

Abstract
Proteins can be very tolerant to amino acid substitution, even within their core. Understanding the factors responsible for this behavior is of critical importance for protein engineering and design. Mutations in proteins have been quantified in terms of the changes in stability they induce. For example, guest residues in specific secondary structures have been used as probes of conformational preferences of amino acids, yielding propensity scales. Predicting these amino acid propensities would be a good test of any new potential energy functions used to mimic protein stability. We have recently developed a protein design procedure that optimizes whole sequences for a given target conformation based on the knowledge of the template backbone and on a semiempirical potential energy function. This energy function is purely physical, including steric interactions based on a Lennard-Jones potential, electrostatics based on a Coulomb potential, and hydrophobicity in the form of an environment free energy based on accessible surface area and interatomic contact areas. Sequences designed by this procedure for 10 different proteins were analyzed to extract conformational preferences for amino acids. The resulting structure-based propensity scales show significant agreements with experimental propensity scale values, both for α-helices and β-sheets. These results indicate that amino acid conformational preferences are a natural consequence of the potential energy we use. This confirms the accuracy of our potential and indicates that such preferences should not be added as a design criterion.
Predicting protein structure requires an understanding of how tertiary structure depends on the primary amino acid sequence. An intuitive approach to that problem is to explore the wealth of information in experimental structures solved at atomic resolution and stored in protein databanks such as the PDB (1). It is observed that the three-dimensional structure of a protein is hierarchical, with a local organization of the amino acids into secondary structure elements (α-helices and β-sheets), which are themselves organized in space to form the tertiary structure. The same hierarchy is used in most ab initio protein structure prediction protocols. Secondary structures are predicted first, usually based on statistical analysis of known protein structures and multiple sequence alignments. Then various close-packing arrangements of these helices and strands are tested, either systematically or by deterministic optimization tools [for a recent review, see Koehl and Levitt (2)]. It is therefore important to understand the physical basis for the correlation between sequence and the presence of an α-helix or a β-sheet in the structure. In this paper, we derive structure-based secondary-structure propensity scales for amino acids, using a physical all-atom potential energy function.
Statistical surveys of proteins of known structures (3–7) revealed that amino acids have clear conformational preferences for one type of secondary structure. The whole field of secondary structure prediction is concerned with analyzing how these preferences determine whether a sequence segment is an α-helix, a β-sheet, or neither. This has led to the development of different methods, based either directly on these statistical data (3, 4, 8, 9), on physicochemical properties of amino acids (10, 11), on multilayered neural networks (12–15), or on evolutionary information (16) and/or on multiple sequence alignments (17–19). Prediction methods reach 68% accuracy when derived from a single sequence (20) and 75% when derived from multiple sequences (21, 22). Although these figures have increased steadily over the years, the present methods are not expected to reach average prediction accuracy better than 85% (23). While there is hope that a completely new method will emerge to break this barrier, it is clear that much still needs to be learned about the forces stabilizing secondary structures.
Experimental studies of β-sheet propensities have focused on proteins [mainly the B1 domain of protein G (24–26)]. The various propensity scales derived from these experiments do not correlate well with each other (27). Plausible reasons for these differences include position and stability effects. In contrast, studies on α-helix propensities are based on short peptides and complete proteins as host molecules [for review, see Pace and Scholtz (28)]. The various scales that have been derived from these data correlate well with each other as well as with statistical scales derived from known protein structures. An attempt to rationalize these scales for α-helix propensities based on thermodynamics was developed by Luque and coworkers (29). In brief, they propose a structural parameterization of folding energetics in which the free energy of folding is expressed as a linear combination of the changes in solvent-accessible surface areas of all atoms of the molecule, following an idea originally proposed by Eisenberg and McLachlan (30). This structure-based thermodynamic analysis was performed to study the effects of a single mutation in a helix of T4 lysozyme, barnase, a synthetic leucine zipper, and a synthetic peptide, for all of which experimental data were available. The corresponding data provide a structure-based helix propensity scale, which was found to correlate well with experimental scales. In all four cases, included in their study, the mutated amino acids are at solvent-exposed locations; this minimizes the effects of the mutations on internal interactions in the native state and maximizes the effects of changes in solvent accessibility from which the free energies are derived. As such, it cannot be directly applied to study buried residues. Furthermore, because the amino acids most commonly found in β-sheets are hydrophobic, another approach is needed for studying β-sheet conformations.
Although it is clear that the local amino acid sequence determines the secondary structure of a protein, general attempts to predict amino acid preferences from their chemical structure have had limited success. Direct calculations of the effect of sequence on α-helix and β-sheet stability is difficult because of the intrinsic problem of estimating the thermodynamic stability of any structure. In this paper, we derive secondary structure preferences by changing the sequence rather than the protein conformation. Our method is derived from the sequence design strategy we have developed recently (69), in which the complete sequence of a protein is optimized based on its target backbone, and a specified amino acid composition. The design is based on a Monte Carlo minimization of the free energy of the model protein built from the sequence and the target conformation. The free energy is computed as the sum of van der Waals interactions, electrostatics, and a free energy of environment. A statistical survey of 100 designed sequences for a set of 10 proteins is used to compute the frequency of occurrence of each amino acid in an α-helix or a β-sheet, and the results are compared with existing scales. We show that protein topology and a simple physicochemical potential are enough to explain amino acid conformational preferences.
Methods
Protein Sequence Design.
A complete description of the protein design procedure has been given elsewhere (69). In brief, the program starts from the backbone, B, of a template protein structure. A random sequence, S0, is generated, based on a given amino acid composition (usually, the native composition for the chosen backbone). An all-atom model of the chimeric protein obtained by threading this sequence on the backbone B is built by using a self-consistent mean field method to position side-chains. The self-consistent mean field method is fast and was shown to generate low energy models as accurate as other side-chain modeling methods (31). The energy, E0, of this model is then computed by using our physical potential energy function. Next, a new sequence, S1, is generated by choosing two positions in S0 at random and exchanging the corresponding amino acid types. The energy, E1, of the new model derived from sequence S1 is calculated, and the change is accepted or rejected, using the classical Metropolis scheme (32): The move is accepted if a random number drawn from a uniform distribution between 0 and 1 is lower than exp [(E0 − E1)/kT]. This optimization scheme converges to a stable sequence for the target fold. Specificity for the template fold is enforced by keeping the amino acid composition constant, in accordance with the random energy model (33, 34).
Free Energy Evaluation.
The stability of a sequence S is measured by the difference ΔG(S) in free energy between its native state, N, and an unfolded state, U:
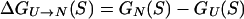 |
1 |
The total free energy, G, can be partitioned into three terms:
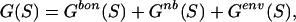 |
2 |
where Gbon(S) and Gnb(S) are the covalently bonded and nonbonded interactions, respectively, and GEnv(S) is the free energy of environment of the sequence S in the template.
The bonded interactions are local interactions and, to the first approximation, only depend on the amino acid composition of the sequence of the protein of interest. With fixed amino acid composition, the contribution of bonded interactions to ΔG is neglected.
The nonbonded interactions are described by the sum of a Lennard-Jones potential and a Coulomb potential for van der Waals and electrostatics interactions, respectively. For any conformation C,
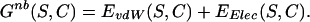 |
3 |
The Lennard-Jones potential of a protein with sequence S and conformation C is given by
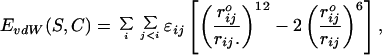 |
4 |
where the summation extends over all pairs of atom (i, j), rij is the interatomic distance between i and j, and ɛij and rijo are constants that depend on the chemical nature of atoms i and j.
The electrostatics potential energy is given by
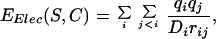 |
5 |
where qi and qj are the partial charges of i and j, respectively. The solvent plays a significant role in determining the electrostatic energy of a protein, most notably through screening of the electrostatic interactions. As a first approximation, this screening is included in the calculation by damping EElec with a distance dependent dielectric constant:
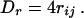 |
6 |
To account for hydrophobic interactions within a protein, as well as contacts with the solvent, Koehl and Delarue (35) introduced a free energy of environment, GEnv, which takes in account the full environment of each atom of the protein (solvent and other protein atoms):
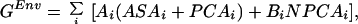 |
7 |
where the summation extends over all atoms i of the protein. ASAi is the accessible surface area of i, and PCAi and NPCAi are the surface areas of i occluded by polar and nonpolar atoms, respectively (also known as the polar and nonpolar contact areas of i). The solvent accessible area, ASAi, measures the contact with solvent, which is considered as part of polar contact area because water is intrinsically polar. Ai and Bi are surface tension factors for polar and nonpolar interactions. Let us define TASAi, the total accessible surface area of atom i in the presence of local interactions only, as
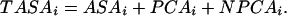 |
8 |
The free energies of a sequence, S, in the native structure, N, and in the denatured state, U, respectively, are therefore given by
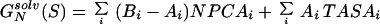 |
9 |
and
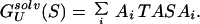 |
10 |
The coefficients Ai and Bi are derived from experimental values of transfers of amino acid analogs from n-octanol to water (36).
Checking the Specificity of the Designed Sequences.
Specificity should be a major concern of computational approaches to protein design. Simply modifying a sequence such as to optimize its stability when threaded on the template backbone may not result in a successful design unless this sequence remains incompatible with competing folds. Specificity is implicitly taken into account in our approach by maintaining the amino acid composition constant. A designed sequence can be tested for specificity by threading it on a large collection of folds: The design is considered successful if threading detects the template fold as the most probable fold for the sequence. To test our sequences, we have used threader, the fold recognition program developed by David Jones (37). Results from threader are given as a Z-score, Z(S, C), which defines how well a given fold, C, recognizes a sequence, S, compared with all other folds. Here we define the relative Z-score of sequence S as the ratio of the Z-score of S for the native conformation, C, to the Z-score of the native sequence, N, for the same native conformation, C.
Computing Amino Acid Conformational Preferences.
The conformational preference CP(j,k) of an amino acid of type j for a secondary structure k is defined as the ratio of the probability, Pj,k, of finding the j residue in secondary structure k to the probability, Pj, of finding the j amino acid anywhere in the protein sequence (4):
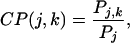 |
11 |
in which
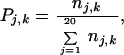 |
12 |
where nj,k is the number of residues of type j in secondary structure k, and
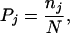 |
13 |
where nj is the number of residue of type j in all of the sequences, and N is the total number of residues.
It is worth mentioning that CP(j,k) is not a probability; it measures the bias of finding the amino acid type j in state k, compared with the average occurrence of any type of amino acid in state k. As such, CP(j,k) will take values >1 for residues that favor conformation k, and <1 otherwise.
Most of the experimental propensity scales are based on free energy differences. We will therefore report preferences as an energy-like term, using
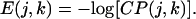 |
14 |
Identification of Secondary Structure Elements.
The positions of the secondary structure elements of each protein considered here were assigned by running stride (38) on each corresponding PDB file. Only helices and β-sheets were considered. Because it is often difficult to identify the beginning and ending residues of a secondary structure segment, we did not consider as part of the secondary structure the first and last residues identified by stride.
Test Cases.
A set of 10 different proteins was considered: the C terminal fragment of the L7/L12 ribosomal protein [PDB code 1CTF (39)]; the chymotrypsin inhibitor 2 from barley seed [PDB code 2CI2-I (40)]; the SH3 domain of the human phosphoric diester hydrolase [PDB code 2HSP (41)]; the bovine calbindin D9k [PDB code 4ICB (42)]; the DNA binding protein of the bacteriophage λ [PDB code 1LMB-3 (43)]; sperm whale myoglobin [PDB code 5MBN(44)]; green alga plastocyanin [PDB code 7PCY (45)]; the bovine pancreatic trypsin inhibitor [PDB code 5PTI (46)]; the B1 domain of protein G [PDB code 1PGB (47)]; and chicken triose phosphate isomerase [PDB code 1TIM-A (48)]. Table 1 summarizes the secondary structure contents of these proteins.
Table 1.
Secondary structure composition of the 10 proteins included in our database
| PDB ID code | Size | α-helix, % | β-sheet, % | Secondary structure, % |
|---|---|---|---|---|
| 1PGB | 56 | 27 | 43 | 70 |
| 5PTI | 58 | 14 | 24 | 38 |
| 2CI2 | 65 | 17 | 29 | 46 |
| 1CTF | 68 | 53 | 26 | 79 |
| 2HSP | 71 | 0 | 28 | 28 |
| 4ICB | 76 | 58 | 0 | 58 |
| 1LMB | 92 | 63 | 0 | 63 |
| 7PCY | 98 | 4 | 48 | 52 |
| 5MBN | 153 | 74 | 0 | 74 |
| 1TIM | 247 | 46 | 17 | 63 |
| Total | 1,154 | 36 | 18 | 54 |
Results
Conformational Preferences of Amino Acids.
Ten sequences were designed for each of the 10 proteins in our data set (Table 1), yielding a total of 100 model proteins (we exclude the native protein with its native sequence in each case). Determination of conformational preferences of each amino acid type was performed according to the procedure of Chou and Fassman (4), as described in Methods. This analysis was applied to 600 nonhomologous native protein sequences, yielding an updated set of statistical conformational preferences: Rev_C&F. A direct comparison between these updated Chou and Fasman propensities and those extracted from our designed sequences is illustrated in Fig. 1 for α-helix and β-sheet. Correlations with other propensity scales are given in Tables 2 and 3, respectively.
Figure 1.

Comparison of the amino acid secondary structure propensity scales of Chou and Fasman (4) (revised in this work) with those derived from computer designed sequences. (A) α-helix. (B) β-sheet. The position of secondary structures was computed by using stride (38), and only α-helices and β-strands are considered.
Table 2.
Comparison of α-helix propensity scales
| Amino acid | N | Design | RAN | Pace | Agadir | Rev_C&F | Luque |
|---|---|---|---|---|---|---|---|
| G | 84 | 1.24 | 0.02 | 1.00 | 1.10 | 1.03 | 0.79 |
| A | 344 | −0.04 | −0.05 | 0.00 | 0.00 | 0.00 | 0.00 |
| V | 257 | −0.06 | 0.09 | 0.61 | 0.46 | 0.58 | 0.36 |
| I | 224 | −0.26 | 0.04 | 0.41 | 0.35 | 0.45 | 0.48 |
| L | 381 | −0.38 | −0.21 | 0.21 | 0.19 | 0.12 | 0.15 |
| F | 127 | −0.01 | 0.09 | 0.54 | 0.47 | 0.51 | 0.35 |
| M | 49 | −0.09 | 0.39 | 0.24 | 0.21 | 0.18 | 0.18 |
| W | 32 | 0.21 | 0.07 | 0.49 | 0.47 | 0.48 | 0.35 |
| C | 25 | 0.57 | 0.81 | 0.68 | 0.60 | 0.72 | 0.57 |
| S | 136 | 0.15 | −0.16 | 0.50 | 0.52 | 0.70 | 0.48 |
| T | 100 | 0.39 | 0.20 | 0.66 | 0.57 | 0.71 | 0.59 |
| N | 90 | 0.25 | 0.25 | 0.65 | 0.60 | 0.67 | 0.52 |
| Q | 107 | −0.02 | −0.09 | 0.39 | 0.32 | 0.14 | 0.30 |
| H | 75 | −0.11 | −0.30 | 0.53 | 0.47 | 0.55 | 0.62 |
| Y | 101 | 0.05 | 0.16 | 0.56 | 0.62 | 0.50 | 0.46 |
| D | 148 | 0.27 | 0.08 | 0.69 | 0.59 | 0.61 | 0.47 |
| E | 392 | −0.33 | −0.13 | 0.40 | 0.34 | 0.09 | 0.37 |
| K | 114 | −0.18 | 0.02 | 0.21 | 0.06 | 0.23 | 0.11 |
| R | 419 | −0.30 | −0.04 | 0.26 | 0.15 | 0.31 | 0.15 |
| Correlation coefficients for all 19 amino acids | |||||||
| Design | 1.00 | 0.40 | 0.79 | 0.83 | 0.79 | 0.70 | |
| RAN | 0.40 | 1.00 | 0.29 | 0.24 | 0.31 | 0.19 | |
The helix propensity scales are from the following: Design, structure-based propensity scale based on computer generated sequences (this work); RAN, propensity scale based on random sequences with native amino acid composition (this work); Pace (28); AGADIR (52); Rev_C&F [revised statistical scale based on Chou and Fasman (4)]; Luque (29). N is the total number of the corresponding amino acid type in α-helices, in the pool of 100 designed sequences.
Table 3.
Comparison of β-sheet propensity scales
| Amino acid | N | Design | RAN | Minor | Minor2 | Smith | Kim | Rev_C&F |
|---|---|---|---|---|---|---|---|---|
| G | 49 | 0.76 | −0.11 | −1.20 | 1.21 | −0.85 | 0.00 | 0.14 |
| A | 139 | −0.12 | −0.22 | 0.00 | 0.00 | 0.00 | −0.35 | 0.24 |
| V | 178 | −0.70 | −0.14 | 0.82 | −0.94 | 0.17 | −0.53 | −0.49 |
| I | 140 | −0.77 | 0.20 | 1.00 | −1.25 | 0.02 | −0.56 | −0.45 |
| L | 85 | 0.15 | 0.39 | 0.51 | −0.45 | −0.24 | −0.48 | −0.16 |
| F | 84 | −0.67 | 0.32 | 0.86 | −1.08 | 0.16 | −0.55 | −0.21 |
| M | 33 | −0.71 | 0.39 | 0.72 | −0.90 | −0.02 | −0.46 | −0.01 |
| W | 17 | −0.14 | −0.20 | 0.54 | −1.04 | −0.17 | −0.48 | −0.22 |
| C | 70 | −0.63 | −0.14 | 0.52 | −0.78 | 0.08 | −0.47 | −0.07 |
| S | 12 | 1.45 | 0.29 | 0.70 | −0.87 | 0.63 | −0.39 | 0.06 |
| T | 107 | −0.70 | −0.35 | 1.10 | −1.36 | 0.83 | −0.48 | −0.29 |
| N | 13 | 1.05 | −0.17 | −0.08 | −0.52 | −0.24 | −0.38 | 0.42 |
| Q | 7 | 1.67 | 0.28 | 0.23 | −0.38 | 0.04 | −0.40 | −0.00 |
| H | 7 | 1.34 | 0.29 | 0.96 | −1.63 | 0.11 | −0.46 | 0.19 |
| Y | 49 | −0.49 | 0.04 | −0.02 | −0.37 | −0.01 | −0.50 | −0.27 |
| D | 20 | 1.12 | −0.15 | −0.94 | 0.85 | −0.10 | −0.41 | 0.42 |
| E | 41 | 0.91 | 0.07 | 0.01 | −0.23 | 0.31 | −0.41 | 0.67 |
| K | 26 | 0.29 | 0.11 | 0.45 | −0.40 | −0.43 | −0.41 | 0.31 |
| R | 81 | 0.34 | 0.06 | 0.27 | −0.35 | −0.40 | −0.44 | 0.06 |
| Correlation coefficients for all 19 amino acids | ||||||||
| Design | 1.00 | 0.18 | −0.43 | 0.35 | −0.12 | 0.46 | 0.67 | |
| RAN | 0.18 | 1.00 | 0.33 | −0.26 | −0.00 | −0.23 | −0.05 | |
| Correlation coefficients for 15 amino acids (excluding S, N, Q, and H) | ||||||||
| Design | 1.00 | −0.11 | −0.80 | 0.82 | −0.48 | 0.59 | 0.82 | |
| RAN | −0.11 | 1.00 | 0.25 | −0.18 | −0.22 | −0.27 | −0.05 | |
The β-sheet propensity scales are from the following: Design, structure-based propensity scale based on computer generated sequences (this work); RAN, propensity scale based on random sequences with native amino acid composition (this work); Minor (25); Minor2 (24); Smith (26); Kim (63); and Rev_C&F [revised statistical scale based on Chou and Fasman (4)]. The β-sheet propensity values defined in this work (Design and RAN) are compared to the five other scales, and the correlation coefficients are shown for the entire set of 19 residues (excluding P) and for a subset of 15 residues (excluding the four amino acids S, N, Q, and H, under-represented).
α-Helix propensity.
Because its existence was suggested by Pauling (49), the α-helix has been the center of most studies on protein structure and protein folding. The α-helix can be studied both as a secondary structure element of a large protein or as an isolated structural entity (50). Peptides are well suited to the experimental studies of helix propensity that measure how a given residue type affects helix stability. Such studies have not been limited to peptides. Experimental helix propensity scales have been based on data obtained from peptides, coiled-coils of α-helices, and intact proteins. Compilation of these results also has led to a peptide-specific scale [AGADIR (51)], as well as to a global scale including all data (28). Both agree well with each other and are in reasonable agreement with a structure-based propensity scale developed by Luque et al. (29) and with propensity scales derived from statistical analysis of known protein structures. We have chosen to compare our own scale with the two compiled scales of Serrano and Munoz (51) and Pace and Scholtz (28), as well as with the structure-based scale of Luque et al. (29) and the statistical scale derived from known proteins [revised Chou and Fasman scale (4); see above]. The results are given in Table 2.
The agreements between our scale and the experimental scales for α-helix propensity are good for 19 amino acids (excluding proline for lack of data in the other scales) with correlation coefficients between 0.70 and 0.83. It is worth noticing the very good correlation (0.79) between our scale and the revised Chou and Fasman scale (4) (see Fig. 1A). In energy terms, the difference observed between the best and worst helix formers in our scale is 1.6 RT, which is close to the 1 kcal/mol difference observed for all experimental scales. Our scale indicates that Ala stabilizes helices but that other residues are better helix formers (including L, E, R, and I). This differs from most other scales in which Ala has the greatest helix propensity. Myers et al. (52, 53) also observed that Ala is not the best helix former in their scale derived from experiments on a full protein.
The amino acid composition of the protein is held fixed during our design procedure. To assess the extent to which this affects our helix propensity scale, we repeated the procedure described above, using an equal number of random sequences of native amino acid composition (see Table 2, column RAN). The correlation between this baseline scale and experimental scales is poor. This clearly indicates that the correlation found between calculated and experimental propensities results from our design procedure and not from the compositional constraint.
An α-helix is stabilized both by enthalpic contributions (formation of backbone hydrogen bonds, favorable van der Waals interactions compared with a coil configuration, electrostatic interactions such as salt bridges, etc.) as well as entropic contributions (the difference between the Ala and Gly propensities can be explained by a large reduction in conformational space available when the side-chain H of a glycine is replaced by a CH3 in Ala) (54–58). Many of these effects can be accounted for by a semiempirical free energy of solvation (59). Based on these results, Luque et al. (29) have derived a structure-based scale, which correlates well with experimental scales. Hydrophobic effects alone, however, cannot explain the behavior of all amino acids: Entropic effects such as those described for Ala and Gly, and specific interactions within a protein, have to be taken into account (52, 60, 61). The structure-based propensity scale described in this study is derived from a complete physical potential, including van der Waals, electrostatics, and hydrophobic interactions. It is found to correlate better with experimental scales than with the structure-based scale of Luque et al. (29) (see Table 2).
β-Sheet propensity.
Experimental studies of β-sheet preferences have been addressed mainly in two protein model systems, a zinc-finger peptide (62), and the B1 domain of protein G (24–26). Other model systems based on homooligopeptide have proven inappropriate for the study of β-sheet propensities.
We compare our structure-based scale with these four experimental scales, as well as with the revised statistical scale of Chou and Fasman (4) in Table 3. The correlation of our structure-derived preferences with the experimental scales is poor if we include all residues except proline (no experimental data). Note that the correlation with the revised statistical scale of Chou and Fasman (4) is good, with a correlation coefficient of 0.74. Four residues have been observed with very low counts: Ser, Asn, Gln, and His, yielding very small preferences and consequently unreasonably high values for the effective energy-like values (Table 3). If these four residues are not included, the correlation between our scale and the other scales become more reasonable (correlation coefficients ranging between 0.48 and 0.82; there is a change of signs for the correlation because of different definitions of ΔΔG in the experimental studies). Chou and Fasman (4) originally found that Ala destabilizes more β-sheet than Gly whereas the revised Chou and Fasman scale shows an opposite behavior. It should be noted that all other scales identify Ala to be more stable than Gly in sheets. This was confirmed experimentally by a systematic mutation study of chymotrypsin inhibitor 2 (63). The low counts we observe are understandable for Asn, Gln, and His, which have been described as poor β-sheet stabilizers. It is not clear why Ser has low count because it is found to favor β-sheet, both experimentally and in known protein structures. Experimental scales based on the B1 domain of protein G find Thr to be the greatest stabilizer of β-sheet. In the Zinc-finger data, however, certain large hydrophobic residues (Val, Ile, Phe) have more stabilizing propensities, and, in our scale derived from designed sequences, Val and Ile favor β-sheet more than Thr. The β-sheet propensities are in fact context-dependent (63). We present results that have been averaged over several positions whereas all experimental scales correspond to one given environment. Considering these limitations, it is encouraging that we observe such good correlation.
To test the influence of a fixed amino acid composition in our design procedure, the same statistical analysis of β-sheet preferences was performed on random sequences with the same amino acid composition as the designed sequences (see Table 3, column RAN). The corresponding random scale shows poor correlation with experimental scales, for both subsets of amino acids (19 or 15). This confirms that our design procedure leads to statistically significant correlation well with experimental β-sheet propensity scales.
Hydrophobic interactions are considered to be the primary factor stabilizing β-sheet (25, 26, 64), explaining why large nonpolar amino acids have the largest sheet-forming propensities. There have been attempts to rationalize these propensity values by relating it to changes in solvent-accessible surface area, packing density, and statistical potentials based on backbone conformations (63); no significant correlation, however, could be detected. This is in contrast with helix propensities, which can be reproduced by changes in solvent accessibility (29, 65). It is therefore encouraging that we could reproduce experimental scales in a procedure based on a physical semiempirical potential, including steric and electrostatic effects, as well as environment interactions. Note that the environment free energy included here is based on solvent accessible surface area as well as interatomic contact areas within the protein.
Secondary Structures Affect Protein Design.
For each protein in our data set, we computed the average distance between the designed sequences and the native sequence by using sequence identity as a metric, and we compared that number with the secondary structure content of the protein, SSC, as measured by stride (38). Results are shown in Fig. 2. Though the correlation is weak (0.62), there is a clear trend. Sequences designed on protein templates with high levels of secondary structure have greater similarities to the native sequences. The same trend is observed when we compare the designed sequences among themselves, i.e., the average sequence identity over the (10 × 9)/2 = 45 different pairs of sequences designed for a given protein correlates positively (0.63) with the secondary structure content (result not shown). Both results indicate that residues involved in an α-helix or a β-sheet are more constrained than loop residues and are consequently less prone to mutation. In a protein, the amino acids most sensitive to substitution are located in the buried, rigid parts of the structure whereas changes on the surface generally have little effect (65, 66). For the 10 proteins in our data set, the average solvent accessibility of residues in helices, strands, and coils are 20, 30, and 43%, respectively [accessible surface area were computed with the program environ (35)]. Residues in proteins with low secondary structure contents are therefore more exposed to solvent on average, which would explain the correlation we observed here.
Figure 2.

(A) The average similarity in sequence between 10 designed sequences and the native sequence for their target backbone is plotted versus the secondary structure content of the protein. The straight line represents a least square fit to the data. The corresponding Pearson correlation coefficient is 0.62. (B) For each protein, the specificity of the designed sequences was tested by using threader, a fold recognition program. The score of the native fold is computed for each designed sequence, SD, and for the native sequence, SN. The ratio ZR = SD/SN defines a relative Z-score: when ZR is close to 1, the designed sequence is as specific as the native sequence. The average ZR over all designed sequences for a given protein is plotted versus the secondary structure content of the protein. The straight line represents a least square fit to the data. The corresponding Pearson correlation coefficient is 0.82.
In Fig. 2B, we report the relative Z-score, ZR, averaged over all designed sequences versus the secondary structure content, SSC, of the protein. Interestingly, 〈ZR〉 and SSC are highly correlated (0.82). There are at least two plausible reasons for this correlation: (i) the way we include specificity in the design procedure is less efficient at low secondary structure content, or (ii) threader itself is not as reliable under these conditions. These two reasons are obviously not exclusive and both are considered to be valid.
Conclusion
Proteins can be very tolerant to amino acid substitution, even within their core (65). Furthermore, the response to the same type of mutation can vary significantly, depending on its position in the structure of the protein. Understanding the factors responsible for these behaviors is consequently of crucial importance for protein engineering and design. Mutations in protein have been quantified in terms of the changes in stability they induce. For example, guest residues in specific secondary structures have been used as probes of conformational preferences of amino acids, yielding propensity scales (67, 68). Predicting amino acid propensities is therefore a good test of any new potential energy functions designed to mimic protein stability. Our protein design procedure uses a semiempirical physical potential, which includes steric interactions, electrostatics, and an environment free energy. The sequences designed by this procedure were used to derive conformational preferences for amino acids. The resulting structure-based propensity scales show significant agreements with experimental ΔG values, both for α-helices and β-sheets. Our results indicate that amino acid conformational preferences should be a natural consequence of the sequence design procedure rather than an input to such a program.
Acknowledgments
This work was supported by Department of Energy Grant DE-FG03-95ER62135. It was carried out while P.K. was on leave of absence from the Centre National de la Recherche Scientifique (Strasbourg, France), partially funded by a long term fellowship from the Union Internationale Contre le Cancer (Geneva).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bernstein F C, Koetzle T F, Williams G, Meyer D J, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 2.Koehl P, Levitt M. Nat Struct Biol. 1999;6:108–111. doi: 10.1038/5794. [DOI] [PubMed] [Google Scholar]
- 3.Nagano K. J Mol Biol. 1973;75:401–420. doi: 10.1016/0022-2836(73)90030-2. [DOI] [PubMed] [Google Scholar]
- 4.Chou P Y, Fasman G D. Biochemistry. 1974;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka S, Scheraga H A. Macromolecules. 1976;9:142–159. doi: 10.1021/ma60049a026. [DOI] [PubMed] [Google Scholar]
- 6.Chou P Y, Fasman G D. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 7.Levitt M. Biochemistry. 1978;17:4277–4284. doi: 10.1021/bi00613a026. [DOI] [PubMed] [Google Scholar]
- 8.Garnier J, Osguthorpe D, Robson B. J Mol Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- 9.Gibrat J, Garnier J, Robson B. J Mol Biol. 1987;198:425–443. doi: 10.1016/0022-2836(87)90292-0. [DOI] [PubMed] [Google Scholar]
- 10.Lim V. J Mol Biol. 1974;88:857–872. doi: 10.1016/0022-2836(74)90404-5. [DOI] [PubMed] [Google Scholar]
- 11.Ptitsyn O, Finkelstein A. Biopolymers. 1983;22:15–25. doi: 10.1002/bip.360220105. [DOI] [PubMed] [Google Scholar]
- 12.Bohr H, Bohr J, Brunak S, Cotterill R M, Lautrup B, Norskov L, Olsen O H, Petersen S B. FEBS Lett. 1988;241:223–228. doi: 10.1016/0014-5793(88)81066-4. [DOI] [PubMed] [Google Scholar]
- 13.Qian N, Sejnowski T. J Mol Biol. 1988;202:865–884. doi: 10.1016/0022-2836(88)90564-5. [DOI] [PubMed] [Google Scholar]
- 14.Holley H, Karplus M. Proc Natl Acad Sci USA. 1989;86:152–156. doi: 10.1073/pnas.86.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bossa F, Pascarella S. Comput Appl Biosci. 1990;5:319–320. doi: 10.1093/bioinformatics/5.4.319. [DOI] [PubMed] [Google Scholar]
- 16.Goldman N, Thorne J, Jones D. J Mol Biol. 1996;263:196–208. doi: 10.1006/jmbi.1996.0569. [DOI] [PubMed] [Google Scholar]
- 17.Maxfield F, Scheraga H. Biochemistry. 1979;18:697–704. doi: 10.1021/bi00571a023. [DOI] [PubMed] [Google Scholar]
- 18.Zvelebil M, Barton G, Taylor W, Sternberg M. J Mol Biol. 1987;195:957–961. doi: 10.1016/0022-2836(87)90501-8. [DOI] [PubMed] [Google Scholar]
- 19.Salamov A, Solovyev V. J Mol Biol. 1995;247:11–15. doi: 10.1006/jmbi.1994.0116. [DOI] [PubMed] [Google Scholar]
- 20.Yi T, Lander E. J Mol Biol. 1993;232:1117–1129. doi: 10.1006/jmbi.1993.1464. [DOI] [PubMed] [Google Scholar]
- 21.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 22.Frishman D, Argos P. Proteins. 1997;27:329–335. doi: 10.1002/(sici)1097-0134(199703)27:3<329::aid-prot1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Frishman D, Argos P. Fold Des. 1997;2:159–162. doi: 10.1016/S1359-0278(97)00022-9. [DOI] [PubMed] [Google Scholar]
- 24.Minor D L, Kim P S. Nature (London) 1994;371:264–267. doi: 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- 25.Minor D L, Kim P S. Nature (London) 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 26.Smith C K, Withka J M, Regan L. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 27.Sotomatsuniwa T, Ogino A. THEOCHEM. 1997;419:155–160. [Google Scholar]
- 28.Pace C N, Scholtz J M. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luque I, Mayorga O L, Freire E. Biochemistry. 1996;35:13681–13688. doi: 10.1021/bi961319s. [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg D, McLachlan A. Nature (London) 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 31.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 32.Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 33.Shakhnovich E I, Gutin A M. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shakhnovich E I, Gutin A M. Protein Eng. 1993;6:793–800. doi: 10.1093/protein/6.8.793. [DOI] [PubMed] [Google Scholar]
- 35.Koehl P, Delarue M. Proteins. 1994;20:264–278. doi: 10.1002/prot.340200307. [DOI] [PubMed] [Google Scholar]
- 36.Fauchere J-L, Pliska V. Eur J Med Chem-Chim Ther. 1983;18:369–375. [Google Scholar]
- 37.Jones D T, Taylor W R, Thornton J M. Nature (London) 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 38.Frishman D, Argos P. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 39.Leijonmarck M, Liljas A. J Mol Biol. 1987;195:555–580. doi: 10.1016/0022-2836(87)90183-5. [DOI] [PubMed] [Google Scholar]
- 40.McPhalen C A, James M N G. Biochemistry. 1987;26:261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 41.Kohda D, Hatanaka H, Odaka M, Mandiyan V, Ullrich A, Schlessinger J, Inagaki F. Cell. 1993;72:953–960. doi: 10.1016/0092-8674(93)90583-c. [DOI] [PubMed] [Google Scholar]
- 42.Svensson L A, Thulin E, Forsen S. J Mol Biol. 1992;223:601–606. doi: 10.1016/0022-2836(92)90976-q. [DOI] [PubMed] [Google Scholar]
- 43.Beamer L J, Pabo C O. J Mol Biol. 1992;227:177–196. doi: 10.1016/0022-2836(92)90690-l. [DOI] [PubMed] [Google Scholar]
- 44.Takano T. J Mol Biol. 1977;110:569–584. doi: 10.1016/s0022-2836(77)80112-5. [DOI] [PubMed] [Google Scholar]
- 45.Collyer C A, Guss J M, Sugimura Y, Yoshizaki F, Freeman H C. J Mol Biol. 1990;211:617–632. doi: 10.1016/0022-2836(90)90269-R. [DOI] [PubMed] [Google Scholar]
- 46.Wlodawer A, Walter J, Huber R, Sjolin L. J Mol Biol. 1984;180:301–329. doi: 10.1016/s0022-2836(84)80006-6. [DOI] [PubMed] [Google Scholar]
- 47.Gallagher T, Alexander P, Bryan P, Gilliland G L. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
- 48.Banner D W, Bloomer A C, Petsko G A, Phillips D C, Pogson C I, Wilson I A, Corran P H, Furth A J, Milman J D, Offord R E, et al. Nature (London) 1975;255:609–614. doi: 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- 49.Pauling L, Corey R, Branson H. Proc Nat Acad Sci USA. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown J, Klee W. Biochemistry. 1971;10:470–476. doi: 10.1021/bi00779a019. [DOI] [PubMed] [Google Scholar]
- 51.Munoz V, Serrano L. J Mol Biol. 1995;245:275–296. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]
- 52.Myers J K, Pace C N, Scholtz J M. Proc Natl Acad Sci USA. 1997;94:2833–2837. doi: 10.1073/pnas.94.7.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myers J K, Pace C N, Scholtz J M. Biochemistry. 1997;36:10923–10929. doi: 10.1021/bi9707180. [DOI] [PubMed] [Google Scholar]
- 54.Nemethy G, Leach S, Scheraga H. J Phys Chem. 1966;70:998–1004. [Google Scholar]
- 55.Wang J, Purisima E O. J Am Chem Soc. 1996;118:995–1001. [Google Scholar]
- 56.Yang A S, Honig G. J Mol Biol. 1995;252:351–365. doi: 10.1006/jmbi.1995.0502. [DOI] [PubMed] [Google Scholar]
- 57.Vila J A, Ripoll D R, Villegas M E, Vorobjev Y N, Scheraga H A. Biophys J. 1998;75:2637–2646. doi: 10.1016/S0006-3495(98)77709-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rohl C A, Baldwin R L. Methods Enzymol. 1998;295:1–26. doi: 10.1016/s0076-6879(98)95032-7. [DOI] [PubMed] [Google Scholar]
- 59.Blaber M, Zhang X J, Lindstrom J D, Pepiot S D, Baase W A, Matthews B W. J Mol Biol. 1994;235:600–624. doi: 10.1006/jmbi.1994.1016. [DOI] [PubMed] [Google Scholar]
- 60.Blaber M, Zhang X J, Matthews B W. Science. 1993;260:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- 61.Vila J A, Williams R L, Grant J A, Wojcik J, Scheraga H A. Proc Natl Acad Sci USA. 1992;89:7821–7825. doi: 10.1073/pnas.89.16.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim C W A, Berg J M. Nature (London) 1993;362:267–270. doi: 10.1038/362267a0. [DOI] [PubMed] [Google Scholar]
- 63.Otzen D, Fersht A. Biochemistry. 1995;34:5718–5724. doi: 10.1021/bi00017a003. [DOI] [PubMed] [Google Scholar]
- 64.Yang A, Honig B. J Mol Biol. 1995;252:366–376. doi: 10.1006/jmbi.1995.0503. [DOI] [PubMed] [Google Scholar]
- 65.Matthews B W. Curr Opin Struct Biol. 1993;3:589–593. [Google Scholar]
- 66.Rennell D, Bouvier S E, Hardy L W, Poteete A R. J Mol Biol. 1991;222:67–87. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 67.Scheraga H A. Pure Appl Chem. 1978;50:315–324. doi: 10.1351/pac196715030503. [DOI] [PubMed] [Google Scholar]
- 68.Sueki M, Lee S, Powers S P, Denton J B, Konishi Y, Scheraga H A. Macromolecules. 1984;17:148–155. [Google Scholar]
- 69.Koehl, P. & Levitt, M. (1999) J. Mol. Biol., in press.