

Abstract
Surmises of how myosin subfragment 1 (S1) interacts with actin filaments in muscle contraction rest upon knowing the relative arrangement of the two proteins. Although there exist crystallographic structures for both S1 and actin, as well as electron microscopy data for the acto–S1 complex (AS1), modeling of this arrangement has so far only been done “by eye.” Here we report fitted AS1 structures obtained using a quantitative method that is both more objective and makes more complete use of the data. Using undistorted crystallographic results, the best-fit AS1 structure shows significant differences from that obtained by visual fitting. The best fit is produced using the F-actin model of Holmes et al. [Holmes, K. C., Popp, D., Gebhard, W. & Kabsch, W. (1990) Nature (London) 347, 44–49]. S1 residues at the AS1 interface are now found at a higher radius as well as being translated axially and rotated azimuthally. Fits using S1 plus loops missing from the crystal structure were achieved using a homology search method to predict loop structures. These improved fits favor an arrangement in which the loop at the 50- to 20-kDa domain junction of S1 is located near the N terminus of actin. Rigid-body movements of the lower 50-kDa domain, which further improve the fit, produce closure of the large 50-kDa domain cleft and bring conserved residues in the lower 50-kDa domain into an apparently appropriate orientation for close interaction with actin. This finding supports the idea that binding of ATP to AS1 at the end of the ATPase cycle disrupts the actin binding site by changing the conformation of the 50-kDa cleft of S1.
The contraction of muscle occurs when the heads of thick-filament-based myosin interact with F-actin (FA) in adjacent thin filaments. This interaction is preceded by the hydrolysis of MgATP by detached myosin heads. The crystallography of actin (1, 2), the generation of a plausible model for FA (3–6), and the crystallography of the myosin head (subfragment 1; S1) (7) have led to the visual “docking” of S1 on FA using these structures and electron microscopy (EM) data (8, 9). This docking led to the first detailed model of the acto–S1 (AS1) interface in the “rigor” (no-ATP) state.
Here we report the quantitative docking of S1 on FA using an extension of an approach used to study FA structure (5). In this method quantitative fitting is done computationally in reciprocal space rather than visually in real space. Thus the fitting is sensitive to all of the electron density in the EM data, not just the more readily visualized surface. Reciprocal space fitting also allows the removal of the least-well determined portion of the AS1 Fourier–Bessel transform—the equator. This removal accentuates higher spatial frequencies making the fitting more sensitive to small changes in the model. Our model resembles, but also significantly differs from, that obtained visually. These differences have consequences for the understanding of the molecular mechanism of muscle contraction.
METHODS
Fitting Procedure.
The fitting of AS1 model structures to the cryo-EM data of Milligan and colleagues (8) was achieved by minimizing a goodness-of-fit parameter (R) computed from reciprocal-space amplitudes and phases (5). The orientation of S1 relative to a model FA filament was varied exhaustively. Using known helical parameters, the Fourier–Bessel transform, F(R,Θ = −π/2,Z) (10), of a model AS1 was computed.§ This transform was then compared with the experimental transform using,
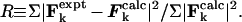 |
Here the sums are over points on layer-lines characteristic of helical Fourier transforms.
Searches for the best fit were performed by systematically varying the relative orientation of S1 and FA by rigid-body movements from an (arbitrary) starting orientation. The six search parameters used for defining the orientation of FA and S1 were as follows: the three Euler angles (α,β,γ), which rotate S1 about its center of mass (c.m.); θact and zact, which position FA azimuthally and longitudinally relative to S1; and rs1, which positions S1 radially. For each set of model parameters a transform was calculated which was phase shifted to optimally match the experimental data. The best-fitting set of parameters from an initial search (4° and 0.4 nm steps) was used as a starting point for the next search at a reduced step size. Parameter step sizes were twice decreased 2-fold to final values of 0.1 nm in translation and 1° in rotation (2,187 parameter sets). To ensure that the minimum was not on a parameter boundary, the entire procedure was then begun afresh at the current best-fitting orientation and repeated until the improvement in R between successive runs was less than 0.0002. This method was found to be quite robust in that the final best-fitting parameters were independent of their starting values.
To account for the diminution of the experimental amplitudes with increasing reciprocal space dimension s (s ≡ [R2 + Z2]½), resulting from limited resolution, calculated amplitudes were modulated by an isotropic low-pass Gaussian filter. In most cases a “temperature factor” (B value) of 25 nm2 was employed; however, the precise value of B did not significantly affect the orientations or conclusions drawn here.
Data Employed in Fitting.
The vertebrate AS1 cryo-EM data used in fitting were identical to those used for visual docking (8) except that the equator was omitted. EM data points from the first 21 layer lines (54/25 helix), extending to a resolution of 2.7 nm, were included. An electron density threshold was set at 175–250% of the theoretical volume to suppress noise (5).
As for the visual docking (8), no adjustments to the data were made to compensate for the effects of the contrast transfer function (ctf). At the current resolution phases are unaffected by the ctf; however, at very low s amplitudes are underemphasized. In this region of the transform the equator’s amplitude is dominant. Thus, elimination of the equator removes the data most severely affected by the ctf. Attempts to compensate for residual ctf effects were made (11) to the data over the s range of 0.133–0.357 nm−1. No significant improvement in R was observed, and the resultant orientations were not significantly different.
We used the nucleotide-free crystallographic structure of chicken S1 for fitting. The S1 used in the EM data came from chymotryptic S1 that was devoid of light chain 2 (LC2). We found that omission of the (denuded) heavy chain in the region of LC2 (residues 803–842) consistently improved the fits. This effect is consistent with the idea that this (single α-helix) portion of the heavy chain becomes disordered when devoid of stabilizing LC2. For this reason these residues were also omitted in the searches.
RESULTS
Fits Using the Unaltered Nucleotide-Free S1 Structure.
For initial fitting we used the unaltered crystal structure of S1 (7) and various models of the FA filament based on fits to x-ray fiber diffraction patterns. We initially assumed, as did Rayment et al. (8), that conformational changes occurring in S1 and FA, when forming a complex, were sufficiently small to be neglected. Initial fits also neglected the relatively small mass missing from the S1 crystallographic structure (6.7%).
Table 1 shows R factors for the visually derived AS1 model (8) and our search results. A significant decrease in R was observed when the positions of S1 and FA were systematically altered to improve the fit. An examination of the parameters shows that for the Lorenz et al. FA model, there is an increase in rs1 of 0.2 nm, a rotation of 2° in θact, and an increase in zact of 0.2 nm. Also, there are changes in the angular orientation of S1 as seen in changes in the Euler angles. The result of these changes on the juxtaposition of residues at the AS1 interface will be discussed below.
Table 1.
Fits to acto–(chymo)S1 cryo-EM data using the unaltered Rayment et al. (8) S1 structure
| FA model employed | R factor* (α, β, γ, θact, zact, rs1) | d144-543,† nm | dz144-543,‡ nm | Pro-543 radius, nm | d333-408,§ nm | Val-408 radius, nm | d96-571,¶ nm |
|---|---|---|---|---|---|---|---|
| Rayment et al. [Lorenz et al. FA] | 0.101 (0, 0, 0, 0, 0, 0) | 0.56 | 0.41 | 3.17 | 0.48 | 3.19 | 0.74 |
| Rayment et al. without actin residues 38-55 | 0.094 (0, 0, 0, 0, 0, 0) | ||||||
| Rayment et al. refined | 0.074 (42, −10, −39, 2, 0.2, 0.2) | 0.69 | −0.11 | 3.47 | 0.84 | 3.59 | 1.07 |
| Rayment et al. refined without residues 38-55 | 0.070 (39, −9, −37, 2, 0.2, 0.2) | 0.68 | −0.05 | 3.48 | 0.84 | 3.66 | 1.05 |
| Holmes et al. | 0.065 | 0.90 | 0 | 3.54 | 1.17 | 3.69 | 1.44 |
| Holmes et al. without residues 38-55 | 0.063 | 0.87 | 0.05 | 3.51 | 1.12 | 3.65 | 1.43 |
| Tirion et al. | 0.066 | 0.88 | 0.20 | 3.62 | 1.02 | 3.75 | 1.63 |
| Tirion et al. without residues 38-55 | 0.065 | 0.74 | 0.14 | 3.51 | 0.88 | 3.64 | 1.56 |
All angles are in degrees and distances are in nm. The z direction is downward in Fig. 2.
Distance between Ala-144 of actin and Pro-543 of the adjacent S1.
Z distance between Ala-144 of actin and Pro-543 of the adjacent S1.
Distance between Pro-333 of actin and Val-408 of the adjacent S1.
Distance between Val-96 of an actin monomer and Ala-571 of S1 one monomer up the long-pitch helix.
Because the structure of FA has not been determined at atomic resolution, other FA models, in addition to that of Lorenz et al. (which was used in both visual docking studies), were examined. The Lorenz et al. FA was obtained by refining the original Holmes et al. FA against high-resolution x-ray fiber-diffraction amplitudes. However, the original Holmes et al. FA was almost identical to that found (5) to give the best fit to FA cryo-EM data (12) as well as to other EM data. Consistent with these fits, we find here that a significantly lower R is obtained for AS1 fits when the original Holmes et al. FA is used. Furthermore, the recently published FA model based on a normal-mode refinement (6) also gives a somewhat better fit for AS1 than does the Lorenz et al. FA¶.
A notable common feature of these improved fits is that residues near the primary actin binding site of S1 are at a higher radius from the helical axis than was found previously. For example, the radius of Pro-543 increases by 0.3–0.45 nm whereas the radius of Val-408, which is positioned axially at the opposite end of the AS1 interface, increases by 0.4–0.56 nm. This slight difference is caused by a rotation of S1 about its long axis relative to the visual fitting. The increase in S1 radius at the AS1 interface reduces clashing between FA and S1 interface residues associated with the earlier AS1 model. It is noteworthy that residues further removed from the interface remain close to their original radii; for example, Glu-802 at the C terminus has virtually the same helical radius (11.9 nm) in the visual and quantitative fits.
In all FA models considered S1 is shifted axially and azimuthally from its position relative to FA in the visual fitting. For example, alignment of AS1 models (0.26 nm rms) using actin residues 70–372 shows that Pro-543 has rotated azimuthally by ≈4° between the visual fit and the fit using the Holmes et al. FA. Here, the axial distance between Ala-144 of FA and Pro-543 of S1 (dz144–543) increases by 0.35–0.41 nm so that S1 is positioned nearer to subdomain 2 of the neighboring actin subunit. For these distances, as well as for helical radii, the greatest changes from the visual fit are generally seen for the FA structures that best fit the AS1 data. This strengthens the significance of the changes.
The Lorenz et al. FA exhibits the greatest fractional decrease in R when the “DNase loop” (residues 38–55) is deleted. Likewise, in our fitting of FA cryo-EM data (5) this loop, manually repositioned prior to fitting of the x-ray data, was a surprisingly strong determinant of the goodness-of-fit to the FA data (12). Omission of the DNase loop in either the Tirion et al. or Holmes et al. FA produced less improvement in the fit to AS1 data. This result suggests that this loop, which appears to be largely disordered in conventionally prepared FA (5) and in crystallography of G-actin not complexed to DNase (2), may have a structure in the AS1 complex that is more similar to the loop found in actin-DNase than that found in FA.
Fits Using the S1 Structure with Added Loops.
In our work on FA, we found that R could be quite sensitive to small loops which extend away from the protein surface (5). For S1 defined by chymotryptic digestion, most of the mass (6% of the total) missing from the crystallographic structure arises from such loops that are presumably disordered in the S1 crystals. We have estimated the effect that such loops would have on R of various AS1 models, using S1 loops generated by a homology-search structure-prediction method (13). Typically 18–20 structures were predicted for each missing loop in S1. Predicted structures for the short loops of S1 are qualitatively similar, but there is considerable heterogeneity in the predictions of the large Gly-627 to Phe-646 loop, which spans the 50- to 20-kDa domain junction (14). This loop is of particular importance as it has been implicated in FA binding by various biochemical methods (15–18).
Inclusion of any of the 19 Gly-627 to Phe-646 loops improved the fit. When R was plotted as a function of the position (d) of the c.m. of the loop across the FA binding “face” of S1 (Fig. 1) there was a significant decrease in R with increasing d. This decrease was independent both of the FA employed and of the presence of the actin DNase loop (not shown). Difference electron-density maps (not shown) calculated between the resultant best-fitting structure without the loop and the experimental data showed a clear peak in the loop region thus verifying the validity of the fitting procedure.
Figure 1.

Results of searches using S1 with the missing loop (Gly-627 → Phe-646) joining 50-kDa and 20-kDa domains supplied from results of homology searches. Plotted on the abscissa is the left-to-right position of the c.m. of the trial loop when S1 is viewed end-on looking roughly at the actin interface region (toward the C terminus of the heavy chain). Smaller loops, which exhibit much less heterogeneity, were not varied. Fits with different 50- to 20-kDa loops (▪) are shown for the Lorenz et al. (- - -) and Holmes et al. (——) FA models. Horizontal lines (□) are the best fits without added loops.
The three dimensional structure of the best fitting AS1 with all loops is shown in Fig. 2A. The best-fitting Gly-627 to Phe-646 loop, which is largely planar and ring-like, protrudes into actin between the actin N terminus residues (near Glu-4), the loop containing Ala-22, and (above) Gly-342. The plane of the loop is roughly normal to the helix axis. Although the position and shape of this loop are approximate, its geometry allows the hydrophobic residues and charged residues of actin and S1 in this region to interact without major rearrangement. Other models of this loop tend to interact with FA less intimately or not at all.
Figure 2.

Stereoviews of fitted AS1 complex. (A) Best fit obtained using Holmes et al. FA and the unaltered crystallographic S1 structure viewed in the same orientation as figure 3b of ref. 8. FA (green with right-angle fold, Tyr-337 → Met-355, in gold) is on the left hand side with its central axis parallel to and located close to the left hand edge of the box. The FA polarity is such that the Z band would be located below the bottom of the figure. S1 extends toward and beyond the right hand edge of the figure. The S1 heavy chain is yellow except for the lower jaw (residues 467–601), which is red, and predicted loops, which are cyan. The 627–646 loop is marked with an asterisk. LC is colored blue. (B) Comparison of best fit with unaltered S1 (yellow heavy chain, cyan LC1) with improved fit in which the lower jaw is reoriented (red heavy chain, blue LC1). (C and D) Detailed views of AS1 interface rotated 180° about filament axis from A and B such that FA is now on right hand side. Colors as for A except that the lower jaw region of S1 is white whereas the helix–loop–helix region (516–558) is red. (C) Original visual alignment (8). (D) Refined fit using modified S1 as in B. Note that here the helix–loop–helix region of S1 has become more closely associated with the right-angled fold of FA. This figure was generated using molscript (19).
Fig. 2A also serves to illustrate other features of fits done without added loops. Whereas the addition of the S1 loops can decrease R significantly, the resultant positions of residues at the AS1 interface are little changed (see Table 2). This is also true for DNase loop removal. However, compared with the visual fit, there are significant changes. The U-shaped helix–loop–helix structure (helix from Gly-516 to Phe-542, loop from Pro-543 to Thr-546, helix from Asp-547 to His-558) at the base of the lower 50-kDa domain of S1 (red in Fig. 2A) is always closer to a right-angle fold in subdomain-1 of actin (helix from Tyr-337 to Ser-348, bend at Leu-349–Ser-350, and helix from Thr-350 to Met-355), as well as to the N terminus of actin, than surmised from the visual docking.
Table 2.
Representative fits to AS1 cryo-EM data S1 with loops and altered lower 50-kDa domain
| FA model | R factor (α, β, γ, θact, zact, rs1) | d144-543, nm | dz144-543, nm | Pro-543 radius, nm | d333-408, nm | Val-408 radius, nm | d96-571, nm |
|---|---|---|---|---|---|---|---|
| Lorenz et al. FA with best loops and without actin residues 38-55 | 0.056 (36, −9, −35, 1, 0.2, 0.2) | 0.725 | −0.02 | 3.52 | 0.84 | 3.66 | 1.0 |
| Lorenz et al. with best loops, without actin residues 38-55 and with S1 lower jaw rotated | 0.053 | 1.36 | −0.56 | 4.02 | 0.85 | 3.66 | 1.23 |
| Holmes et al. with best loops and without actin residues 38-55 | 0.052 | 0.92 | 0.14 | 3.56 | 1.08 | 3.70 | 1.44 |
| Homes et al. with best loops, without actin residues 38-55 and with S1 lower jaw rotated and translated (0.25 nm) | 0.047 | 1.23 | −0.33 | 3.80 | 1.08 | 3.72 | 1.37 |
Fits with Altered S1 Structures.
The visual fitting (8, 9) has suggested (20) that when S1 binds to FA, the upper and lower 50-kDa domains of S1 may move, bringing conserved, exposed hydrophobic residues on the inner surfaces of these “jaws” into closer proximity. In the present work, we examined R when moving a 40-residue deletion though the S1 sequence (data not shown), as for FA (21). We obtained a broad minimum in R when these deletions were centered near residue 480.‖ This minimum appears to be related to the hypothesized jaw closure. In addition to closing by rotation, it has been postulated (20) that the lower jaw (residues 467–601) is translated toward actin, and away from the reactive cysteine (SH697, SH707) residues. Because a detailed model of the jaw closure is not yet available, we have tested this and other similar possibilities by investigating several relative movements of the jaws. In one, AS1 was refined with the upper portion of S1 (Asp-4–Ala-465) translated relative to the lower portion (Gly-466–Glu-802 + LC3) with the jaw opening held fixed, and also closed by 10°. In another, AS1 was refined with the lower jaw visually moved toward the upper jaw with the aim of optimizing jaw closure. Neither of these changes significantly improved R.
In a more fruitful approach, the lower jaw alone was systematically translated and reoriented. The lower jaw was translated in 0.25 nm steps along an axis roughly parallel to the remainder of S1. At each step this domain was wobbled about an origin located at the Cα of Gly-466 until no further improvement in R resulted. The overall position of this new S1 was then refined again. For the Holmes et al. FA these movements resulted in a very broad minimum in R, centered in the range 0.25–0.5 nm, as the lower jaw was translated. Here, R dropped from 0.052 to 0.047—approximately a 10% improvement over the best fit using unmodified S1, and over a 100% improvement from the original visual docking. The resulting structure is shown overlaid with the refined alignment using undistorted S1 (Fig. 2B): it is apparent that these two alignments are quite close with the greatest departure located in the lower 50-kDa domain. The broad minimum in the deletion plot near residue 480 (mentioned above) was eliminated in this alignment in agreement with the notion that it arose due to a conformational change of S1 on binding to FA. However, only for the cases of no translation (R = 0.048) and 0.25 nm translation (R = 0.047, Fig. 2D) was the structure of these modified S1’s clearly more “closed” than the crystallographic S1. In these cases the end of the conserved 475–508 helix proximal to actin “scissored” toward the upper jaw so that there was a shortening of the Cα–Cα chords between conserved hydrophobic residues on opposing jaws (e.g. Leu-269 → Phe-475; Leu-269 → Phe-480). This closed structure bore some resemblance to the structures of S1 catalytic domains with bound nucleotide analogs: S1Dc⋅MgADP⋅MgAlF4 (20) and S1Dc⋅MgADP⋅VO4 (22) (where S1Dc is the catalytic domain of Dictyostelium discoideum myosin II). However, no outward movement of the base of the lower jaw was found; substitution of the lower jaw from S1Dc⋅MgADP⋅VO4 visually aligned with the S1 upper 50-kDa domain, upon refinement gave at best R = 0.052 when using the Holmes et al. FA. When the lower jaw was translated by more than 0.25 nm, the most significant change was a reorientation of the portion of the 475–508 helix proximal to FA away from the viewer (when looking through the jaws side-on). Here there was very significant clashing at the AS1 interface.
The improvement to fits by such alterations of the lower jaw is a general feature of the fitting. Similar fractional improvements in R and similar overall orientations were obtained with Lorenz et al. FA (Table 2) when using either the Rayment et al. orientation or refined orientation as starting points, and also with or without the DNase loop (data not shown). The improvement seen when altering the structure of the lower jaw was not affected by inclusion of the loop connecting jaws.
The solvent-inaccessible surface area of the new primary AS1 interaction site (Fig. 2D) with the 50- to 20-kDa loop is 10.1 nm2. Without this loop the area is 7.5 nm2, which is somewhat smaller than the original visual-docking value of 9.4 nm2. These areas are comparable to what has been observed experimentally (23), and formation of an interface of this magnitude could account for the large decrease in free energy that occurs upon formation of the acto-myosin bond (24).
DISCUSSION
In the visually docked AS1 structure (8), clashing at the AS1 interface was observed. It was suggested that these clashes could be relieved either by moving the myosin molecule “a few angstroms” away from FA or by the closing of the narrow cleft separating lower and upper 50-kDa domains. The latter possibility was favored as S1 in the former “would no longer be able to be contained within the envelope of the reconstruction.” The method used herein, which eliminates the (poorly determined) equator and thus the surface envelope, produced models with less clashing without internal rearrangement of S1. However, some lower jaw rearrangement further improves the fits and reduces clashing. Such closure harmonizes with the recent crystallography of S1Dc with MgADP⋅AlF4 and MgADP⋅VO4, which suggests that in the transition state, and also probably in the subsequent ADP·Pi state (25), the γ phosphate of ATP interacts with residues near Gly-466 (chicken) and in doing so moves the lower jaw and the remainder of S1 relative to the upper 50-kDa domain.
A recent phylogenetic study of all known members of the myosin family (26) reveals surprisingly little residue conservation in the putative FA-binding regions. In the “stereo-specific” (U-shaped helix–loop–helix) binding region (Gly-516 → His-558) only Leu-525, Glu-527, Gly-531, and Leu-536 are highly (>90%) conserved. Two residues, Pro-543 and Lys-553, are well conserved (83–84%) and Pro-543 is highly conserved in myosin IIs. Mutational studies of residues near the highly conserved residues (27, 28) support involvement of this region in FA binding. In the visual docking (Fig. 2C) the most highly conserved residues are rather far from FA. However, in the refined models here, these residues are now closer to FA. For example, the actin Leu-349 to S1 Leu-536 Cα–Cα distance decreases from 1.0 nm in the visual fitting to 0.78 nm in the refined structure. With a lower jaw cleft closure and translation of 0.5 nm, this distance decreases to 0.66 nm. The Cα–Cα distance between FA Leu-349 and S1 Gly-531 drops from 1.43 nm in the visual fitting to 0.83 nm after refinement. In addition to the highly conserved Gly-531 and Leu-536, residues 532, 533, and 535 are hydrophobic in all known myosins (26). Thus, for all myosins there is a cluster of hydrophobic residues between 531 and 536 that could well be interacting with the lower portion of the hydrophobic-rich right-angle fold in actin (Fig. 2, B and D). In addition to (or perhaps instead of) interactions of these residues in the first helix of the helix–loop–helix motif, there could also be interactions of the same portion of the actin right-angle fold with hydrophobic residues in the (doubled-back) second helix (Fig. 2D). Cope et al. (26) found residues 550 and 554 to be almost always (>97%) hydrophobic. The Cα–Cα distance from Leu-349 to residue 550 and from Phe-352 to residue 554 are both here near 0.9 nm whereas in the visual fitting they are both about 1.5 nm. Thus the residues in either or perhaps both helices could be oriented to interact with FA by the conformational change of the lower jaw occurring at the time of ATP hydrolysis and could perhaps undergo a further change upon product release. (Local conformational changes would likely be required to expose buried hydrophobic residues for interaction.) It seems likely, as previously suggested (8, 20), that the binding of ATP to S1 in AS1 would promote movement of the jaws and thereby disrupt the myosin binding site on FA.
It has been proposed (8) that residues in the Arg-405–Lys-415 loop of S1 interact with FA near Pro-332. Residue 405 is almost exclusively a (positively charged) arginine or a lysine and thus this residue, and possibly an exposed hydrophobic residue normally found at position 402, are the most likely candidates for interaction with FA in this otherwise poorly conserved region (26). In the present work we find that Val-408 is always found at a higher radius (Tables 1 and 2) and nearer to the actin N terminus (data not shown) than found in the visual docking. However, examination of distances from the end of the Arg-405 side chain to the side chains of the nearest negatively charged residues (Asp-25 and Glu-334) indicates that in both the visual and quantitative fits significant atomic rearrangement are required to bring these side chains close enough to allow interaction. It may be that this coulombic “weak” interaction occurs transiently during an early stage of the docking of S1 on FA.
The visual docking has suggested that residues in the S1 Lys-567 → His-578 loop might interact with residues Tyr-91 → Glu-100 and form a secondary binding site on a second actin below the primary binding site. In particular, the positively charged residues Lys-572 and Lys-574 might interact with negatively charged Glu-99 and Glu-100. However, there appears to be little conservation of these S1 residues. Further, in the present work the Cα–Cα distance between Ala-571 and Val-96 has increased from 0.74 nm to as much as 1.6 nm (Tables 1 and 2). For Holmes et al. FA the Ala-571 → Glu-99 distance increases from 1.19 to 1.49 nm. If the lower jaw position is optimized, this distance increases to 1.9 nm. Thus, rigor AS1 interactions in this region seem unlikely. We suggested above that the binding of the DNase loop of an adjacent actin to the stereo-specific helix–loop–helix motif (Fig. 2 B and D) could supplant the originally proposed secondary binding site. There are (tropomyosin-dependent) cross-linking data supporting this suggestion (29). Actin residue 43, which is always hydrophobic (30), may interact with S1 residue 554 (>97% hydrophobic).
The refined AS1 structure has implications for thin filament-linked regulation. The most extensive interactions in AS1 appear to occur between the S1 stereo-specific site and actin near residues 335–372. Recent evidence suggests that this site is covered by tropomyosin in relaxed vertebrate thin filaments (31); thus regulation of its interaction with S1 may well occur by steric blocking. The putative blocking of other FA sites seems less clear. In particular actin residues 144–148 and 332–334 which also appear somewhat covered by tropomyosin are further from S1 in the refined AS1 structure and thus may not be involved in direct interaction with S1. Conversely, the N-terminal region of actin is now close to the S1 50- to 20-kDa loop, but not to the relaxed tropomyosin position. On this basis this interaction may be involved in unregulated weak S1 binding.
In summary, quantitative fitting procedures generate a new model for the AS1 rigor complex that, while broadly similar to the visually determined model, improves the R factor by 94%. Here the AS1 interface is significantly rearranged bringing a number of conserved hydrophobic residues into close proximity. The loop between the 50- to 20-kDa junction, which is missing in the crystal structure, is placed near the N terminus of actin. Further improvement in the fits, obtained by partial closure of the 50- to 20-kDa cleft, indicates that the reversal of this closure may be involved in the nucleotide-induced dissociation of S1 from FA.
Acknowledgments
We thank Drs. R. Cooke, J. Cope, R. Fletterick, R. Milligan, M. Morales, and I. Rayment for helpful conversations. We acknowledge use of the University of California, San Francisco, Computer Graphics Lab. This work was supported by National Science Foundation (MCB-9404705) and National Institutes of Health (R01-AR39710 and P01-AR42895) grants to R.A.M.
ABBREVIATIONS
- AS1
acto-S1
- c.m.
center of mass
- FA
F-actin
- LC
light chain
- S1
myosin subfragment one
- S1Dc
catalytic domain of Dictyostelium discoideum myosin II
- EM
electron microscopy
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY (reference 1ALM).
(R,Θ,Z) are cylindrical coordinates in reciprocal space. Corresponding real-space coordinates are (r,θ,z).
This FA also generates a better fit to the FA cryo-EM data (12) than does the Lorenz et al. FA, but this fit is also less good than the Holmes et al. FA (unpublished observations).
An additional minimum centered near residue 65 may be related to varying orientations of the β-barrel (8) in AS1. In extensive computations we find that deletion of residues 38–78 generally reduces the R factor by about 10%, lowers S1 on actin by 0.1 nm or less, and changes the angular coordinates only slightly. Thus the differences in orientation of S1 on FA with and without these residues are negligible. However, as there is no compelling reason for deletion of this feature of S1 crystallography, we report here only results obtained with these residues included.
References
- 1.Kabsch W, Mannherz H G, Suck D, Pai E F, Holmes K C. Nature (London) 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin P J, Gooch J T, Mannherz H G, Weeds A G. Nature (London) 1993;364:685–692. doi: 10.1038/364685a0. [DOI] [PubMed] [Google Scholar]
- 3.Holmes K C, Popp D, Gebhard W, Kabsch W. Nature (London) 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz M, Popp D, Holmes K C. J Mol Biol. 1993;234:826–836. doi: 10.1006/jmbi.1993.1628. [DOI] [PubMed] [Google Scholar]
- 5.Mendelson R A, Morris E. J Mol Biol. 1994;240:138–154. doi: 10.1006/jmbi.1994.1428. [DOI] [PubMed] [Google Scholar]
- 6.Tirion M M, ben-Avraham D, Lorenz M, Holmes K C. Biophys J. 1995;68:5–12. doi: 10.1016/S0006-3495(95)80156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rayment I, Rypniewski W R, Schmidt-Base K, Smith R, Tomchick D R, Benning M M, Winkelmann D A, Wesenberg G, Holden H M. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 8.Rayment I, Holden H M, Whittaker M, Yohn C B, Lorenz M, Holmes K C, Milligan R A. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 9.Schroder R R, Manstein D J, Jahn W, Holden H, Rayment I, Holmes K C, Spudich J A. Nature (London) 1993;364:171–174. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- 10.Cochran W, Crick F H C, Vand V. Acta Crystallogr. 1952;5:581–586. [Google Scholar]
- 11.Toyoshima C, Unwin N. Ultramicroscopy. 1988;25:279–292. doi: 10.1016/0304-3991(88)90003-4. [DOI] [PubMed] [Google Scholar]
- 12.Milligan R A, Whittaker M, Safer D. Nature (London) 1990;348:217–221. doi: 10.1038/348217a0. [DOI] [PubMed] [Google Scholar]
- 13.Ring C S, Kneller D G, Langridge R L, Cohen F E. J Mol Biol. 1992;224:685–699. doi: 10.1016/0022-2836(92)90553-v. [DOI] [PubMed] [Google Scholar]
- 14.Stone D B, Schneider D K, Huang Z, Mendelson R A. Biophys J. 1995;69:767–776. doi: 10.1016/S0006-3495(95)79973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mornet D, Pantel P, Audemard E, Kassab R. Biochem Biophys Res Commun. 1979;89:925–932. doi: 10.1016/0006-291x(79)91867-9. [DOI] [PubMed] [Google Scholar]
- 16.Sutoh K. Biochemistry. 1983;22:1579–1585. doi: 10.1021/bi00276a009. [DOI] [PubMed] [Google Scholar]
- 17.Chaussepied P, Morales M F. Proc Natl Acad Sci USA. 1988;85:7471–7475. doi: 10.1073/pnas.85.20.7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uyeda T Q, Ruppel K M, Spudich J A. Nature (London) 1994;368:567–569. doi: 10.1038/368567a0. [DOI] [PubMed] [Google Scholar]
- 19.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 20.Fisher A J, Smith C A, Thoden J, Smith R, Sutoh K, Holden H M, Rayment I. Biophys J. 1995;68:19S–26S. ; 27S–28S. [PMC free article] [PubMed] [Google Scholar]
- 21.Mendelson R A, Morris E. Adv Exp Med Biol. 1994;358:13–23. doi: 10.1007/978-1-4615-2578-3_2. [DOI] [PubMed] [Google Scholar]
- 22.Smith C A, Rayment I. Biochemistry. 1996;35:5404–5417. doi: 10.1021/bi952633+. [DOI] [PubMed] [Google Scholar]
- 23.Highsmith S, Duignan K, Cooke R, Cohen J. Biophys J. 1996;70:2830–2837. doi: 10.1016/S0006-3495(96)79852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horton N, Lewis M. Protein Sci. 1992;1:169–181. doi: 10.1002/pro.5560010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendelson R A, Schneider D K, Stone D B. J Mol Biol. 1996;256:1–7. doi: 10.1006/jmbi.1996.0063. [DOI] [PubMed] [Google Scholar]
- 26.Cope M, Whisstock J, Rayment I, Kendrick-Jones J. Structure. 1996;4:969–987. doi: 10.1016/s0969-2126(96)00103-7. [DOI] [PubMed] [Google Scholar]
- 27.Onishi H, Morales M F, Katoh K, Fujiwara K. Proc Natl Acad Sci USA. 1995;92:11965–11969. doi: 10.1073/pnas.92.26.11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patterson B, Spudich J A. Genetics. 1995;140:505–515. doi: 10.1093/genetics/140.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonafe N, Mathieu M, Kassab R, Chaussepied P. Biochemistry. 1994;33:2594–2603. doi: 10.1021/bi00175a031. [DOI] [PubMed] [Google Scholar]
- 30.Sheterline P, Clayton J, Sparrow J. Protein Profile. 1995;2:1–103. [PubMed] [Google Scholar]
- 31.Lehman W, Vibert P, Uman P, Craig R. J Mol Biol. 1995;251:191–196. doi: 10.1006/jmbi.1995.0425. [DOI] [PubMed] [Google Scholar]