

Abstract
Muscle contraction is the result of myosin cross-bridges (XBs) cyclically interacting with the actin-containing thin filament. This interaction is modulated by the thin filament regulatory proteins, troponin and tropomyosin (Tm). With the use of an in vitro motility assay, the role of Tm in myosin’s ability to generate force and motion was assessed. At saturating myosin surface densities, Tm had no effect on thin filament velocity. However, below 50% myosin saturation, a significant reduction in actin–Tm filament velocity was observed, with complete inhibition of movement occurring at 12.5% of saturating surface densities. Under similar conditions, actin filaments alone demonstrated no reduction in velocity. The effect of Tm on force generation was assessed at the level of a single thin filament. In the absence of Tm, isometric force was a linear function of the density of myosin on the motility surface. At 50% myosin surface saturation, the presence of Tm resulted in a 2-fold enhancement of force relative to actin alone. However, no further potentiation of force was observed with Tm at saturating myosin surface densities. These results indicate that, in the presence of Tm, the strong binding of myosin cooperatively activates the thin filament. The inhibition of velocity at low myosin densities and the potentiation of force at higher myosin densities suggest that Tm can directly modulate the kinetics of a single myosin XB and the recruitment of a population of XBs, respectively. At saturating myosin conditions, Tm does not appear to affect the recruitment or the kinetics of myosin XBs.
Striated muscle contraction is the result of cyclic interactions between two contractile proteins, actin and the molecular motor myosin. This interaction is regulated by the actin-associated proteins, troponin and tropomyosin (Tm), with calcium binding to troponin considered the trigger for activation. However, evidence suggests that a significant component of thin filament activation is the result of myosin binding strongly to actin (1–4). This conclusion is based on the observation that both myosin binding and ATPase activity are a cooperative function of the number of strong-binding myosin cross-bridges (XBs) that interact with the thin filament (3, 4). Although cooperative activation is enhanced with troponin–Tm plus calcium, a significant degree of cooperativity is observed with Tm alone (2, 5), supporting an integral role for Tm in this cooperative process. However, the dependence of cooperative activation on the presence of both Tm and myosin strong binding is complex, because Tm inhibits actomyosin ATPase activity at low myosin concentrations (6) and potentiates ATPase activity at high myosin concentrations relative to actin and myosin alone (2).
To account for the complex effects of calcium and myosin binding on thin filament activation, McKillop and Geeves (7) proposed a three-state model of thin filament activation. In this model, the absence of calcium results in Tm sterically blocking myosin binding to actin (i.e., the “blocked state”). In the presence of calcium, Tm shifts on actin, occupying a “closed state” in which myosin weak binding can occur but the transition to the strongly bound state is inhibited. Finally, full activation of the thin filament occurs when Tm shifts further on the thin filament as a function of myosin strong binding, resulting in the “open state.” Tm movement on the thin filament has been observed with high-resolution electron microscopy (8), with the positional states of Tm on actin corresponding to the theoretical states of the McKillop and Geeves model (9, 10). In a simplified regulatory system that contains actin and Tm without the calcium-sensitive regulation of troponin, the McKillop and Geeves model can be reduced to two states, closed and open (11). In this context the McKillop and Geeves model is similar to the two-state model of thin filament regulation proposed by Hill et al. (1), with its weak and strong myosin binding states analogous to the closed and open states, respectively. In both models, myosin strong binding cooperatively activates the thin filament.
Although biochemical and structural data suggest that myosin strong binding facilitates thin filament cooperative activation, the effect of myosin binding on actomyosin mechanical performance at the molecular level has not been determined. In this study, thin filament activation by myosin strong binding is investigated by using in vitro techniques that determine the force and velocity of individual actin or actin–Tm filaments interacting with a myosin-coated surface. From the data we hypothesize that at subsaturating myosin conditions, Tm cooperatively modulates actomyosin interaction as a function of myosin strong binding by affecting both the recruitment of XBs and the kinetics of the XB cycle but that, at saturating conditions, neither recruitment nor XB kinetics is affected by Tm.
Methods
Actin and myosin were purified from chicken pectoralis skeletal muscle by standard techniques (12, 13). Tm was isolated from rabbit skeletal muscle by the methods of Smillie (14), with further purification by DEAE chromatography. Actin–Tm thin filaments were reconstituted as described by Homsher et al. (15). In brief, 20 μM actin was combined with 5 μM Tm in a low salt buffer (25 mM KCl/25 mM imidazole/5 mM MgCl2/10 mM DTT/1 mM EGTA, pH 7.4) and stored overnight at 4°C. The actin–Tm solution was diluted 1:10 before labeling with rhodamine-phalloidin. Actin filaments with or without Tm were then labeled with rhodamine-phalloidin in low salt buffer at a 1:1 actin/phalloidin ratio. After labeling, stoichiometric binding of Tm to actin was determined by centrifugal sedimentation (35,000rpm in a TLA 100.1 rotor; Beckman Instruments) for 30 min. The proteins in the pellet and supernatant were electrophoresed by SDS/PAGE, and the actin–Tm binding stoichiometry was determined by densitometric analysis (Fig. 1). Immediately before the assay either actin–Tm or actin was diluted to 8–20 nM. With actin–Tm, an additional 100 nM Tm was added to the motility solutions to ensure continued Tm binding to actin (15).
Figure 1.

SDS/PAGE and densitometry of a representative pellet and supernatant obtained after a sedimentation assay of an actin–Tm reconstitution. The amount of Tm pelleted with actin implies a 1:6 actin–Tm binding stoichiometry.
The motility assay and microneedle force assay were used as described (16). In brief, myosin molecules were adhered to a nitrocellulose-coated coverslip. The myosin concentration in the loading buffer (300 mM KCl/25 mM imidazole/5 mM MgCl2/10 mM DTT/1 mM EGTA, pH 7.4) was varied from 12.5 μg/ml to 250 μg/ml. Myosin binding to the surface is a linear function of the myosin concentration up to surface saturating conditions of myosin (100 μg/ml) (17). All experiments were performed at 30°C in the above-described low salt buffer with the addition of 0.375% methycellulose, 2 mM ATP, and an oxygen scavenger system (glucose oxidase at 0.1 mg/ml, catalase at 0.0018 mg/ml, and glucose at 2.3 mg/ml). In the in vitro motility assay, individual thin filaments were observed moving across the myosin-coated surface. Thin filament velocity and length were determined as described (18). Velocity of actin or actin-Tm filaments as a function of the myosin surface density was determined.
The force exerted by a population of myosin molecules on a single actin filament was determined by attaching an actin or actin–Tm filament to a calibrated glass microneedle. Force measurements were performed at myosin surface loading conditions of 25, 50, and 250 μg/ml. Assay conditions were the same as for the velocity experiments. Calibration of microneedles and measurement of steady-state force from the deflection of the microneedle as the thin filament interacts with the myosin surface have been described (16). To normalize the observed force to the length of actin in contact with the myosin surface, least squares linear regression of force versus actin filament length in contact with the surface was determined for each myosin concentration studied.
Results
Stoichiometric binding of Tm to actin was determined by sedimentation as described above. By inspection of the SDS/PAGE gel, virtually all of the Tm was bound to actin, as demonstrated by its cosedimentation with actin. A representative densitometric scan of the lane containing a sample of the pellet provided an estimate that binding of Tm to actin was at a 1:6 stoichiometry (Fig. 1), consistent with reported data (19). Tm binding to actin was also confirmed at the level of a single thin filament as demonstrated by the profound effect Tm had on force and velocity in the in vitro motility assay (see below).
In the absence of Tm, actin filament velocity was independent of the myosin loading concentrations in the range of 12.5–250 μg/ml, as reported (17). The addition of Tm to actin did not affect thin filament velocity at myosin loading concentrations of greater than 50 μg/ml (Fig. 2). However, at myosin concentrations of less than 50 μg/ml, a reduction in velocity for actin–Tm was observed, with complete inhibition of velocity occurring at a myosin concentration of 12.5 μg/ml. When filament motility was completely inhibited, filaments remained attached to the motility surface. This attachment was presumably to myosin because the filaments did not demonstrate reptation (i.e., longitudinal Brownian motion), which is characteristic of freely diffusing filaments that remain close to the motility surface due to methylcellulose in the assay buffer. The similarity in thin filament velocities for actin and actin–Tm at saturating myosin surface conditions (i.e., >100 μg/ml) is similar to the results of Fraser and Marston (20) but in contrast to Honda et al. (21), where enhanced thin filament velocity was reported after the addition of skeletal Tm.
Figure 2.

Thin filament velocity of actin (○) and actin–Tm (▴) as a function of myosin loading concentration, which in turn determines the motility surface density. Each data point represents an individual experiment in which the mean of at least 20 individual filament velocities are reported. The regression lines for actin–Tm (solid) and actin (dashed) were obtained by fitting the data to the hyperbolic function: y = y0 + (ax)/(b + x); the fit is solely intended to delineate the velocity differences between actin and actin–Tm. The parameters of the fit for actin and actin–Tm were as follows: actin (y0 = 4.71; a = 0.96; b = 19.72); actin–Tm (y0 = −346.91; a = 353.02; b = 0.21).
Three potential explanations for the reduction in actin–Tm filament velocity at low myosin concentrations are that (i) Tm binds to the motility surface and can thus attach to actin filaments and act as a load; (ii) Tm reduces the number of binding sites on actin and thus limits the number of myosins that can interact with actin; or (iii) Tm modulates the kinetics of the XB cycle.
The fact that excess Tm (100 nM) is added to the assay buffer could potentially allow free Tm to bind to the motility surface. Because of Tm’s actin-binding capacity, it could then act as a load to reduce filament velocity. As a control, thin filament velocity was measured as a function of myosin concentration but with various amounts of excess free Tm (i.e., 50–200 nM). If one assumes that the load created by Tm is proportional to the concentration of excess Tm, then the concentration of myosin at which thin filament velocities are inhibited should also vary with the concentration of excess Tm. This was not the case (data not shown), with inhibition occurring at the same myosin concentration regardless of the concentration of excess Tm. This finding eliminates a trivial explanation that the reduction in filament velocities was due to a drag force brought about by excess Tm.
To determine whether the reduction in velocity with actin–Tm is the result of Tm reducing the effective number of cycling myosin XB heads (i.e., derecruitment), we plotted the relationship between velocity and thin filament length at a myosin concentration of 22.5 μg/ml for both actin and actin Tm (Fig. 3). At this myosin concentration, actin–Tm filament velocity was significantly reduced. Therefore, if Tm effectively reduced the number of myosin binding sites on actin so that fewer heads were available to interact with the filament (i.e., analogous to effectively lowering surface myosin density), then this might account for the observed reduction in velocity. If so, then comparing the relationships of filament velocity versus filament length for actin versus actin–Tm filaments should reveal a strong dependence on filament length for actin–Tm (see Fig. 3 dashed curve) with shorter filaments (i.e., fewer number of heads interacting with the filament) moving slower than longer filaments (i.e., greater number of heads interacting with the filament) (see Fig. 3 for details). This was not the case because actin–Tm filament velocity was independent of filament length as both short and long actin-Tm filaments moved at the same slow velocity over the observed range of lengths (Fig. 3). Simply stated the reduction in velocity that is observed with actin–Tm is independent of the number of myosin XBs available to interact with the thin filament. This strongly suggests that Tm does not cause a reduced velocity by limiting the effective number of binding sites on actin (i.e., XB derecruitment) but rather the reduction of velocity for actin–Tm is the result of a modulation of XB kinetics. The fact that thin filaments are still bound to the surface at a myosin concentration of 12.5 μg/ml demonstrates that a sufficient number of myosins are available to bind to the thin filament even in the presence of Tm but suggests that their cycling and thus kinetics are inhibited under these conditions.
Figure 3.

Velocity of individual actin (□) and actin–Tm filaments (●) as a function of thin filament length and the predicted relationship if the presence of Tm effectively reduces the number of available XBs (dashed curve). The mean velocity of actin and actin–Tm filaments was 5.0 and 1.8 μm/s, respectively, at a myosin concentration of 22.5 μg/ml. At low myosin surface densities, velocity is a function of the number of XBs that can potentially interact with the filament (22). Under these conditions, the determinants of velocity are filament length (i.e., the total number of myosin heads that can interact with the thin filament) and duty cycle (i.e., the fraction of the XB cycle that myosin is strongly bound to actin). The dependence of velocity on filament length is hyperbolic with the asymptote of the relationship achieved, when a sufficient number of XBs interact with the filament to move it at its maximal velocity (17). For illustrative purposes, the solid curves that depict this relationship for actin and actin–Tm are presented and were generated with a defined relationship (17): vmax = avo[1 − (1 − fxb)n], where vmax is the filament velocity, avo is the filament velocity when at least one XB is attached to actin at all times and undergoing its power stroke, fxb is the XB duty cycle, and n is the number of XBs available to interact with the filament. For this model (solid curves), we assumed the avo to be equal to the mean velocities (see above), with fxb = 0.038 and n = 20 heads per micrometer of filament length based on previous studies (17). If Tm effectively reduces the number of available XB heads without having any effect on the kinetics of the XB cycle (i.e., no change in fxb or avo), then one can create a relationship in which the parameters fxb and avo are set at 0.038 and 5.0, respectively, and n is adjusted so that the mean velocity of the fit (over the range of thin filament lengths measured) equals the mean velocity for the actin–Tm data. The dashed curve is this relationship and requires Tm to effectively reduce the number of available XB heads per micrometer of filament length by 92%. If so, then there should have been a profound dependence of velocity on filament length, which was not observed.
The maximum isometric force for a population of myosins interacting with a single actin or actin–Tm filament was determined at myosin concentrations of 25, 50, and 250 μg/ml (Fig. 4). Force was normalized to the thin filament length in contact with the motility surface by determining the slope of the linear regression through the force versus filament length data (Fig. 4 and Table 1). From this analysis, the force per micrometer of actin filament length is a linear function of the myosin density on the surface (Fig. 5 and Table 1). These data indicate that force generation is proportional to the number of available XBs. As mentioned above, myosin surface density saturates at 100 μg/ml, therefore, it was not surprising that at suprasaturating myosin concentrations (250 μg/ml), the normalized force was not more than twice that at 50 μg/ml.
Figure 4.

Maximal isometric steady-state force per micrometer of thin filament in contact with a myosin-coated surface for actin (▴) and actin–Tm (□) filaments. Experiments were performed with myosin concentrations of 250, 50, and 25 μg/ml. The data were fit by linear regression (solid lines), with 95% confidence limits for the regression indicated by the dashed lines.
Table 1.
Force for actin and actin–Tm as a function of myosin surface density
| Myosin, μg/ml | Force, (pN/μm)*
|
|
|---|---|---|
| Actin | Actin-Tm | |
| 25 | 2.9 ± 0.9 | 3.8 ± 0.8 |
| 50 | 5.6 ± 0.8 | 12.2 ± 1.7 |
| 250 | 12.4 ± 1.0 | 14.5 ± 2.3 |
Force is the slope and the standard error of the estimate for the linear regressions in Fig. 3.
Figure 5.

Normalized force data for actin ( ● and actin–Tm (▴) as a function of myosin loading concentration, which is related to the surface density. Force for actin–Tm at a myosin concentration of 12.5 μg/ml is extrapolated from the complete inhibition of velocity at this myosin concentration. Saturating myosin surface data were those obtained at a myosin concentration of 250 μg/ml. We have shown (17) that the myosin surface is fully saturated at 100 μg/ml and hence additional myosin loading does not result in additional surface density. In light of this fact, the actin data were fit to a linear regression (solid line) with the surface saturated force measurement being set at a myosin concentration of 100 mg/μl. The actin–Tm data were fit to a sigmoidal regression (dashed line).
In contrast to the linear dependence of force on myosin concentration, actin–Tm demonstrates a sigmoidal dependence on myosin concentration (Fig. 5). With myosin at 25 and 250 μg/ml, normalized force for actin–Tm was similar to that of actin alone but, at 50 μg/ml, force was nearly twice that for actin (Fig. 4 and Table 1). Given that actin–Tm filament velocity was completely inhibited at a myosin concentration of 12.5 μg/ml, we have assumed that force would be similarly inhibited since the microneedle deflection requires that myosin be capable of movement against a load. Based on the sigmoidal relationship between force and myosin surface-loading concentration for actin–Tm (Fig. 5), it appears that the addition of Tm confers cooperativity, which was not exhibited for actin alone.
Discussion
This study demonstrates that Tm directly affects the mechanical performance of the actomyosin interaction in a cooperative manner. The effects of Tm are most notable at low myosin concentrations with complete inhibition of thin filament velocity and at moderate myosin concentrations where a 2-fold enhancement of force is observed. Thus, Tm is able to inhibit or activate actomyosin activity as a function of the number of myosin XBs available to interact with the thin filament. In addition, at saturating myosin concentrations, Tm does not affect the kinetics or recruitment of XBs. Given that the in vitro motility assay is well suited to control the activation level of the thin filament, by varying the myosin density on the surface and, thus, altering the number of XBs that bind to the thin filament, direct assessment of thin filament activation by myosin strong binding was determined.
Based on the observed effects of Tm on myosin’s average force (Favg) and actin filament velocity (vmax), is it possible to understand the molecular mechanism by which Tm modulates the interaction of actin and myosin? At the molecular level,
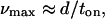 |
1 |
where d is the myosin step size and ton is the step duration so that 1/ton is the rate at which myosin detaches from actin (23). In addition, for a population of myosin molecules,
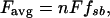 |
2 |
where n is the number of myosin molecules that can interact with the thin filament, F is the force generated by a single cross bridge, and fxb is the duty cycle or the fraction of the entire XB cycle during which the myosin XB is strongly bound and generating force (23). For any myosin, the duty cycle under isometric conditions is a function of the kinetics of the XB cycle.
If we assume these molecular definitions for vmax and Favg, the presence of Tm on actin can affect these mechanical indices by one of two nonexclusive mechanisms as follows: (i) XB recruitment in which Tm modulates the number of cycling XBs that can interact with the thin filament, thus an effect on n; (ii) XB kinetics in which Tm directly affects the rates of individual XB attachment and detachment from the thin filament and thus an effect on ton and/or fxb. It should be noted that the parameters of ton and fxb are strain dependent and thus can vary with load (24).
Attempting to determine which aspects of muscle activation that can be attributed to XB recruitment and XB kinetics has been the focus of several prior studies (25–28). These investigations, using muscle fibers, suggest that thin filament activation is the result of XB recruitment and/or the modulation of XB kinetics (25–28). Sorting out recruitment from kinetics issues is a difficult task because neither parameter is easily dissected from the other (Fig. 6). Studies that examine the binding of myosin to the thin filament with and without Tm may be, in its simplest form, a measure of the number of binding sites available on the thin filament. Myosin subfragment 1 (S1) binding to actin with different ligands in the presence of Tm is cooperative, and a greater fraction of actin sites are occupied by S1 when compared with actin alone at all S1/actin ratios (5). Thus, Tm facilitates XB binding to the thin filament, which suggests that Tm may be able to enhance XB recruitment over actin alone, possibly by altering the conformation of the myosin-binding interface on actin (29). Alternatively, data to support a change in XB kinetics during muscle activation have been reported in skinned fully regulated muscle fibers through measurements of the rate of tension redevelopment and shortening velocity (28). By integrating the results of prior studies with the data presented herein, insight may be achieved into the role of XB recruitment and XB kinetics in the activation of the thin filament as a function of myosin strong binding. This study and actomyosin ATPase data (2, 6, 30) demonstrate that the role of Tm is dependent on the extent of myosin strong binding, such that Tm either inhibits or enhances actomyosin mechanical performance at subsaturating myosin concentrations. These two functions are discussed separately below.
Figure 6.

Illustration of how a 2× increase in Favg (an ensemble force measurement), resulting from a change in XB recruitment due to the presence of Tm on actin, could be misinterpreted as a change in XB kinetics. In this case, we assume that the total XB population (n) equals 4 and that the XB duty cycle (fxb) is constant at 50% and unchanged in the presence of Tm. In addition, weakly bound XBs are depicted with open heads and strongly bound force-generating XBs are depicted with solid heads and contribute 1 unit of force. With actin alone (Left), it is modeled that the conformation of actin (shaded actin segment) prevents half of the XBs from attaching (shaded detached XBs). Given that only two XBs can interact with actin, and with fxb = 50%, then one XB will generate an Favg =1. However, if one estimates fxb based on Favg and n, then the population-based estimate of fxb is fpop = Favg/n or 25%. With the addition of Tm (Right), all XBs are capable of interacting with actin and Favg = 2. Even though fxb is unchanged, the population-based estimate has increased with fpop = 50%. Without knowing the true XB duty cycle, one could misconstrue the 2× increase in the population-based duty cycle estimate as an effect of Tm on XB kinetics rather than on recruitment. Thus, single molecule studies in the laser trap may be needed to definitively determine the contributions of changes in XB recruitment and kinetics to the observed effects of Tm on actomyosin mechanics.
Thin Filament Inhibition.
At low myosin surface densities, we observed a reduction in actin–Tm filament velocity with complete inhibition of myosin’s mechanical activity (i.e., force and velocity) at the lowest myosin concentration studied (i.e., 12.5 μg/ml). Both the binding of actin–Tm filaments to the myosin surface and the lack of a dependence of velocity on filament length (i.e., XB number) at these low myosin concentrations suggest that Tm does not prevent or limit the number of myosins that can attach to the thin filament. These data also suggest that as the myosin density on the surface is decreased, the number of XBs that are available to activate the thin filament also is decreased. Because XBs are still capable of binding at low myosin concentrations, the reduction in filament velocity is most likely due to Tm’s ability to modulate the kinetics of the XB cycle eventually arresting the thin filaments on the surface at the lowest myosin concentration. This complete inhibition of velocity would argue that the thin filament is in a closed state according to the McKillop and Geeves model (7). As such, the bound myosin would be in a weak binding state (1), because the weak-to-strong isomerization would be inhibited by Tm (31). This shift to a predominance of weakly bound XBs could be the result of altering the kinetics of a specific step or steps in the XB cycle. For instance, Tm is thought to inhibit Pi release (32), thus slowing the transition between weak and strong myosin binding states (33). However, this transition is not considered to be rate limiting for filament velocity but rather the detachment step, i.e., MgADP release from myosin at saturating MgATP. Therefore, at myosin concentrations where vmax is reduced in the presence of Tm, Tm may also be affecting MgADP release (34). Modulation of ADP release from myosin as a function of Ca2+ activation has been demonstrated in fully regulated thin filaments in solution (35). With full activation, ADP release is not altered (35), which is consistent with our observation that velocities for actin and actin–Tm were similar (see below).
If the weak-to-strong isomerization is inhibited with the deactivation of the thin filament, then, as the number of XBs interacting with the thin filament is reduced, an increasing proportion of XBs would be in the weakly bound state. In the motility assay, weakly bound XBs can create an internal load against which the remaining cycling XBs must operate (36). Thus, a secondary cause for the reductions in vmax observed with Tm could be that as the weakly bound XB population increases with thin filament inactivation so will the internal load. This load would slow the remaining cycling XBs, which would be perceived as an effect of Tm on XB kinetics. Whether Tm affects XB transitions that directly contribute to the reduction in velocity or indirectly by internal loading of the thin filament by weakly bound XBs cannot be resolved with the present data.
Thin Filament Activation.
As the myosin density increases, actin–Tm filament velocity increases as a hyperbolic function of the myosin surface density, with the maximal velocity for actin–Tm and actin filaments being similar (Fig. 2). Force also increases as a function of the myosin density on the surface. However, unlike velocity, force measured at a myosin concentration of 50 μg/ml for actin–Tm is double that measured for actin and, interestingly, is equal to actin alone at saturating myosin (i.e., 250 μg/ml). The actin–Tm filament is presumably fully activated to the open (7) or strong binding (1) state at a myosin concentration of 50 μg/ml, as further increases in myosin surface density does not result in further increases in force or velocity. Thus at this myosin density the critical number of XBs needed to fully activate the actin–Tm has been achieved. Once again, the apparent activating effect of Tm on force production could be due to alterations to XB recruitment, n, and/or kinetics, fxb, as described above (see Eq. 2). Velocity is insensitive to increases in the number of myosins interacting with the actin filament once the minimum number of heads that are needed to generate maximum velocity is attained. However, the velocity data do suggest that the XB detachment rate and thus XB kinetics under unloaded conditions is unchanged at a myosin concentration of ≥50 μg/ml in the presence of Tm. This does not preclude a change in XB kinetics (i.e., fxb) under loaded conditions, given the known dependence of XB kinetics on XB strain. However, if we assume that XB kinetics are not affected by Tm at a myosin concentration of 50 μg/ml, then the enhanced force observed for actin–Tm could be accounted for by a greater number of myosin binding sites being available on actin–Tm relative to actin and hence a recruitment of XBs. In addition, force as a function of myosin concentration reaches an asymptote at lower myosin concentrations for actin–Tm than for actin alone. This suggests that myosin binding to the thin filament in the presence of Tm saturates near a myosin concentration of 50 μg/ml and that actin–Tm is incapable of binding additional XBs regardless of an increase in myosin surface density.
Even though these motility data provide clear evidence for Tm having direct effects on actomyosin mechanical function, the relative contribution of XB recruitment and/or kinetics at any given level of thin filament activation cannot be definitively determined with the data at hand. As illustrated in Fig. 6, distinguishing between Tm’s effects on the kinetics of an individual XB versus changes in XB recruitment is difficult when studying the mechanical response from a population of myosin XBs. For example, if the 2-fold increase in force observed at a myosin concentration of 50 μg/ml for actin–Tm was due to a 2-fold increase in the number of available XBs (i.e., recruitment), one could misinterpret the force data as evidence for a change in the duty cycle, fxb, of an individual XB rather than XB recruitment (see Fig. 6 for details).
In contrast to subsaturating myosin conditions, our motility data and solution biochemical studies (37, 38) suggest that both XB kinetics and XB recruitment are not altered by Tm under saturating myosin conditions. This conclusion is based on the similarities observed when comparing actin and actin–Tm for the attachment rate of S1 strong binding to the thin filament (37) and the maximally activated myosin ATPase activity (38). This is supported by our motility data for actin and actin–Tm, demonstrating no effect of Tm on filament velocity or force at myosin concentrations of ≥50 μg/ml.
Conclusions.
The sigmoidal relationship between force and myosin concentration in the presence of Tm confirms that cooperativity of actomyosin function is conferred by Tm on actin. From our motility data, it is possible to estimate the number of actin monomers associated with an activated Tm-based cooperative unit. From our previous studies, the number of available myosin heads on the motility surface per micrometer of actin filament length at which the actin–Tm filament is fully activated (i.e., 50 μg/ml) is approximately 44 (17). Dividing this number into the number of actin monomers per micrometer of thin filament, assuming 5.5 nm per actin monomer, gives an estimated four monomers per head. This is close to the estimate of between five and six monomers per cooperative unit for actin–Tm determined by S1 binding in solution (39), thus suggesting that one myosin XB head per cooperative unit is sufficient to fully activate the thin filament or 1.7 heads per turn of the actin helix, which is similar to other predictions (40). Although in good agreement, it may not be appropriate to compare cooperative unit estimates based on in vitro filament velocities generated by cycling XBs to those from binding studies in solution with rigor bound XBs (39). In addition, although the data reported herein have direct applicability to muscle activation, a fully calcium regulated system with the addition of troponin would bring another level of complexity to the interpretation of thin filament function. As such, it would be of interest to extend these studies to troponin–Tm thin filaments to determine whether the cooperative unit length increases as predicted by solution studies (39). The effects of different Tm isoforms and familial hypertrophic cardiomyopathy mutations on the actomyosin interaction and cooperative unit length would also be of significant importance.
In summary, the data presented herein provide strong evidence that the presence of Tm can modulate the actomyosin mechanical performance in the in vitro motility assay. In addition, myosin strong binding is essential to activate the thin filament in the presence of Tm. Although the precise molecular mechanism for this modulation cannot be definitively characterized at present, it is clear that Tm has a dramatic effect on actomyosin performance as a function of activation. The change in force and motion observed in the presence of Tm is the result of either an alteration to the kinetics of an individual XB and/or the recruitment of a population of XBs. The laser trap assay may provide the answer by assessing whether or not alterations in the kinetics of a single XB result from the presence of Tm on actin.
Acknowledgments
We thank Eric Hayes, Kelly Begin, and Shari Alix for their superb technical assistance and Joe Haeberle for Tm samples. Norman Alpert and Martin LeWinter for their comments on this manuscript. This work was supported by the National Institutes of Health, Grant HL59408 to D.M.W., Grant HL07647 to K.A.P., and the Burroughs-Wellcome Fund Career Award to P.V.B.
Abbreviations
- XB
cross-bridge
- Tm
tropomyosin
- S1
myosin subfragment 1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hill T L, Eisenberg E, Greene L E. Proc Natl Acad Sci USA. 1980;77:3186–3190. doi: 10.1073/pnas.77.6.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehrer S S, Morris E P. J Biol Chem. 1982;257:8073–8080. [PubMed] [Google Scholar]
- 3.Lehrer S S. J Muscle Res Cell Motil. 1994;15:232–236. doi: 10.1007/BF00123476. [DOI] [PubMed] [Google Scholar]
- 4.Swartz D R, Moss R L, Greaser M L. Biophys J. 1996;71:1891–1904. doi: 10.1016/S0006-3495(96)79388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams D L J, Greene L E. Biochemistry. 1983;22:2770–2774. doi: 10.1021/bi00280a027. [DOI] [PubMed] [Google Scholar]
- 6.Eaton B L, Kominz D R, Eisenberg E. Biochemistry. 1975;14:2718–2725. doi: 10.1021/bi00683a025. [DOI] [PubMed] [Google Scholar]
- 7.McKillop D F, Geeves M A. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehman W, Craig R, Vibert P. Nature (London) 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- 9.Vibert P, Craig R, Lehman W. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 10.Holmes K C. Biophys J. 1995;68:2S–5S. [PubMed] [Google Scholar]
- 11.Schaertl S, Lehrer S S, Geeves M A. Biochemistry. 1995;34:15890–15894. doi: 10.1021/bi00049a003. [DOI] [PubMed] [Google Scholar]
- 12.Pardee, J. D. & Spudich, J. A. (1982) Methods Enzymol.85, Pt. B, 164–181. [DOI] [PubMed]
- 13.Margossian, S. S. & Lowey, S. (1982) Methods Enzymol.85, Pt. B, 55–71. [DOI] [PubMed]
- 14.Smillie, L. B. (1982) Methods Enzymol.85, Pt. B, 234–241. [DOI] [PubMed]
- 15.Homsher E, Kim B, Bobkova A, Tobacman L S. Biophys J. 1996;70:1881–1892. doi: 10.1016/S0006-3495(96)79753-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanBuren P, Work S S, Warshaw D M. Proc Natl Acad Sci USA. 1994;91:202–205. doi: 10.1073/pnas.91.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris D E, Warshaw D M. J Biol Chem. 1993;268:14764–14768. [PubMed] [Google Scholar]
- 18.Work S S, Warshaw D M. Anal Biochem. 1992;202:275–285. doi: 10.1016/0003-2697(92)90106-h. [DOI] [PubMed] [Google Scholar]
- 19.Hill L E, Mehegan J P, Butters C A, Tobacman L S. J Biol Chem. 1992;267:16106–16113. [PubMed] [Google Scholar]
- 20.Fraser I D, Marston S B. J Biol Chem. 1995;270:7836–7841. doi: 10.1074/jbc.270.14.7836. [DOI] [PubMed] [Google Scholar]
- 21.Honda H, Kitano Y, Hatori K, Matsuno K. FEBS Lett. 1996;383:55–58. doi: 10.1016/0014-5793(96)00218-9. [DOI] [PubMed] [Google Scholar]
- 22.Uyeda T Q, Kron S J, Spudich J A. J Mol Biol. 1990;214:699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 23.Guilford W H, Dupuis D E, Kennedy G, Wu J, Patlak J B, Warshaw D M. Biophys J. 1997;72:1006–1021. doi: 10.1016/S0006-3495(97)78753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huxley A F. Prog Biophys Chem. 1957;7:255–317. [PubMed] [Google Scholar]
- 25.Wolff M R, McDonald K S, Moss R L. Circ Res. 1995;76:154–160. doi: 10.1161/01.res.76.1.154. [DOI] [PubMed] [Google Scholar]
- 26.Podolsky R J, Teichholz L E. J Physiol (London) 1970;211:19–35. doi: 10.1113/jphysiol.1970.sp009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Julian F J. Biophys J. 1969;9:547–570. doi: 10.1016/S0006-3495(69)86403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenner B. Proc Natl Acad Sci USA. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Squire J M, Morris E P. FASEB J. 1998;12:761–771. doi: 10.1096/fasebj.12.10.761. [DOI] [PubMed] [Google Scholar]
- 30.Sobieszek A. J Mol Biol. 1982;157:275–286. doi: 10.1016/0022-2836(82)90234-0. [DOI] [PubMed] [Google Scholar]
- 31.Lehman W, Vibert P, Uman P, Craig R. J Mol Biol. 1995;251:191–196. doi: 10.1006/jmbi.1995.0425. [DOI] [PubMed] [Google Scholar]
- 32.Chalovich J M, Chock P B, Eisenberg E. J Biol Chem. 1981;256:575–578. [PMC free article] [PubMed] [Google Scholar]
- 33.Chalovich J M, Greene L E, Eisenberg E. Proc Natl Acad Sci USA. 1983;80:4909–4913. doi: 10.1073/pnas.80.16.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siemankowski R F, Wiseman M O, White H D. Proc Natl Acad Sci USA. 1985;82:658–662. doi: 10.1073/pnas.82.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenfeld S S, Taylor E W. J Biol Chem. 1987;262:9994–9999. [PubMed] [Google Scholar]
- 36.Warshaw D M, Desrosiers J M, Work S S, Trybus K M. J Cell Biol. 1990;111:453–463. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trybus K M, Taylor E W. Proc Natl Acad Sci USA. 1980;77:7209–7213. doi: 10.1073/pnas.77.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams D L J, Greene L E, Eisenberg E. Biochemistry. 1988;27:6987–6993. doi: 10.1021/bi00418a048. [DOI] [PubMed] [Google Scholar]
- 39.Geeves M A, Lehrer S S. Biophys J. 1994;67:273–282. doi: 10.1016/S0006-3495(94)80478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swartz D R, Moss R L. J Biol Chem. 1992;267:20497–20506. [PubMed] [Google Scholar]