

Abstract
The contribution of several individual ribozyme⋅substrate base pairs to binding and catalysis has been investigated using hammerhead ribozyme substrates that were truncated at their 3′ or 5′ ends. The base pairs at positions 1.1–2.1 and 15.2–16.2, which flank the conserved core, each contribute 104-fold in the chemical step, without affecting substrate binding. In contrast, base pairs distal to the core contribute to substrate binding but have no effect on the chemical step. These results suggest a “fraying model” in which each ribozyme⋅substrate helix can exist in either an unpaired (“open”) state or a helical (“closed”) state, with the closed state required for catalysis. The base pairs directly adjacent to the conserved core contribute to catalysis by allowing the closed state to form. Once the number of base pairs is sufficient to ensure that the closed helical state predominates, additional residues provide stabilization of the helix, and therefore increase binding, but have no further effect on the chemical step. Remarkably, the >5 kcal/mol free energy contribution to catalysis from each of the internal base pairs is considerably greater than the free energy expected for formation of a base pair. It is suggested that this unusually large energetic contribution arises because free energy that is typically lost in constraining residues within a base pair is expressed in the transition state, where it is used for positioning. This extends the concept of “intrinsic binding energy” from protein to RNA enzymes, suggesting that intrinsic binding energy is a fundamental feature of biological catalysis.
A fundamental challenge in enzymology is to understand how transition state stabilization is achieved without commensurate stabilization of bound substrates. An “enzyme” that stabilizes the reaction’s ground state and transition state to the same extent provides no rate advantage, as the barrier for reaction to form products is not reduced (1). These energetic requirements can be analyzed by using the concept of “intrinsic binding energy,” introduced by Jencks (2). Intrinsic binding energy is not a molecular explanation for catalysis, but rather provides a conceptual tool for analyzing the energetics of enzyme action from structural and chemical contexts.
Intrinsic binding energy refers to the total interaction energy between substrate and enzyme functional groups. Not all of the intrinsic binding energy is expected to be observed or expressed in the ground state, because aspects of the binding process will be energetically unfavorable relative to the situation in aqueous solution. An example of this is the loss in entropy associated with fixation of the substrate in the active site. This loss reduces the binding energy expressed in the ground state, but fixing the substrate in the ground state can be used to position the substrate with respect to catalytic groups in the active site. Thus, the interactions between the enzyme and substrate that provide this intrinsic binding energy contribute to catalysis, although the amino acids involved are not those typically referred to as “catalytic residues.” Interactions that provide intrinsic binding energy that is not expressed in the ground state because of substrate desolvation, distortion, and electrostatic destabilization can also contribute to catalysis.
Ribozymes may be particularly amenable for investigations designed to uncover energetic principles of catalysis, in part because of the simplicity of duplex behavior and the systematic information available about the energetics of base-pairing interactions. Here we address the energetics of catalysis by an RNA enzyme, the hammerhead ribozyme (Fig. 1A; refs. 3–5). The contribution of several base pairs to binding and catalysis is evaluated and placed within the context of a structural model. These results allow the concept of intrinsic binding energy to be extended from protein to RNA enzymes, as do analogous recent results with the Tetrahymena group I ribozyme (10).
Figure 1.

The hammerhead ribozyme HH16 (A) and the kinetic and thermodynamic framework for its reaction (B). (A) The ribozyme is depicted with bound full-length substrate, S (empty and filled block letters). The arrow depicts the cleavage site, such that the filled block letters correspond to the 5′ product, P1, and the empty block letters correspond to the 3′ product, P2. The three helical regions and residues referred to in the text are numbered in accord with standard hammerhead nomenclature (6). The disposition of these helices and interactions of residues in the conserved core follow schematically from the x-ray crystal structure of complexes of the ribozyme with oligonucleotide substrate analogs (7, 8). The dashes refer to Watson–Crick base pairs, and dots refer to noncanonical base pairs observed. The ribozyme⋅substrate base pairs bordering the conserved core that give the large energetic effects discussed herein are boxed. (B) The reaction steps for the hammerhead ribozyme reaction (9). The ribozyme (E) cleaves its substrate, S, to give a 5′ product P1 with a 2′,3′-cyclic phosphate and a 3′ product P2 with a 5′-hydroxyl group. P1 and P2 are bound in helices III and I, respectively, as shown in A for S.
METHODS
RNA Synthesis.
Unless stated otherwise, all oligonucleotides were synthesized chemically by standard solid-phase protocols (11). Hammerhead ribozymes with single deoxyribose residues were a kind gift from L. Beigelman of Ribozyme Pharmaceuticals (Boulder, CO). Ribozymes containing all-ribose residues were generated by in vitro transcription with T7 RNA polymerase using synthetic DNA templates (Fig. 1A) (12). The substrates P1-G, P1-A, P1-C, and P1-U were prepared by ligation of oligonucleotide P1 with pGp, pAp, pCp, and pUp, respectively, using T4 RNA ligase (13). Oligonucleotides were 5′- or 3′-end-labeled, purified, and stored as previously described (9). A mixture of 5′- or 3′-end-labeled substrate oligonucleotides was generated by partial alkaline hydrolysis of the appropriately labeled full-length substrate, as previously described (14).
Kinetics.
Single-turnover reactions were performed in 50 mM Tris⋅HCl buffer, pH 7.5/10 mM MgCl2 with 200–2,000 nM HH16 ribozyme and ≈1 nM 32P-end-labeled oligonucleotide substrate or ≈1 nM of a ladder of end-labeled truncated substrates and analyzed as described previously (9, 14). All reactions were followed to completion, except for the substrates P1-N, NUC-P2, UC-P2, and C-P2, for which rate constants were obtained from initial rates, corresponding to the first 10–15% of the reaction. The observed cleavage rate constant was independent of the tested ribozyme concentration in all cases, indicating that all of the substrate was bound to the ribozyme. Thus, the reported rate constants (k2) reflect the cleavage rate constant of the ribozyme⋅substrate complex.
Nonenzymatic Cleavage.
To determine the background rate of phosphodiester bond cleavage, the oligonucleotide substrate was incubated in 50 mM Tris⋅HCl buffer, pH 7.5/10 mM MgCl2 at 25°C in the absence of ribozyme. Aliquots were removed every few days over a period of 3 weeks and were analyzed by denaturing gel electrophoresis and quantification with a PhosphorImager (Molecular Dynamics) (9). The disappearance of substrate was linear over this time, with 15% cleaved in the slowest reactions. Dividing the observed rate constant for loss of substrate by the number of phosphodiester bonds in the substrate (16 for the full-length substrate) gave the average rate constant for cleavage of a single phosphodiester bond (kuncat = 5 × 10−7 min−1). The pattern of products appeared relatively uniform, so this average value was used in the analysis herein. Control experiments showed that kuncat is not significantly affected by the buffer (50 mM Tris⋅HCl, Na⋅Hepes, Na⋅Epps, or Bistris propane⋅HCl, each at pH 7.5), but raising the pH to 8.1 (Tris⋅HCl) increased the cleavage rate ≈4-fold. Omitting Mg2+ from the reaction mixture reduced the rate of substrate cleavage by >10-fold, whereas increasing the [Mg2+] from 10 to 100 mM increased the observed cleavage rate ≈3-fold. Nonradioactive carrier substrate and the total amount of radioactive substrate did not affect the observed rate constants.
Kd Determinations.
Equilibrium dissociation constants (Kd) of oligonucleotide⋅ribozyme complexes were measured using nondenaturing gel electrophoresis, essentially as described previously (9, 15). Seven ribozyme concentrations (0.1–100 nM) and 0.05 nM 5′-end-labeled substrate were allowed to reach equilibrium at 25°C in 50 mM sodium Mes, pH 6.5/10 mM MgCl2. Incubations of 1–5 hr gave the same apparent Kd, suggesting that equilibration was complete. Ribozyme concentrations spanned the observed Kd values by at least 4-fold in both directions for each determination, and Kd values were confirmed in multiple independent determinations. The binding reactions were conducted at pH 6.5 to reduce the rate of ribozyme-catalyzed cleavage (16) because several of the tested oligonucleotides could potentially cleave during the long incubations. To determine if conversion to products during the preincubation could have altered the Kd measurements, a fraction of the same binding reaction mixture was analyzed for cleavage by denaturing polyacrylamide electrophoresis. Even after the longest incubation period (5 hr), there was <5% cleavage of each oligonucleotide. The error in Kd introduced by this small amount of cleavage is negligible compared with the experimental uncertainty in Kd values of ≈50% estimated from the range of independent determinations of the individual dissociation constants.
A ligation-equilibrium experiment was performed to determine KdP2′, the dissociation constant of the oligonucleotides P2–1 and P2–2 from the E⋅P1 complex (50 mM Tris⋅HCl, pH 7.5/10 mM MgCl2, 25°C; Fig. 1B). (P2–1 and P2–2 are 3′-truncated forms of the cleavage product P2, lacking the last one or two 3′-nucleotides, respectively.) The equilibrium extent of ligation of the two products was determined as a function of P2–2 or P2–1 concentration (20–3,000 nM) with 1 nM 5′-end-labeled P1 and 20 nM HH16, which ensured that essentially all P1 was complexed. [Kd for HH16⋅P1 is 1 nM (9).] Each mixture was allowed to react and equilibrate for 60 min. The extent of ligation as a function of the concentration of P2–1 or P2–2 was determined by denaturing gel electrophoresis and used to obtain KdP2′ for P2–1 and P2–2 dissociation from complexes with E⋅P1 and Keqint (Fig. 1B) for the substrates S-1 and S-2; the data gave good nonlinear least-squares fits (KaleidaGraph) to a single ligand binding curve.
Values of KdS for S-1 and S-2 were determined from the equilibrium constants for the individual steps in the ribozyme reaction: KdP2′ and Keqint, determined as described above, the value of KdP1 ≈ 1 nM from ref. 9 and the known equilibrium constant for the overall cleavage reaction of Keqext = 1.6 M according to the following equation (9), which was derived from Fig. 1B:
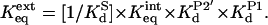 |
These determinations assume that the distal residues do not have a significant effect on the overall equilibrium, Keqext. The uncertainty inherent to the estimation of Keqext (9) contributes to the uncertainty of the absolute values of KdS but does not affect the values of relative values (KdS,rel in Table 3).
Table 3.
Determination of the effect of substrate residues distal from the cleavage site on binding to the ribozyme
| Substrate | KdP2′, nM | Keqint | KdS, nM | KdS,rel | KdS,rel predicted* |
|---|---|---|---|---|---|
| S† | 0.05 | 130 | 4 × 10−9 | (1) | (1) |
| S-1 | 100 | 60 | 3.7 × 10−6 | 1,000 | 2,000 |
| S-2 | 1600 | 200 | 2 × 10−4 | 50,000 | 60,000 |
S-1 and S-2 are substrates that are truncated by 1 and 2 residues, respectively, at their 3′ end. KdP2′, Keqint (= k2/k−2), and KdS are defined in Fig. 1B. Equilibrium constants were determined as described in Methods.
The predicted relative dissociation constants were calculated using the thermodynamic rules of nearest-neighbor contributions (20). This calculation requires information about binding contributions from only the two terminal base pairs and does not require information about the energetics of the internal base pairs that are not changed.
From ref. 9. Note that the form of S used in these experiments included a 3′ dangling pCp. The small binding effect of this overhang has been taken into account in the above comparisons. It affects the binding of S and P2 but has no effect on k2 or Keqint.
RESULTS
The Base Pairs Adjacent to the Conserved Core Contribute in the Chemical Step, but Not in the Binding Step.
The rate constant for reaction of the ribozyme⋅substrate complex, k2, was determined in single-turnover reactions for substrates truncated from the 3′ and 5′ ends. Fig. 2 compares these cleavage rates to one another and to the observed background cleavage rate of 5 × 10−7 min−1. The overall rate enhancement is ≈106-fold. Remarkably, the substrates P1-G or GUC-P2 have cleavage rates of ≈10−2 min−1, 104-fold faster than the background rate and the cleavage rate of UC-P2. This represents an energetic contribution to the chemical step of more than 5 kcal/mol (1 kcal = 4.184 kJ) from the G residue at the 3′ end of P1 or from the G residue at the 5′ end of UC-P2 [ΔΔG = −RTln(k2/kuncat) = −RTln 104 = −5.4 kcal/mol at 25°C]. As the substrate is further extended on the 3′ or 5′ end, the cleavage rate increases to a limiting rate of 1 min−1. Thus, the residues more distal to the conserved core contribute much less to the chemical step, with a contribution of <3 fold from residues beyond the third on either side (P1-GUC and ACGUC-P2).
Figure 2.

Effect of individual substrate residues on cleavage by HH16. The rate constant for cleavage of bound substrate, k2, is shown for a series of oligonucleotide substrates with nucleotides added either to the 3′-end of P1 (A) or to the 5′-end of P2 (B). (That is, the point above the leftmost G in part A represents the rate constant for reaction of P1-G, and the point that follows above the U represents the rate constant for reaction of P1-GU. Analogously, the point above the leftmost C in part B represents the rate constant for reaction of C-P2, and the point immediately to its right represents the rate constant for reaction of UC-P2.) The arrow demarks the cleavage site. The rate constants were obtained in 50 mM Tris⋅HCl, pH 7.5/10 mM MgCl2 at 25°C as described in Methods, except for the rate constant above P1 in part A; P1 does include the cleavage site, so this rate constant was taken from the cleavage rate with oligonucleotides with mismatches in helix I: P1-A and P1-C (Table 1; note that P1-U may be slightly faster due to formation of a wobble pair). The broken line represents the background cleavage rate of 5 × 10−7 min−1. For P1-GUCGU, k2 is less than that for substrates shorter or longer by one residue; this decrease has been attributed to nonproductive binding of this oligonucleotide in an alternative duplex with HH16 (14).
To determine if base pairing with the ribozyme is required for the large rate enhancements observed, the identity of the first residue adjacent to the conserved central region was varied. Only the residues that can pair with the ribozyme, G in P1-N and G in NUC-P2, give rate enhancements significantly above background (Table 1). The base pair requirement was further tested by comparing cleavage of the four P1-N substrates by a mutant ribozyme, HH16′, in which the pairing partner for N was changed from C to A (Fig. 1A, position 2.1). The complementary substrate of HH16′, P1-U, is cleaved ≈300-fold faster than the background rate, whereas its cleavage is not catalyzed by HH16 (Table 2). In contrast, HH16 cleaves its complementary substrate, P1-G, ≈100 fold faster than HH16′, which lacks the complementary residue. These comparisons and the inability of both ribozymes to catalyze the cleavage P1-A and P1-C confirm that base pairing is required for the large observed rate effects.
Table 1.
Effect of base identity on cleavage rate and binding affinity
| Oligonucleotide | k2, min−1 | krel | Kd, nM | Kdrel |
|---|---|---|---|---|
| P1 alone | 1 × 10−6* | (1) | 10 | (1) |
| P1-G | 0.005 | 10,000 | 7 | 0.7 |
| P1-A | 1 × 10−6 | 1 | — | — |
| P1-U | 5 × 10−6 | 5 | — | — |
| P1-C | 1 × 10−6 | 1 | 10 | 1 |
| P2 alone | 1 × 10−6* | (1) | 6 | (1) |
| C-P2 | 1 × 10−6 | 1 | — | — |
| UC-P2 | 2 × 10−6 | 2 | 4.5 | 0.8 |
| GUC-P2 | 0.02 | 20,000 | 6 | 1 |
| AUC-P2† | ≈1 × 10−6 | 1 | — | — |
| UUC-P2 | 0.5 × 10−6 | 0.5 | — | — |
| CUC-P2 | 0.3 × 10−6 | 0.3 | — | — |
Conditions: 10 mM MgCl2, 25°C.
These oligonucleotides do not contain the cleavage site; the nonenzymatic cleavage rate, determined as described in Methods, is listed. These values are within 2-fold of those obtained for S, and this small difference in background cleavage rates has no effect on the conclusions drawn from these data.
The oligonucleotide AUC-P2 gave a predominant cleavage band after position 16.3, consistent with binding of a fraction of the oligonucleotide in an alternative conformation (with the 5′-terminal A pairing with U15.5 of the ribozyme). The rate constant in the table corresponds to the ≈1% of the reaction that occurs at the normal cleavage position.
Table 2.
The large rate enhancement from introduction of residue 1.1 requires base pairing with the ribozyme
| Oligonucleotide |
k2, min−1
|
|
|---|---|---|
| HH16 (C2.1) | HH16′ (C2.1A) | |
| S | 1.1 | 3 × 10−5 |
| S16′ | 7 × 10−4 | 1.5 |
| P1-G | 5 × 10−3 | ≈3 × 10−5 |
| P1-U | <10−5 | 3 × 10−4 |
| P1-C | <10−5 | <10−5 |
| P1-A | <10−5 | <10−5 |
Conditions: 10 mM MgCl2, 25°C. S is the full-length oligonucleotide substrate shown in Fig. 1A that forms a matched duplex with the recognition sequence of HH16. S16′ (GGGAACGUCUACGUCGC) is the analogous oligonucleotide substrate for HH16′. The mutations in HH16′ relative to HH16 are C2.1 to A and A2.2 to U. The base at position 1.1 probed in this study has been changed from G in S16 to U in S16′ (Fig. 1A). There is also a second change: the neighboring residue of S (1.2) has been changed from U in S16 to A in S16′ to maintain complementarity with the change at position 2.2 of HH16′. This change is not expected to affect the conclusions herein because position 2.2 does not base pair with the P1-N potential substrates and because the two ribozymes give similar cleavage rates.
We next wanted to determine how substrate binding affinity is affected by these base pairs proximal to the conserved core. Native gel electrophoresis was used to determine equilibrium dissociation constants for P1, P1-G, and P1-C under conditions where cleavage is minimal. Surprisingly, the dissociation constant for all three oligonucleotides is virtually the same (Table 1), indicating that the 3′-terminal nucleotide does not affect binding, even when it is a G residue that can potentially pair with the ribozyme. Similar experiments comparing P2, UC-P2, and GUC-P2 gave analogous results (Table 1), indicating that there is no significant binding energy from the G16.2 and U16.1 residues, despite the potential of both of these residues to form base pairs with the ribozyme.¶
The equilibrium binding results demonstrate that the presence of only one complementary base pair beyond the conserved region is insufficient to favor base pair formation in the ground state complex. However, the single base pair enhances the cleavage rate by 104 fold. Thus, this hammerhead base pair provides transition state stabilization without commensurate ground state stabilization.
Base Pairs Distal from the Conserved Core Contribute to Binding, but Not to the Chemical Step.
Unlike the residues proximal to the conserved core, the distal residues have no significant effect on the chemical step (Fig. 2). This is consistent with previous observations that hammerhead ribozymes with various numbers of base pairs in helices I and III give the same cleavage rate (refs. 17 and 19 and references therein; T. Stage & O.C.U., unpublished results).
However, the distal residues do have a substantial effect on binding. The binding affinities of P1-GUCGUCC (S-1) and P1-GUCGUC (S-2) were obtained by a quantitative, though indirect, approach, because these substrates are expected to bind much too tightly to allow Kd determinations by native gel electrophoresis. This approach, detailed in Methods, is analogous to that used previously to obtain the binding affinity for S (9). Substrates truncated by one (S-1) or two (S-2) residues from the 3′ end bind 103- and 5 × 104-fold weaker than the full-length substrate (Table 3). These relative values are in good agreement with values predicted by nearest-neighbor free energy rules for base pairs in simple RNA⋅RNA duplexes (20). Thus, the distal base pairs contribute significantly to substrate binding but do not contribute in the chemical step. This is in contrast to the internal base pairs, which do not contribute to binding but are critical in the chemical step.
The 2′-Hydroxyl Groups of the Proximal Base Pairs Do Not Contribute Significantly to Catalysis.
The large rate enhancement in the chemical step upon formation of the proximal base pairs raised the possibility that there were contributions to the rate effects from interactions in addition to base pairing. 2′-Hydroxyl groups are a reasonable candidate for such additional interactions, as these groups have previously been to shown to be involved in RNA tertiary interactions and to be important recognition elements in other ribozymes (e.g., see refs. 15 and 21–23). However, removal of any single 2′-hydroxyl group from the proximal base pairs has only minor effects (<3-fold) on the cleavage rate (Table 4). In addition, removal of all of the 2′-hydroxyl groups of the HH16 substrate, except the 2′-hydroxyl group at the cleavage site, gives only a 10-fold decrease in the cleavage rate (N. Baidya and O.C.U., unpublished results). These results suggest that the 2′-hydroxyl groups of the proximal base pairs do not provide significant catalytic interactions.
Table 4.
Effect of individual 2′-hydroxyl groups on the chemical step
| Oligonucleotide | k2, min−1 | krel |
|---|---|---|
| Substrate* | ||
| All-ribose | 0.7 | (1) |
| d1.1 | 0.3 | 0.4 |
| d1.2 | 0.7 | 1.0 |
| d16.1 | 0.9 | 1.3 |
| d16.2 | 0.4 | 0.6 |
| d16.3 | 0.6 | 0.8 |
| Ribozyme | ||
| All-ribose | 1.4† | (1) |
| d2.1 | 1.8 | 1.3 |
| d15.2 | 0.5 | 0.4 |
Conditions: 25°C, 10 mM MgCl2, and pH 7.5. d refers to a single deoxyribose residue at the position indicated. The numbering is shown in Fig. 1A.
The full-length substrate depicted in Fig. 1A and single deoxyribose variants of it were used in these experiments.
The HH16 ribozyme used in the comparisons of the different substrates was synthesized by in vitro transcription, whereas a synthetic HH16 ribozyme was used for comparisons involving the deoxyriboribozymes, as these were also synthetic. A 2-fold difference in k2 was observed for the all-ribose ribozymes.
DISCUSSION
Addition of residues close to the conserved core contributes in the chemical step of the hammerhead reaction, but not in the binding step, whereas the residues tested in the periphery contribute solely to binding. These and other observations can be accounted for by the fraying model shown in Fig. 3. According to this model, both helices I and III must be formed for the ribozyme to cleave its substrate. However, when either helix is too short, inactive “open” ribozyme⋅substrate complexes form in which one of the helices is base paired but the other is not. The observed rate of cleavage can be described by Eq. 1, derived from Fig. 3, in which kchem represents the rate of cleavage from the closed complex. This description assumes that kchem is unaffected by the number of base pairs in helices I and III. While this assumption is likely to be correct to a first approximation (refs. 17 and 19; T. Stage and O.C.U., unpublished data; see also below), small helix I length effects of ≈5-fold have been reported (ref. 24; B. Cloet-d’Orval and O.C.U, unpublished data).
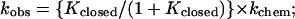 |
1 |
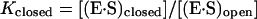 |
The fraying model provides a consistent explanation for the data in Fig. 2 and Tables 1 and 2. The 102-fold slower observed cleavage rate for P1-G and GUC-P2 compared with the maximal cleavage rate obtained with longer substrates is consistent with the open forms being favored by ≈100-fold (i.e., Kclosed ≈ 0.01). This equilibrium in turn explains why the binding affinity for P1-G is the same as that for P1: the G1.1⋅C2.1 pair is not substantially formed in the ribozyme⋅substrate complex. Analogously, the lack of substantial formation of the A15.1⋅U16.1 and G15.2⋅C16.2 base pairs with UC-P2 and GUC-P2 accounts for the similar binding affinity of GUC-P2, UC-P2, and P2. The inability to form these base pairs proximal to the conserved core is expected because the unfavorable free energy of ΔGcore ≈ +5 to +7 kcal/mol for closing the asymmetrical internal loop that constitutes the hammerhead core (9, 25) is considerably larger than the favorable free energy of ΔGG⋅C ≈ −2 kcal/mol for addition of a G⋅C base pair to a helix (20). With substrates that are one residue longer, P1-GU and CGUC-P2, the observed cleavage rates give values of Kclosed ≈ 0.1 according to Eq. 1, consistent with a further stabilization of the closed complex by the added base pair.‖ For even longer substrates, the observed cleavage rate essentially matches the maximal rate of k2, suggesting that three base pairs in helix I and four base pairs in helix III are sufficient to overcome the unfavorable free energy of ΔGcore required to close the internal loop.
Figure 3.

Fraying model with open and closed ribozyme⋅substrate helices. Formation of the closed complex is suggested to be required for the chemical step. When either helix I (below) or helix III (above) has sufficiently reduced stability, one or the other of the open complexes [(E⋅S)open] will accumulate at equilibrium.
The results with the mutant hammerhead, HH16′, are also consistent with the fraying model. The wild-type ribozyme cleaves its cognate substrate, P1-G, ≈10-fold faster than HH16′ cleaves its cognate substrate, P1-U. This presumably reflects the ability of the wild-type G⋅C base pair to provide greater stabilization to the closed complex than the A⋅U base pair of the mutant, as expected from simple base pairing energetics (20).
There are reports of substantial rate decreases and increases upon deoxyribose substitution in the hammerhead ribozyme recognition arms (refs. 7 and 26–33; N. Baidya and O.C.U., unpublished results). However, the results summarized in Table 4 indicate that the effects on the chemical step from deleting individual 2′-hydroxyl groups are small in the context of HH16, providing no indication of important functional interactions. Instead, larger rate decreases from deoxyribose substitutions (ref. 7; N. Baidya and O.C.U., unpublished results) could arise because the DNA⋅RNA duplexes, which are generally weaker than the corresponding RNA⋅RNA duplexes (20, 34), fray to give open complexes (Fig. 3). This is expected to occur with short recognition arms that allow deoxyribose substitution to weaken helix stability sufficiently to give predominantly the open complex. Increased rates upon deoxyribose substitution (28–30, 33) could arise from an increase in the rate of dissociation of the oligonucleotide product in multiple turnover experiments with ribozymes that are limited by product release, and from a decrease in the formation of alternative, nonproductive structures (35, 36).
Use of the Intrinsic Binding Energy of Base Pair Formation for Catalysis.
Though the absence of a binding effect from substrate residues proximal to the conserved core is simply explained by the fraying model (Fig. 3), the presence of the 3′ or 5′ G of P1-G or GUC-P2, respectively, results in ≈104-fold faster cleavage than that for oligonucleotides one residue shorter (Fig. 2). This was initially surprising because the rate enhancements of ≈104 correspond to ≈5 kcal/mol of stabilization energy, considerably larger than the free energy contribution that would be expected from the formation of a single base pair. For comparison, a G⋅C base pair that stacks onto a preformed helix contributes only ≈2–3 kcal/mol to helix stability (20). This section describes how these large energetic contributions from the proximal base pairs can be understood in terms of the concept of intrinsic binding energy (2).
The role of intrinsic binding energy in ribozyme catalysis is illustrated in Fig. 4, which compares the cleavage reaction with and without formation of the proximal base pair of helix I, G1.1⋅C2.1 (horizontal comparison). The reactive phosphoryl group and the conserved core are brought together by formation of this base pair, thereby aligning the phosphoryl group with respect to catalytic residues and metal ions. This base pair may be especially effective in bringing about proper transition state alignment because it is directly adjacent to the reactive phosphoryl group.
Figure 4.

Energetics of base pair formation and catalytic contributions for the G1.1⋅C2.1 base pair proximal to the conserved core. The dashes represent hypothetical interactions that are present only in the transition state. The rate constants in the figure were obtained, in accord with the fraying model, as follows: kobs represents the observed rate of cleavage of P1-G by HH16 of ≈10−2 min−1; the rate of cleavage of ≈10−6 min−1 for the reaction without the base pair formed was taken from the observed rate constants in Table 1 for oligonucleotides that cannot form this proximal base pair; and the maximal rate constant for cleavage of ≈1 min−1 (Fig. 2) is taken as the rate constant for the reaction from the closed complex (“reaction with base pair”). These values allow estimation of Keq and Keq≠ for base pair formation in the ground state and transition state, respectively, by completion of the thermodynamic cycles in the figure. The rate constants are rounded off for simplicity.
A different, but equivalent, thermodynamic description of the effect of this base pair can be made by comparing its formation in the ground state and in the transition state (Fig. 4, vertical comparison). In the ground state, the loss of entropy or motional freedom of the reactive phosphoryl group and of the conserved core region upon duplex formation lessens the observed free energy of base pair formation. In the transition state, however, the reactive phosphoryl group and conserved residues are already positioned with respect to one another. This positioning assists formation of the proximal base pair.
The analysis summarized in Fig. 4 also indicates that the proximal base pair of helix I is ≈106-fold stronger in the transition state than in the ground state (vertical comparison). This corresponds to stabilization of the base pair by 8 kcal/mol in the transition state relative to the ground state. The increase in base pair strength is presumably a manifestation of the greater order of the transition state complex relative to the ground state complex. Enzymes bind transition states more strongly than ground states in part because this greater order, imposed by partial covalent bonds that are present in the transition state but not in the ground state, results in a smaller loss of entropy upon binding (2). Stronger or additional transition state interactions with active site groups that arise from charge redistributions presumably also contribute to transition state ordering. For the hammerhead ribozyme, the formation of these transition state interactions is apparently accompanied by a large conformational rearrangement, which is depicted schematically in Fig. 4 (refs. 7 and 37; A.P., L. Beigelman, E. Scott, O.C.U. & D.H., unpublished results).
The 5 kcal/mol contribution to transition state stabilization from the G1.1⋅C2.1 proximal base pair represents a lower limit for the intrinsic binding energy of this base pair, because the slow reaction in the absence of this pair could proceed via an alternate transition state conformation. The value of 8 kcal/mol, although an approximate value, represents a direct comparison of the strength of the G1.1⋅C2.1 proximal base pair in the transition state vs. the ground state and is therefore used as a measure of the intrinsic binding energy of this base pair. This intrinsic binding energy is in excess of the free energy observed for addition of a base pair to a helix [ΔG ≈ 2–3 kcal/mol (20)] by ≈5–6 kcal/mol, but this excess energy is physically reasonable. If all of this excess free energy were used for positioning within the active site, it would correspond to a purely entropic contribution of ΔS ≈ −20 cal/mol⋅deg. (The negative sign denotes that the base pair aids in positioning.) For comparison, such a loss of entropy corresponds roughly to that from fixation about six freely rotatable bonds (2, 38). It is also worth noting that the observed change in enthalpy for addition of a single base pair to a helix is similar to or greater than the intrinsic binding energy of 8 kcal/mol [ΔH° = −6 to −14 kcal/mol (20)]. Although the above values provide some useful measures for comparison, it is important to note that observed thermodynamic parameters are typically affected by substantial contributions from solvent, ions, and rearrangements that accompany a binding process.
What is the physical basis for the intrinsic binding energy in excess of the free energy of base pair formation? Experiments with phosphorothioate substitutions have provided evidence for transition state interactions of an active site metal ion with the reactive phosphoryl group, which is adjacent to the G1.1⋅C2.1 base pair of helix I (refs. 39 and 40; A.P., D.H., E. Scott, and O.C.U., unpublished results). Additional transition state interactions presumably involve the nucleophilic 2′-hydroxyl group and/or the 5′-oxygen leaving group. These additional interactions could permit the proximal bases of helix I to be better positioned to form a base pair in the transition state than in a simple duplex. In contrast, the small rate effects from removal of the 2′-OH groups of this base pair (Table 4) and from changing the identity of these pairing partners (refs. 19 and 41–43; T. Stage and O.C.U., unpublished results) suggest that there are no base or 2′-hydroxyl-specific interactions with this base pair.
In contrast to the situation for the proximal base pair of helix I, it is harder to imagine a simple physical model that accounts for a large intrinsic binding energy of the proximal base pair of helix III, G15.2⋅C16.2, as it is not immediately adjacent to the reactive phosphoryl group. Since only small changes in the cleavage rate are observed when the identity of this base pair is changed, it is unlikely that specific contacts are made (44–46). As observed for the G1.1⋅C2.1 base pair of helix I, the effect of removing 2′-hydroxyl groups is small, providing no indication of tertiary interactions involving the 2′-hydroxyl groups of the G15.2⋅C16.2 base pair. It is possible that interactions between the phosphoryl groups of this base pair and functional groups of the core help align the active site; this is consistent with the qualitative observation obtained with a different hammerhead construct of a “thio-effect” upon replacing the pro-SP oxygen 5′ of position 15.1 and 16.2 with sulfur (47). The energetic effect could then be analogous to that described above for the proximal base pair of helix I. An alternative explanation for the large intrinsic binding energy is that the proximal base pair of helix III derives more energy from stacking interactions than in a standard duplex. This could arise, for example, if the conserved A⋅U pair had an additional partner in the transition state and formed a base triple, thereby providing a larger aromatic surface for stacking of the G15.2⋅C16.2 base pair. Finally, the large energetic contribution of the G15.2⋅C16.2 base pair of helix III could arise from the sum of several modest effects. The models described above provide a physical context to consider the intrinsic binding energy, although there are as yet no data to distinguish between these and other possibilities.
Contextual Contributions to Catalysis.
Creation of a mismatch at either base pair proximal to the conserved core decreases the rate of the chemical step when the base pair is contained within a long helix I (18), just as is observed herein with truncated substrates. However, the mismatch within the longer helix also decreases binding (18), whereas binding is unaffected with the truncated substrates. This difference is readily accounted for by the fraying model in Fig. 3. Removal or mutation of the last potential base pairing residue from a truncated substrate has no effect on binding because the substrate is already in the open state; i.e., the base pair is not formed even with the cognate substrate. In contrast, creation of a mismatch at the same position within a longer substrate weakens binding (18) because base pairing interactions that were present in the closed state are lost. Thus, depending on the context, the same interaction can, in some cases, contribute solely in the chemical step, can contribute in both the binding and chemical steps, or can contribute solely to binding; the constellation of other interactions that are present determines which reaction step is affected (see ref. 48). Hence, it is not always possible to classify a particular catalytic interaction as “uniform binding” or “specific transition state stabilization” (49). Analogous contextual energetics have been observed and characterized for the Tetrahymena group I ribozyme (G. J. Narlikar and D.H., unpublished results).
Acknowledgments
We thank G. Narlikar for stimulating discussions, L. Beigelman for a generous gift of synthetic ribozymes, T. Stage for advice, and C. Fierke, G. Joyce, J. Klinman, W. P. Jencks, and D. Knitt for critical review of the manuscript. This work was supported by National Institutes of Health Grant GM49243 to D.H. and Grant GM36944 to O.C.U.; D.H. is a Lucille P. Markey Scholar in the Biomedical Sciences; and A.P. was supported by a Human Frontier postdoctoral fellowship.
ABBREVIATIONS
- Tris
tris(hydroxymethyl)aminomethane
- Epps
N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid
- Hepes
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- Bistris propane
1,3-bis[tris(hydroxymethyl)methylamino]propane. P1 and P2 are the 5′- and 3′-cleavage products, respectively. The nomenclature used to describe the truncated substrates is as follows: “P1-” and “-P2” are used to represent truncated sequences that contain the entire sequence of the P1 and P2 products along with additional 3′ or 5′ residues, respectively (Fig. 1A). The additional nonconserved residues that base pair to the ribozyme to form helices I and III (Fig. 1A) and are varied in this study are underlined
Footnotes
It was previously suggested that binding of full-length oligonucleotides substrates to the hammerhead ribozyme is weaker than binding of products, consistent with a “substrate destabilization” mechanism (17). However, the support for this mechanism appears to have arisen from an experimental artifact in the measurement of dissociation rate constants (18). The binding data presented in Table 1 provide further evidence against thermodynamic destabilization that weakens binding of substrate relative to products.
The rate increases observed upon adding the U of P1-GU and the C of CGUC-P2 are within a factor of 3 of that predicted from the fraying model and nearest-neighbor calculations that account for the free energy contributions from the added base pair and stacking interactions (ref. 20; calculations not shown).
References
- 1.Jencks W P. Cold Spring Harbor Symp Quant Biol. 1987;52:65–73. doi: 10.1101/sqb.1987.052.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Jencks W P. Adv Enzymol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 3.Symons R H. Annu Rev Biochem. 1992;61:641–671. doi: 10.1146/annurev.bi.61.070192.003233. [DOI] [PubMed] [Google Scholar]
- 4.Long D M, Uhlenbeck O C. FASEB J. 1993;7:25–30. doi: 10.1096/fasebj.7.1.8422971. [DOI] [PubMed] [Google Scholar]
- 5.Bratty J, Chartrand P, Febeyre G, Cedergren R. Biochim Biophys Acta. 1993;1216:345–359. doi: 10.1016/0167-4781(93)90001-t. [DOI] [PubMed] [Google Scholar]
- 6.Hertel KJ, Pardi A, Uhlenbeck OC, Koizumi M, Ohtsuka E, Uesugi S, Cedergren R, Eckstein F, Gerlach W, Hodgson R, Symons RH. Nucleic Acids Res. 1992;20:3252. doi: 10.1093/nar/20.12.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pley H W, Flaherty K M, McKay D B. Nature (London) 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 8.Scott WG, Finch JT, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 9.Hertel K J, Herschlag D, Uhlenbeck O C. Biochemistry. 1994;33:3374–3385. doi: 10.1021/bi00177a031. [DOI] [PubMed] [Google Scholar]
- 10.Narlikar G J, Khosla M, Usman N, Herschlag D. Biochemistry. 1997;36:2465–2477. doi: 10.1021/bi9610820. [DOI] [PubMed] [Google Scholar]
- 11.Scaringe S A, Francklyn C, Usman N. Nucleic Acids Res. 1990;18:5433–5441. doi: 10.1093/nar/18.18.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milligan J F, Uhlenbeck O C. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 13.England T E, Uhlenbeck O C. Biochemistry. 1978;17:2069–2076. doi: 10.1021/bi00604a008. [DOI] [PubMed] [Google Scholar]
- 14.Hertel K J, Herschlag D, Uhlenbeck O C. EMBO J. 1996;15:3751–3757. [PMC free article] [PubMed] [Google Scholar]
- 15.Pyle A M, McSwiggen J A, Cech T R. Proc Natl Acad Sci USA. 1990;87:8187–8191. doi: 10.1073/pnas.87.21.8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 17.Fedor M J, Uhlenbeck O C. Biochemistry. 1992;31:12042–12054. doi: 10.1021/bi00163a012. [DOI] [PubMed] [Google Scholar]
- 18.Werner M, Uhlenbeck O C. Nucleic Acids Res. 1995;23:2092–2096. doi: 10.1093/nar/23.12.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fedor M J, Uhlenbeck O C. Proc Natl Acad Sci USA. 1990;87:1668–1672. doi: 10.1073/pnas.87.5.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freier S M, Kierzek R, Jaeger J A, Sugimoto N, Caruthers M H, Neilson T, Turner D H. Proc Natl Acad Sci USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bevilacqua P C, Turner D H. Biochemistry. 1991;30:10632–10640. doi: 10.1021/bi00108a005. [DOI] [PubMed] [Google Scholar]
- 22.Strobel S A, Cech T R. Biochemistry. 1993;32:13593–13604. doi: 10.1021/bi00212a027. [DOI] [PubMed] [Google Scholar]
- 23.Abramovitz D L, Friedman R A, Pyle A M. Science. 1996;271:1410–1413. doi: 10.1126/science.271.5254.1410. [DOI] [PubMed] [Google Scholar]
- 24.Hendry P, McCall M. Nucleic Acids Res. 1996;24:2679–2684. doi: 10.1093/nar/24.14.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hertel K. Ph.D. thesis. Boulder: University of Colorado; 1993. [Google Scholar]
- 26.Dahm S C, Uhlenbeck O C. Biochimie. 1990;72:819–823. doi: 10.1016/0300-9084(90)90191-i. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Perreault J-P, Labuda D, Usman N, Cedergren R. Biochemistry. 1990;29:11156–11160. doi: 10.1021/bi00503a002. [DOI] [PubMed] [Google Scholar]
- 28.Hendry P, McCall M J, Santiago F S, Jennings P A. Nucleic Acids Res. 1992;20:5737–5741. doi: 10.1093/nar/20.21.5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimayama T. Gene. 1994;149:41–46. doi: 10.1016/0378-1119(94)90410-3. [DOI] [PubMed] [Google Scholar]
- 30.Taylor N R, Kaplan B E, Swiderski P, Li H, Rossi J J. Nucleic Acids Res. 1992;17:4559–4565. doi: 10.1093/nar/20.17.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCall M J, Hendry P, Jennings P A. Proc Natl Acad Sci USA. 1992;89:5710–5714. doi: 10.1073/pnas.89.13.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawata S, Shimayama T, Komiyama M, Kumar P K R, Nishikawa S, Taira K. Nucleic Acids Res. 1993;21:5656–5660. doi: 10.1093/nar/21.24.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendry P, McCall M J. Nucleic Acids Res. 1995;23:3928–3936. doi: 10.1093/nar/23.19.3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M. Biochemistry. 1995;34:11211–11216. doi: 10.1021/bi00035a029. [DOI] [PubMed] [Google Scholar]
- 35.Herschlag D. J Biol Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 36.Uhlenbeck O C. RNA. 1995;1:4–6. [PMC free article] [PubMed] [Google Scholar]
- 37.McKay D B. RNA. 1996;2:395–403. [PMC free article] [PubMed] [Google Scholar]
- 38.Page M I, Jencks W P. Proc Natl Acad Sci USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dahm S C, Uhlenbeck O C. Biochemistry. 1991;30:9464–9469. doi: 10.1021/bi00103a011. [DOI] [PubMed] [Google Scholar]
- 40.Slim G, Gait M J. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forster A C, Symons R H. Cell. 1987;49:211–220. doi: 10.1016/0092-8674(87)90562-9. [DOI] [PubMed] [Google Scholar]
- 42.Pabon-Pena L M, Zhang Y, Epstein L M. Mol Cell Biol. 1991;11:6109–6115. doi: 10.1128/mcb.11.12.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Usman N, Chartrand P, Cedergren R. Biochemistry. 1992;31:5005–5009. doi: 10.1021/bi00136a013. [DOI] [PubMed] [Google Scholar]
- 44.Ruffner D E, Stormo G D, Uhlenbeck O C. Biochemistry. 1990;29:10695–10702. doi: 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- 45.Shimayama T, Nishikawa S, Taira K. Biochemistry. 1995;34:3649–3654. doi: 10.1021/bi00011a020. [DOI] [PubMed] [Google Scholar]
- 46.Zoumadakis M, Tabler M. Nucleic Acids Res. 1995;23:1192–1196. doi: 10.1093/nar/23.7.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knoll R, Bald R, Furste JP. RNA. 1997;3:132–140. [PMC free article] [PubMed] [Google Scholar]
- 48.Jencks WP. Proc Natl Acad Sci U S A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albery WJ, Knowles JR. Angew Chem Int Ed Engl. 1977;16:285–293. doi: 10.1002/anie.197702851. [DOI] [PubMed] [Google Scholar]