sealed assay vials in a ratio (1 mol CO2 to 4 mol bicarbonate) that maintained the pH of the solutions at 7.6. The concentrations of total carbonate species varied between 0 and 50 mM. For assays lacking CO2 and for Km determination studies, residual CO2 was removed by sparging buffers and flushing sealed assay vials with CO2-free nitrogen. For CO2-free assays, a KOH-impregnated filter trap (16) was included in the vials. Assays were initiated by the addition of acetone. Vials were incubated throughout the course of the assay in a shaking water bath at 30°C and 250 cycles per minute. At desired time points, 100 μl samples of the gas phase (for analysis of acetone) and 1 μl samples of the liquid phase (for analysis of acetone plus acetoacetate) were removed and analyzed by gas chromatography as described (15). The time course of consumption of other potential substrates was followed by gas chromatography in the same manner.
Determination of Phosphate.
Phosphate produced during the time course of assays was quantified by a modified molybdophosphoric acid method (17). At desired time points, 25 μl liquid samples were removed from assay vials and added to 175 μl of 100 mM Mops (pH 7.6) containing 3.5 mM MgCl2. The samples were brought to 1 ml total volume by the addition of H2O (600 μl), 2.5 M HClO4 (100 μl), and 0.41 M Na2MoO4 (100 μl) followed by vortexing. After 5-min incubation at room temperature, the A380 of the samples was recorded. The phosphate content of samples was determined by comparison with a standard curve prepared with potassium phosphate.
Determination of Pyrophosphate.
Pyrophosphate was determined after conversion to inorganic phosphate using inorganic pyrophosphatase. Twenty-five microliter samples were removed from assay vials and assayed as described for the phosphate analysis, except that inorganic pyrophosphatase (0.5 unit) was present in the 175 μl of Mops/MgCl2. After 30-sec incubation, the additional reagents were added and the A380 was measured as described above. Standards of pyrophosphate were treated identically and shown to undergo quantitative conversion to inorganic phosphate by this method. Pyrophosphate was calculated on the basis of the difference in phosphate content of the pyrophosphatase-treated and nontreated samples.
Determination of ADP and AMP in Fixed-Time Point Assays.
ADP was determined spectrophotometrically by measuring the ADP-dependent oxidation of NADH to NAD+ using a modification of a coupled enzyme assay (18). Samples (25 μl) were removed from assay vials at desired time points and added to sealed cuvettes containing 0.975 ml of an assay mixture consisting of 2 mM phosphoenolpyruvate, 2 mM MgCl2, 1 mM ATP, 0.2 mM NADH, 21 units each pyruvate kinase and lactate dehydrogenase, and 100 mM Mops buffer at pH 7.6. After mixing, the cuvettes were incubated for 15 sec at 30°C, which was a sufficient time for quantitative phosphorylation of ADP and concomitant oxidation of NADH according to Eqs. 1 and 2:
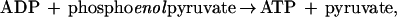 |
1 |
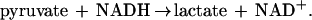 |
2 |
The absorbance at 340 nm was recorded, subtracted from the initial absorbance value, and the difference used to calculate the amount of ADP present in the sample. After recording the A340, adenylate kinase (10 units) was added to cuvettes to convert AMP to ADP according to Eq. 3:
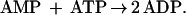 |
3 |
The cuvettes were incubated an additional 15 sec to allow complete reaction of AMP and production of NAD+ according to Eqs. 1–3. The A340 was then recorded, subtracted from the A340 recorded prior to addition of adenylate kinase, and used to calculate AMP present in the samples. To verify the accuracy of these determinations, AMP and ADP were also quantified from standards and samples by HPLC analysis as described by Seefeldt and Mortenson (19). The two methods gave results for AMP and ADP determination that agreed within 2%.
Continuous, Coupled Spectrophotometric Assay for ADP and AMP Formation.
Assays were performed as described above, but in stoppered cuvettes containing the additional components (coupling enzymes, phosphoenolpyruvate, NADH) allowing AMP and/or ADP formation to be coupled to the oxidation of NADH (see Eqs. 1–3). Cuvettes were preincubated for 5 min at 30°C with all assay components except acetone. Assays were initiated by the addition of acetone. Assays were monitored by following the absorbance change at 340 nm in a Shimadzu model UV160U spectrophotometer containing a thermostated cell holder maintained at 30°C.
Protein Characterization.
SDS/PAGE (12% T, 2.7% C running gel) was performed following the Laemmli procedure (20). Electrophoresed proteins were visualized by staining with Coomassie blue. The apparent molecular masses of polypeptides based on SDS/PAGE migration were determined by comparison with Rf values of standard proteins. The standards used were myosin (200 kDa), β-galactosidase (116.2 kDa), phosphorylase b (97.4 kDa), BSA (68 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), soybean trypsin inhibitor (21.5 kDa), lysozyme (14.4 kDa), and aprotinin (6.5 kDa). Polypeptide molecular weights were also determined using matrix-assisted laser desorption ionization–time-of-flight mass spectrometry performed in linear mode with an acceleration voltage of 16 kV. The matrix was α-phenyl-4-acetamido-cinnamic acid, and the internal standard was BSA. The relative subunit proportions were determined by integrated scanning densitometry of SDS/PAGE gels with an Isco model 1312 gel scanner. The native molecular weight was estimated by HPLC gel filtration using an Ultraspherogel TSK 3000SW column (0.75 × 30 cm) developed with 25 mM Mops (pH 7.6) containing 0.2 M NaCl and 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. The column was calibrated with apoferritin (443 kDa), β-amylase (200 kDa), and alcohol dehydrogenase (150 kDa). Metal analysis was performed with an inductively coupled plasma atomic emission spectrophotometer at the Utah State University Soil and Plant Analysis Laboratory. Protein concentrations were determined by a modified biuret assay (21) using BSA as the standard.
Induction of Acetone Carboxylase, 35S-Labeling, and Autoradiography.
Batch cultures (25 ml) grown with glucose as the carbon source (OD600 = 1.5) were induced by the addition of propylene oxide, acetone, or isopropanol (13.5 μmol each), labeled with [35S]methionine and cysteine, lysed, and autoradiographed as described (22).
Data Analysis.
Kinetic constants (Km and Vmax) were calculated by fitting initial rate data to the Michaelis–Menten equation as described by Cleland (23) and using the software sigmaplot.
RESULTS
Purification and Characterization of Acetone Carboxylase.
The soluble fractions of cell extracts prepared from cultures of Xanthobacter strain Py2 grown with acetone or isopropanol consistently exhibited specific activities for ATP-dependent acetone carboxylation of 0.04 to 0.05 unit/mg. These rates are directly comparable to the rates of acetone consumption measured in actively growing cultures or resting-state whole cell suspensions of acetone- or isopropanol-grown Xanthobacter strain Py2 (3). These results demonstrate that acetone carboxylase can be reconstituted in vitro at physiologically relevant rates and suggest that the enzyme will be amenable to purification in an active state.
Acetone carboxylase was purified 4.2-fold from the soluble fraction of cell extracts with a recovery of 56% and specific activity for acetone carboxylation of 0.206 unit/mg of protein (Table 1). As shown in Fig. 1, the purification resulted in the enrichment of three polypeptides that migrated on SDS/PAGE with apparent molecular masses of 78.8, 68.4, and 23.0 kDa. These bands are readily visible in the soluble fraction used as the source of enzyme for the purification (Fig. 1, lane 2), but are not visible in cell extracts prepared from cultures grown under conditions where acetone carboxylase is not expressed, e.g., with glucose or propylene as carbon sources (22). To further confirm the central roles of these polypeptides in acetone metabolism, cultures of glucose-grown Xanthobacter Py2 were induced for acetone carboxylase activity by the addition of acetone or isopropanol, followed by pulse-labeling with 35S amino acids. As shown in the autoradiogram presented in Fig. 2, induction with acetone or isopropanol resulted in the new and high level synthesis of the three polypeptides that purify in association with acetone carboxylase activity. In contrast, no synthesis of these polypeptides was detected in the noninduced, glucose-grown cells (compare lane 1 with lanes 3 and 4). As a control, the gel banding patterns of proteins synthesized in cells exposed to acetone and isopropanol are compared in Fig. 2 with those synthesized in cells exposed to propylene oxide, which induces to high levels a distinct set of enzymes involved in aliphatic alkene and epoxide metabolism (22). It is apparent from these results that acetone carboxylase is highly inducible and represents a sizable percentage of total soluble protein in cultures grown (or induced) with acetone or isopropanol, an observation that explains the low-fold purification required to obtain a homogenous preparation of the enzyme (Table 1). The high level of expression of acetone carboxylase is presumably related to the relatively low specific activity of the enzyme, i.e., high levels of acetone carboxylase are required to support cell growth at the observed rates (3).
Table 1.
Purification of acetone carboxylase
| Step | Volume, ml | Total protein, mg | Total activity, units* | Specific activity, units/mg | Purification, x-fold | Recovery, % |
|---|---|---|---|---|---|---|
| Cell extract | 211 | 4,480 | 222 | 0.0496 | 1 | 100 |
| DEAE-Sepharose | 562 | 1,500 | 208 | 0.139 | 2.8 | 93 |
| Hydroxyapatite | 15 | 1,250 | 188 | 0.151 | 3.0 | 85 |
| Sephacryl S-300 | 121 | 650 | 129 | 0.198 | 4.0 | 58 |
| Q-Sepharose | 223 | 609 | 125 | 0.206 | 4.2 | 56 |
Activity assays were performed as described in Materials and Methods using 0.2–0.6 mg protein, 10 mM ATP, 50 mM CO2 plus KHCO3, and 2 μmol acetone. A unit of activity is defined as 1 μmol of acetone degraded per minute at 30°C.
Figure 1.

SDS/PAGE analysis of acetone carboxylase. Lanes: 1, molecular weight standards (2 μg each); 2, cell extract (35 μg); 3, DEAE-Sepharose fraction (17 μg); 4, hydroxyapatite fraction (15 μg); 5, Sephacryl S-300 fraction (11 μg); 6 and 7, Q-Sepharose fraction (5.1 and 7.2 μg, respectively).
Figure 2.

Gel electrophoretic profiles of proteins synthesized after exposure of glucose-grown Xanthobacter strain Py2 to acetone and isopropanol. The autoradiogram of a 14% SDS gel is shown. Each gel lane contains cell extract equal to 20 μg protein. Lanes: 1, no inducer added; 2, propylene oxide added as inducer; 3, acetone added as inducer; 4, isopropanol added as inducer.
Mass spectrometry was used to provide a more accurate estimation of polypeptide molecular weights. This analysis revealed molecular weights of 85.3, 78.3, and 19.6 kDa for the three subunits. The staining intensities of the three polypeptides on SDS/PAGE gave relative molar ratios of 1.0 (85.3-kDa band), 1.0 (78.3-kDa band), and 1.2 (19.6-kDa band), suggesting a minimal core complex consisting of one each of the three subunits with a molecular mass of 183 kDa. The native molecular weight of the acetone carboxylase complex was determined to be 353 kDa. Therefore, the native enzyme is likely to have an α2β2γ2 subunit configuration.
Acetone carboxylase activity was dependent upon the addition of ATP and an additional divalent metal ion (e.g., Mg2+). The addition of K+ stimulated acetone carboxylase approximately 2-fold over assays performed in its absence. Other nucleoside triphosphates (GTP, CTP, UTP, TTP) did not support acetone carboxylation. Acetone carboxylase activity was stable for several days at 4°C over the pH range 6.5 to 8.0. The pH optimum for activity was 7.6. The biotin-binding protein avidin was not an inhibitor of acetone carboxylation.
The UV/visible absorption spectrum of acetone carboxylase exhibited an absorption maximum at 281 nm. No additional absorbance was present in the wavelength range from 300 to 800 nm. Metal analysis of three separate acetone carboxylase preparations purified from three different batches of cells revealed the presence of significant quantities of Fe, Mn, and Zn. The stoichiometries, averaged for the three preparations and reported as mols of metal per mol of α2β2γ2 complex are: 0.70 ± 0.089 Fe, 1.31 ± 0.061 Mn, and 1.02 ± 0.055 Zn. Dialysis of acetone carboxylase vs. buffers containing 2 mM EDTA or 1 mM α-α′-dipyridyl did not decrease the metal contents, indicating that the metals are tightly bound. Likewise, dialysis of acetone carboxylase vs. buffer containing 5 mM MgCl2 did not decrease the metal contents, demonstrating that the metals are not readily exchangeable. These treatments did not affect the specific activity of acetone carboxylase. The addition of exogenous Fe2+, Mn2+, Zn2+, Ca2+, Co2+, Cu2+, or Ni2+ to assays did not stimulate acetone carboxylase activity above the maximal levels obtained in the presence of Mg2+ alone.
Of a number of other ketones evaluated as possible substrates for acetone carboxylase (butanone, 2-pentanone, 3-pentanone, 2-hexanone, and chloroacetone), only butanone was a substrate under the assay conditions used for acetone consumption. Butanone was consumed in a CO2- and ATP-dependent fashion and with a specific activity of 0.094 unit/mg of protein, which is 46% of the rate observed with acetone. Pyruvate, phosphoenolpyruvate, acetaldehyde, propionaldehyde, and propylene oxide were not substrates for acetone carboxylase.
Acetone Carboxylase–Catalyzed Nucleotide Hydrolysis.
The identity and stoichiometry of an ATP hydrolysis product(s) formed in the course of acetone carboxylation was investigated. As shown in Fig. 3A, the carboxylation of acetone to acetoacetate was coupled to the formation of AMP as a stoichiometric (1:1 mol ratio) product. ADP was also detected as a product, but accumulated only at low, substoichiometric levels. Inorganic pyrophosphate could not be detected as an intermediate or product of nucleotide hydrolysis. Rather, inorganic phosphate was identified as the sole inorganic product of nucleotide hydrolysis. The amount of phosphate produced was equal to the sum of the ADP formed plus twice the sum of AMP formed. Upon complete consumption of acetone, the rates of AMP, ADP, and phosphate formation decreased to the background rates observed when acetone carboxylase was incubated with ATP, but in the absence of acetone and CO2. These background rates were between 5 and 10% of the initial rates shown in Fig. 3A.
Figure 3.

Time course of acetone carboxylase-catalyzed acetone degradation and product formation. Assays were performed as described in Materials and Methods using 0.29 mg of purified acetone carboxylase, 10 mM ATP, 50 mM CO2 plus KHCO3, and 2 μmol acetone. At the indicated time points, individual assays were terminated by removing the assay vial from the water bath and analyzing samples of the liquid and gas phase for substrate and products. Each time point is an average of measurements on duplicate vials. (A) Assays performed in the presence of 50 mM CO2 plus KHCO3. (B) Assays performed in the absence of CO2 and KHCO3. (□), acetone; (▪), acetoacetate; (○), inorganic phosphate; (▵), AMP; (▿), ADP; (•), pyrophosphate.
In the absence of CO2 and KHCO3, acetone carboxylase did not catalyze any detectable consumption of acetone (Fig. 3B). However, the enzyme did catalyze the hydrolysis of ATP to form both ADP and AMP at rates significantly higher that those observed in the absence of acetone. Particularly intriguing was the observation that ADP accumulated to higher levels than AMP under these conditions. As observed for assays with CO2 present, the inorganic product of phosphodiester bond hydrolysis was phosphate.
The addition of pyrophosphate or inorganic pyrophosphatase to assays had no effect on the rate of acetone carboxylation. Acetone carboxylase did not exhibit any detectable pyrophosphatase activity, in either the absence or presence of acetone and CO2.
Continuous Spectrophotometric Assay of Acetone Carboxylase-Catalyzed ATP Hydrolysis.
The assays used to quantify acetone consumption and product formation in Fig. 3 suffer from several limitations, the most significant of which is reliance on fixed time point measurements. A continuous spectrophotometric assay would be superior for measuring initial rates and obtaining steady-state kinetic data. An appropriate assay has been developed that relies on coupling acetone carboxylase-catalyzed AMP and ADP formation to NADH oxidation using the coupling enzymes adenylate kinase (excluded for measurements of ADP formation only; included for measurements of AMP plus ADP formation), pyruvate kinase, and lactate dehydrogenase (see Eqs. 1–3). The rates of NADH oxidation are numerically equivalent to rates of phosphodiester bond hydrolysis since phosphate is the only inorganic hydrolysis product observed.
With acetone and CO2 present, the initial rates of phosphodiester bond hydrolysis measured using this assay were 0.487 μmol min−1⋅mg−1 in the presence of adenylate kinase and 0.0207 μmol min−1⋅mg−1 in the absence of adenylate kinase (Fig. 4, Traces 1A and 2A). These results agree with those presented in Fig. 3 showing AMP rather than ADP to be the predominant nucleotide product formed during acetone carboxylation. The specific activity of AMP formation was determined to be 0.233 μmol AMP formed min−1⋅mg−1, which is directly comparable to the specific activity of 0.206 μmol acetone consumed min−1⋅mg−1 measured in fixed time point assays. In the absence of acetone, phosphodiester bond hydrolysis occurred at significantly lower rates than those observed in the presence of acetone (Traces 3A and 4A).
Figure 4.

Continuous, spectrophotometric assay of acetone carboxylase-catalyzed ATP hydrolysis. NADH-linked, coupled enzyme assays were performed in stoppered cuvettes as described using 0.091 mg of purified acetone carboxylase. All assay cuvettes contained pyruvate kinase and lactate dehydrogenase. Acetone (2 μmol) and adenylate kinase (10 units) were included where noted below. (A) Assays containing 50 mM CO2 plus KHCO3. (B) Assays lacking CO2 and bicarbonate. Traces 1A and 1B, assays with acetone and adenylate kinase; Traces 2A and 2B, assays with acetone and without adenylate kinase; Traces 3A and 3B, assays without acetone and with adenylate kinase; Traces 4A and 4B, assays without acetone and without adenylate kinase.
In the presence of acetone and absence of CO2, phosphodiester bond hydrolysis rates of 0.584 and 0.189 μmol min−1⋅mg−1 were calculated with and without adenylate kinase in the assays, respectively (Traces 1B and 2B). The corresponding initial rates of nucleotide formation are 0.189 μmol ADP formed min−1⋅mg−1 and 0.197 μmol AMP formed min−1⋅mg−1. These results agree with the results presented in Fig. 3, which show both AMP and ADP to be significant products of acetone-dependent, CO2-independent ATP hydrolysis. As observed for the assays performed in the presence of CO2, the rates of ATP hydrolysis were significantly lower in assays performed in the absence of acetone (Traces 3B and 4B).
Kinetic Characterization of Acetone Carboxylase–Catalyzed Reactions.
Fig. 5 shows a plot of the rate of phosphodiester bond hydrolysis vs. acetone concentration in coupled enzyme assays where ATP was saturating, and CO2 plus bicarbonate were either saturating or completely absent. Fitting these data to the Michaelis–Menten equation provided Vmax values of 0.485 ± 0.015 μmol phosphodiester bonds hydrolyzed min−1⋅mg−1 in the presence of CO2 and 0.616 ± 0.012 μmol phosphodiester bonds hydrolyzed min−1⋅mg−1 in the absence of CO2. The corresponding apparent Km values for acetone were 7.80 ± 0.79 μM in the presence of CO2 and 7.68 ± 0.48 μM in the absence of CO2. It is interesting that the rate of acetone-dependent phosphodiester bond hydrolysis is slightly faster in the absence of CO2, whereas the Km for acetone is unchanged by the presence or absence of CO2.
Figure 5.

Effect of acetone concentration on the rate of acetone carboxylase-catalyzed phosphodiester bond hydrolysis. NADH-linked, coupled enzyme assays were performed in stoppered cuvettes as described using 0.026 mg of purified acetone carboxylase. All assay cuvettes contained pyruvate kinase, adenylate kinase, and lactate dehydrogenase. Assays were initiated by the addition of acetone. Rates were derived from the linear portions of progress curves of A340 vs. time (typically within the first 100 sec of reaction). The low rate of nucleotide hydrolysis occurring in the absence of acetone was subtracted for each rate. (▴), assays containing 50 mM CO2 plus KHCO3. (•), assays without CO2 and KHCO3.
A plot of rate of phosphodiester bond hydrolysis vs. ATP concentration in coupled enzyme assays where acetone and CO2 were saturating also followed Michaelis–Menten kinetics, providing a Vmax of 0.463 ± 0.018 μmol phosphodiester bonds hydrolyzed min−1⋅mg−1 and an apparent Km for ATP of 0.122 ± 0.014 mM. This value of Vmax is statistically equivalent to the value of 0.485 μmol min−1⋅mg−1 reported above for assays in which acetone concentrations were varied.
Since nucleotide hydrolysis occurs in the absence of CO2, it was necessary to employ fixed time point assays, where acetone consumption and acetoacetate formation could be monitored, to determine kinetic parameters for CO2. A plot of rate of acetoacetate formation vs. total carbonate species (CO2 plus HCO3−) in assays where acetone and ATP were present at saturating concentrations provided a Vmax of 0.225 ± 0.011 μmol acetoacetate formed min−1⋅mg−1 and an apparent Km for CO2 plus HCO3− of 4.17 ± 0.69 mM. This value of Vmax is slightly less than one-half of the Vmax values obtained for phosphodiester bond hydrolysis using the coupled enzyme assays. This is expected, since each acetoacetate formed requires the hydrolysis of two, rather than one, phosphodiester bonds (Fig. 2).
DISCUSSION
In this study, a soluble, multimeric acetone-utilizing enzyme has been purified and characterized from Xanthobacter strain Py2. The physiological function of acetone carboxylase is to convert acetone, a toxic and recalcitrant organic molecule, to acetoacetate, which is a central metabolite that can undergo further metabolic transformations by conventional and well-characterized biochemical pathways.
Acetone carboxylase exhibited an obligate requirement for ATP as a cofactor. Acetone carboxylation is a thermodynamically unfavorable process (ΔG°′ for acetone carboxylation with bicarbonate is +17.1 kJ/mol), and the hydrolysis of ATP to ADP (ΔG°′ = −31 kJ/mol) would theoretically provide sufficient energy to drive the carboxylation reaction. Interestingly, during the course of acetone carboxylation, AMP and inorganic phosphate form as the products of ATP hydrolysis (Fig. 3A) according to Eq. 4:
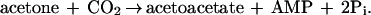 |
4 |
The carboxylation of acetone thus requires the hydrolysis of both the α–β and β–γ phosphodiester bonds of a single ATP molecule.
It is reasonable to speculate that ATP hydrolysis plays a role in activating acetone for CO2 addition through one or more group transfer reactions. Possibly, a phosphoryl or pyrophosphoryl group is transferred directly to acetone, or via the mediation of a phosphoryl- or pyrophosphoryl-enzyme intermediate. Acetone carboxylation presumably involves nucleophilic attack of the carbanion of acetone on CO2 (or bicarbonate). The carbanion might be formed by general base abstraction of a proton, but would be hard to generate and highly unstable due to the high pKa of the methyl group. The carbanion could be stabilized by keto to enol tautomerization, and the enol tautomer could be further stabilized by the transfer of a phosphoryl (or other) group from ATP to the oxygen atom of the enolate. Nucleophilic attack of the enol on CO2 (or bicarbonate) with concomitant hydrolysis of the oxygen–phosphate bond would result in the formation of acetoacetate.
This hypothetical reaction scheme bears similarities to well-characterized reactions involving the glycolytic intermediates pyruvate and phosphoenolpyruvate (24–26). The conversion of pyruvate to phosphoenolpyruvate is catalyzed by phosphoenolpyruvate synthase, which phosphorylates the enol tautomer of pyruvate at the expense of two high-energy phosphoanhydride bonds as shown in Eq. 5 (24, 26):
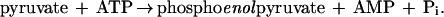 |
5 |
The hydrolysis of two phosphoanhydride bonds is required to drive the phosphorylation of pyruvate, which is an uphill reaction relative to a single ATP hydrolysis (ΔG°′ for phosphoenolpyruvate hydrolysis is −61.9 kJ/mol). Based on the same thermodynamic considerations, the hydrolysis of two phosphoanhydride bonds may be required for the generation of phosphoenolacetone or another activated acetone intermediate as well.
If group transfer from ATP to acetone does occur as part of the catalytic mechanism it would appear that the intermediate is not stable, since no detectable depletion of acetone was observed in assays performed in the absence of CO2 (Fig. 3B). In these assays, acetone stimulated the hydrolysis of ATP by ≈20-fold over assays performed in the absence of acetone, suggesting that a reaction of ATP with acetone is taking place. Possibly, the activated acetone intermediate simply breaks back down to acetone if CO2 is not available for further reaction to form acetoacetate. Alternatively, acetone may induce a conformational change that activates the ATPase activity of the enzyme.
It is interesting that both ADP and AMP accumulated as significant products of acetone-dependent ATP hydrolysis in the absence of CO2 (Fig. 3). ADP was also detected as a minor hydrolysis product in assays conducted in the presence of CO2 (Fig. 2). These results demonstrate that acetone carboxylase can catalyze the hydrolysis of both the α–β and β–γ phosphodiester bonds of ATP. Possibly, ATP hydrolysis occurs by the sequential cleavage of the β–γ and α–β phosphodiester bonds of ATP with the generation of an ADP intermediate rather than by an initial cleavage of the α–β bond, which would generate AMP and pyrophosphate (or a pyrophosphorylated intermediate) as the first hydrolysis products. Acetone carboxylase did not exhibit any pyrophosphatase activity, and we were unable to detect pyrophosphate as an intermediate in the steady-state carboxylation reactions (Figs. 2 and 3). These results argue against pyrophosphate as an intermediate in the reaction. However, we cannot rule out the possibility of a tightly or covalently bound pyrophosphoryl group as an intermediate in the reaction sequence. It is apparent that the elucidation of the mechanistic details of acetone carboxylation and the roles ATP hydrolysis play in this reaction will require the application of a variety of kinetic, mechanistic, and spectroscopic tools.
Schink and coworkers (5, 10, 11, 27, 28) have studied acetone metabolism in several anaerobic bacteria and obtained in vivo and in vitro data supporting the existence of CO2-dependent pathways in which acetone is carboxylated to acetoacetate or an acetoacetyl derivative (e.g., acetoacetyl-CoA). The enzymatic activity believed to be responsible for acetone carboxylation in a denitrifier designated strain BunN was studied in cell extracts. The carboxylation of acetone could not be reconstituted in cell extracts in either the absence or presence of ATP and other cofactors (9, 12). Cell extracts of strain BunN did catalyze two reactions believed to be relevant to acetone carboxylation: the ADP-dependent decarboxylation of acetoacetate and ADP-dependent exchange of 14CO2 into the carboxylate carbon atom of acetoacetate (9, 12). Notably, these enzymatic activities were lacking or greatly reduced in cell extracts prepared from cells grown with other carbon sources, providing evidence that they are associated with the acetone-carboxylating enzyme (9, 12).
Recently, Birks and Kelly (13) demonstrated acetone carboxylase activity in cell extracts prepared from acetone-grown cultures of Rhodobacter capsulatus. This activity required ATP and was stimulated by CoA or acetyl-CoA (these cofactors had no effect on acetone carboxylase activity in Xanthobacter Py2). The levels of activity observed for R. capsulatus extracts were much lower (200- to 1,000-fold) than those observed in cell extracts of Xanthobacter Py2 and, as noted by the authors, too low to be of physiological significance (13). Possibly, an additional component not required by the Xanthobacter system is limiting in the assay, and/or the enzyme is much less stable in vitro. Similar scenarios may apply to strain BunN and other anaerobes for which acetone carboxylation cannot be reconstituted in vitro.
In summary, this paper provides the first reported purification and characterization of a catabolic acetone-metabolizing enzyme. The identification and characterization of this enzyme fills a long-standing gap in our understanding of the microbial acetone cycle. The properties of acetone carboxylase reported in this initial study suggest a novel mechanism of ATP-dependent acetone carboxylation that promises to reveal new insights into biological strategies for adding CO2 to organic substrates.
Acknowledgments
We thank Jinhua Feng and Prof. Robert Brown for performing mass spectrometry of acetone carboxylase. This work was supported by National Science Foundation Grant MCB9630081.
ABBREVIATION
- Mops
4-morpholinepropanesulfonic acid
References
- 1.Davies R, Stephenson M. Biochem J. 1941;35:1320–1331. doi: 10.1042/bj0351320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argilés J P. Trends Biochem Sci. 1986;11:61–65. [Google Scholar]
- 3.Sluis M K, Small F J, Allen J R, Ensign S A. J Bacteriol. 1996;178:4020–4026. doi: 10.1128/jb.178.14.4020-4026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor D G, Trudgill P W, Cripps R E, Harris P R. J Gen Microbiol. 1980;118:159–170. [Google Scholar]
- 5.Janssen P H, Schink B. Arch Microbiol. 1995;163:188–194. doi: 10.1007/BF00305352. [DOI] [PubMed] [Google Scholar]
- 6.Lukins H H, Foster J W. J Bacteriol. 1963;85:1074–1087. doi: 10.1128/jb.85.5.1074-1087.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vestal J R, Perry J J. J Bacteriol. 1969;99:216–221. doi: 10.1128/jb.99.1.216-221.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnet-Smits E M, Robertson L A, Van Dijken J P, Senior E, Kuenen J G. J Gen Microbiol. 1988;134:2281–2289. [Google Scholar]
- 9.Platen H, Schink B. Biodegradation. 1990;1:243–251. doi: 10.1007/BF00119761. [DOI] [PubMed] [Google Scholar]
- 10.Platen H, Temmes A, Schink B. Arch Microbiol. 1990;154:355–361. doi: 10.1007/BF00276531. [DOI] [PubMed] [Google Scholar]
- 11.Janssen P H, Schink B. J Bacteriol. 1995;177:277–282. doi: 10.1128/jb.177.13.3870-3872.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen P H, Schink B. Eur J Biochem. 1995;228:677–682. doi: 10.1111/j.1432-1033.1995.0677m.x. [DOI] [PubMed] [Google Scholar]
- 13.Birks S J, Kelly D J. Microbiology. 1997;143:755–766. doi: 10.1099/00221287-143-3-755. [DOI] [PubMed] [Google Scholar]
- 14.van Ginkel C G, de Bont J A M. Arch Microbiol. 1986;145:403–407. [Google Scholar]
- 15.Allen J R, Ensign S A. J Bacteriol. 1996;178:1469–1472. doi: 10.1128/jb.178.5.1469-1472.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Small F J, Tilley J K, Ensign S A. Appl Environ Microbiol. 1995;61:1507–1513. doi: 10.1128/aem.61.4.1507-1513.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boltz D F, Lueck C H, Jakubiec R J. In: Colorimetric Determination of Nonmetals. Boltz D F, Howell J A, editors. Vol. 8. New York: Wiley; 1978. pp. 342–343. [Google Scholar]
- 18.Kreuzer K N, Jongeneel C V. Methods Enzymol. 1983;100:144–160. doi: 10.1016/0076-6879(83)00051-8. [DOI] [PubMed] [Google Scholar]
- 19.Seefeldt L C, Mortenson L E. Protein Sci. 1993;2:93–102. doi: 10.1002/pro.5560020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Chromy V, Fischer J, Kulhanek V. Clin Chem. 1974;20:1362–1363. [PubMed] [Google Scholar]
- 22.Ensign S A. Appl Environ Microbiol. 1996;62:61–66. doi: 10.1128/aem.62.1.61-66.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleland W W. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 24.Cooper R A, Kornberg H L. In: The Enzymes. Boyer P, editor. Vol. 10. New York: Academic; 1974. pp. 631–649. [Google Scholar]
- 25.Chollet R, Vidal J, O’Leary M H. Annu Rev Plant Physiol Plant Mol Biol. 1996;47:273–298. doi: 10.1146/annurev.arplant.47.1.273. [DOI] [PubMed] [Google Scholar]
- 26.Cooper R A, Kornberg H L. Methods Enzymol. 1969;13:309–315. [Google Scholar]
- 27.Platen H, Schink B. J Gen Microbiol. 1989;135:883–891. doi: 10.1099/00221287-135-4-883. [DOI] [PubMed] [Google Scholar]
- 28.Platen H, Janssen P H, Schink B. FEMS Microbiol Lett. 1994;122:27–32. doi: 10.1111/j.1574-6968.1994.tb07138.x. [DOI] [PubMed] [Google Scholar]