

Abstract
Transport of peptides across the membrane of the endoplasmic reticulum for assembly with MHC class I molecules is an essential step in antigen presentation to cytotoxic T cells. This task is performed by the major histocompatibility complex-encoded transporter associated with antigen processing (TAP). Using a combinatorial approach we have analyzed the substrate specificity of human TAP at high resolution and in the absence of any given sequence context, revealing the contribution of each peptide residue in stabilizing binding to TAP. Human TAP was found to be highly selective with peptide affinities covering at least three orders of magnitude. Interestingly, the selectivity is not equally distributed over the substrate. Only the N-terminal three positions and the C-terminal residue are critical, whereas effects from other peptide positions are negligible. A major influence from the peptide backbone was uncovered by peptide scans and libraries containing d amino acids. Again, independent of peptide length, critical positions were clustered near the peptide termini. These approaches demonstrate that human TAP is selective, with residues determining the affinity located in distinct regions, and point to the role of the peptide backbone in binding to TAP. This binding mode of TAP has implications in an optimized repertoire selection and in a coevolution with the major histocompatibility complex/T cell receptor complex.
Keywords: ABC transporter, antigen presentation, antigen processing, combinatorial chemistry
Cytotoxic T lymphocytes distinguish between self and non-self by monitoring peptides presented in association with major histocompatibility complex (MHC) class I molecules on the cell surface. These peptides are believed to be generated mainly from endogenous proteins in the proteasomal pathway, and they have to cross the membrane of the endoplasmic reticulum (ER) for association with assembling class I molecules. The essential role of the heterodimeric TAP (transporter associated with antigen processing) complex in peptide translocation across the ER membrane became evident from studies with mutant cell lines that were deficient in MHC class I-dependent antigen presentation when, in transfection experiments with tap genes, cytotoxic T cell recognition could be restored (1–3). ATP-dependent peptide translocation into the ER lumen by TAP was demonstrated directly, using streptolysin O-permeabilized cells (4, 5) or microsomal membranes (6, 7). In these experiments, transported peptides were retained in the ER by trapping on MHC class I molecules or through N-linked glycosylation of peptides carrying a consensus recognition site (NXS/T). From these results more detailed questions about the immunological relevance of TAP arose: (i) does TAP put a restriction on the pool of antigenic peptides available for presentation to cytotoxic T cells, and (ii), if so, what is the peptide selectivity of TAP?
Several assays have been developed to study peptide specificity of TAP. Most results came from assays relying on trapping of transported peptides in the ER via glycosylation. Comparing the amount of glycosylated peptide recovered from sets of peptides that differed in one or more residues allowed the investigation of side chain preference and length selectivity of mouse, rat, and human TAP. In particular, the length selectivity with a lower limit of 8 residues and a less discrete upper limit of 13–24 residues could be demonstrated (8, 9), and the only significant preferences for particular amino acids were found for the peptide C terminus (10–13). So far, differences for allelic variants with respect of preference for peptide length and C-terminal residue have been reported only for rat TAP (11, 12). For human TAP, even the C-terminal residue of the peptide substrate appeared to have only a minor influence on the amount of translocated and glycosylated peptides, and human TAP thus was referred to as “nonselective” (14).
An alternative approach to decrypt TAP selectivity has made use of the fact that below room temperature, ATP-independent peptide binding to TAP occurs (15, 16). Peptide affinities of TAP can be determined as binding constants (KD), and screening of peptides is performed through competition experiments. Again, length selectivity as well as side chain preferences were studied. However, a significantly larger spectrum of affinities was found. When the same set of peptides was studied in both assay systems, similar relative affinities but notable differences in absolute values were observed (16–18). Thus, it can be suggested that glycosylation-based transport assays and binding assays are functionally indistinguishable (19). Whereas in a multistep process composed of transport, glycosylation, degradation, and export of peptides, differences between peptides might be underestimated, bimolecular binding assays offer higher resolution without the possible bias from complex kinetic analysis. Thus, we have chosen competition binding assays in combination with complex peptide libraries for detailed analysis of the peptide specificity and recognition principle of TAP. This combinatorial concept uses randomized peptide mixtures sharing one defined amino acid position. It offers the opportunity to determine effects of individual residues at a given position directly, independent from a sequence context, by comparison of stabilizing factors for particular residues, because they represent the difference of the free enthalpies ΔΔG for binding. In addition, by comparison of the totally randomized peptide mixture with the predicted highest and lowest affinity peptides, we can quantify the affinity spectrum covered by TAP. Conversely, our results can also be interpreted from the aspect of TAP structure. TAP accommodates substrates that are readily available from solid-phase synthesis, and that can be permutated by combinatorial chemistry. Therefore, these substrate libraries provide a tool to map the peptide binding site of TAP at high resolution. The binding mode of TAP should elucidate the principles underlying the selection of dominant and subdominant epitopes for antigen presentation and allow us to compare recognition principles of TAP, MHC class I molecules, and the T cell receptor in terms of coevolution and optimized repertoire selection.
MATERIALS AND METHODS
Cell Culture.
The generation of baculoviruses carrying the genes for human TAP1 and TAP2 has been described previously (7). Sf9 (Spodoptera frugiperda) cells were grown in Sf900II medium (Gibco) following standard procedures. Infection was routinely performed with a multiplicity of infection of 3–5.
Preparation of Microsomes.
Sf9 cells were harvested 60–72 h postinfection by centrifugation and washed once with PBS. The preparation of microsomes was performed as described previously (7). In brief, cells were lysed by drawing through a 26-gauge needle, and the cell lysate was loaded onto a step gradient of sucrose buffers. The turbid fraction from the interface of the 1.3-M and 2-M sucrose solutions was collected and recentrifuged. The vesicles were resuspended in PBS/1 mM DTT, snap frozen in liquid nitrogen, and stored at −80°C. The protein concentration was determined using the bicinchoninic acid assay (Pierce).
Synthesis and Characterization of Peptides and Peptide Libraries.
The synthetic peptides, peptide libraries, and peptide sublibraries were prepared by solid-phase peptide synthesis using Fmoc/tBu (fluorenylmethoxycarbonyl/tert.-butyl) chemistry. Solvents, amino acids, and coupling reagents were handled by a robot for multiple peptide synthesis (Syro, MultiSynTech, Bochum, Germany). Peptide libraries and sublibraries were synthesized on an equimolar mixture of Fmoc-l/d-amino acid-p-benzyloxybenzyl alcohol and/or Fmoc-l/d-Pro-2-chlorotrityl polystyrene resins. Fmoc-l/d amino acids were used with the following side chain protecting groups: tert.-butyl ethers for Ser, Thr, and Tyr; tert.-butyl esters for aspartate and Glu; trityl for His, Asn, and Gln; tert.-butyloxycarbonyl for Lys and Trp; and 2,2,5,7,8-pentamethylchroman-6-sulfonyl and 2,2,4,6,7-pentamethyl-dibenzofuran-5-sulfonyl for Arg.
To couple all 19 amino acids (cysteine was excluded) in randomized sequence positions (X) double reactions were performed with equimolar mixtures of Fmoc amino acids used in an equimolar ratio with respect to the coupling sites of the resins. The loading of the resin was analyzed by quantitative Fmoc determination. Resins were distributed in 30-mg aliquots (15 mmol) to filter tubes, which were positioned in the format of a microtiter plate on valve blocks. Fmoc deprotections were carried out two times, 7 min each, with 220 ml 30% piperidine in dimethylformamide (DMF). Resins were washed nine times with 300 ml DMF. Then, the coupling reagent diisopropylcarbodiimide [1.5 M in 50 ml DMF:CH2Cl2 (1:2, vol/vol)], Fmoc amino acids and Fmoc amino acid mixtures were distributed to the reaction vessels. For coupling of defined positions, 0.5 M Fmoc amino acids were dissolved with 0.5 M N-hydroxybenzotriazole in DMF. Equimolarly premixed Fmoc amino acids [0.075 M in DMF:CH2Cl2 (1:7, vol/vol)] were distributed for coupling of randomized positions. Double couplings (3 h each) were carried out in open tubes. After 2 h of coupling, diisopropylethylamine [1 M in 20 ml DMF:CH2Cl2 (1:1, vol/vol)] was added. Coupling reagents were filtered off and the resins were washed three times with DMF. The peptides and peptide mixtures were cleaved from the resins and side chains were deprotected with 1 ml trifluoroacetic acid:phenol:ethanedithiol:thioanisol (96:2:1:2, vol/wt/vol/vol). The cleavage solutions were filtered from the resins, which were washed with 0.3 ml acetic acid. Five milliliters of cold N-heptane:diethylether (1:1, vol/vol) was added to the filtrates. The precipitates (−20°C) were washed twice by sonification with N-heptane:diethylether (1:1, vol/vol) and were lyophilized from acetic acid:water:tert.-butyl alcohol (1:10:50, vol/vol/vol).
The identity of the peptides with defined sequences was confirmed by electrospray mass spectrometry, and the purity was determined by HPLC to be higher than 94%. The amino acid composition of the peptide libraries and sublibraries was determined by pool sequencing (20), electrospray mass spectrometry (21), and amino acid analysis. Deviations from equimolar representation of the amino acids in randomized sequence positions were found to be within the error limits of the analytical methods (22). Peptide concentrations were determined using the bicinchoninic acid assay (Pierce).
Peptides were iodinated as described previously (7). In brief, the reaction was performed with 15 nmol of peptide and 1 mCi Na125I (1 Ci = 37 GBq) using the chloramine T method. Free iodine was removed by gel filtration through a Sephadex G10 (Pharmacia) column. The specific activities for the peptides were 40–65 cpm/fmol.
Peptide Binding and Competition Assays.
The assays were performed as filtration assays, using a multiple filtration manifold (Multiscreen Assay System, Millipore) that is capable of handling 96 samples in parallel. For each reaction performed in the microplate format, unlabeled competitor peptide in the appropriate concentration, or buffer for the controls, was added to microsome suspension in assay buffer (PBS with 1 mg/ml dialyzed bovine serum albumin, 1 mM DTT, 2 mM MgCl2, and final concentration of microsomal protein adjusted to 60 μg/ml and homogenized by drawing through a 23-gauge needle) with radiolabeled peptide *RRYQKSTEL* (final concentration 100 nM or the respective concentrations in the saturation-binding experiments) to a final volume of 120 μl. After incubation on ice for 5 min, the reactions were transferred onto a multiscreen filter (Durapore membrane, 0.65-μm pore size, Millipore). Unbound peptide was removed by washing with 1 ml ice-cold PBS. The filters were air-dried, and bound radioactivity was quantified by γ counting. The peptides examined were not modified or degraded during the assay. The amount of bound, labeled peptide was corrected for unspecific binding, which was determined in the presence of a 1,000-fold molar excess of unlabeled peptide RRYQKSTEL. The data set was fitted by the competition function
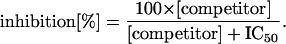 |
1 |
The concentration needed for 50% inhibition was determined from at least 10 measurements for each peptide. The assay was repeated when the correlation coefficient R for the curve-fit was below 0.95. Values were standardized for the total random peptide X9, for RRYQKSTEL, or for the corresponding all-L peptides tested in each set of experiments and given as IC50/IC50;ref.. Affinity constants for particular peptides were calculated from the affinity constant of the reporter peptide (KD,RRYQKSTEL), determined by Scatchard analysis, and from the ratio of IC50 values by
 |
2 |
The selectivity at each peptide position is given by the variance of the IC50 values of the 19 sublibraries X8O:
 |
3 |
RESULTS
The Peptide Binding Motif of TAP as Revealed with Combinatorial Peptide Libraries.
To establish the competition binding assay as a means for screening combinatorial peptide libraries, we performed a saturation binding experiment with radiolabeled peptide *RRYQKSTEL* in the presence of varying concentrations of unlabeled competitor RRYQKSTEL. This peptide displayed an affinity of KD = 146 nM (Fig. 1A; Table 1), and unlabeled peptide acted as a competitive inhibitor, not affecting saturation, but with a rising apparent KD value (Table 1), as can also be seen from the double-reciprocal plot (Fig. 1B). Thus, it is possible to determine the affinity constant (KD) for a particular peptide to TAP from the molar excess of the peptide that is needed for 50% inhibition of binding of a radiolabeled reporter peptide (IC50) and the affinity of the reporter peptide (see Materials and Methods). This eliminates the need to radiolabel each peptide as it would be necessary for direct determination of KD values by Scatchard analysis.
Figure 1.

Mechanism of peptide binding to TAP. Saturation binding to TAP was performed with radiolabeled peptide *RRYQKSTEL* in the absence (•) and presence of the same unlabeled peptide (○, 30 nM; □, 100 nM; ◊, 300 nM; ▵, 1 μM). Error bars represent ±SD (n = 3). Saturation can easily be inspected in the linear plot (A), while in the double-reciprocal plot (B) the linear fits intersect on the y axis, indicating competitive inhibition.
Table 1.
Comparison of apparent affinity constants and saturation values for binding of the radiolabeled peptide *RRYQKSTEL* to human TAP in the presence of the same unlabeled peptide as competitor
| Competitor concentration, nM | Apparent affinity constant KD, nM | Saturation binding, 1/1,000 cpm |
|---|---|---|
| 0 | 146 | 20.3 |
| 30 | 170 | 20.2 |
| 100 | 333 | 22.2 |
| 300 | 535 | 18.8 |
| 1,000 | 1,680 | 18.7 |
To get an overall estimate for the selectivity of TAP, a randomized nonapeptide library (X9) was used as competitor for binding of *RRYQKSTEL*. The averaged KD = 2.4 μM (IC50/IC50;ref. = 17 ± 0.5) indicates that TAP strongly selects for high-affinity peptides like the reporter. We then investigated the side chain preferences at each individual position of a nonamer peptide by comparing X9 with 19 × 9 = 171 sublibraries (termed X8O) containing one defined position (Fig. 2A). The IC50 values, as plotted on a log scale, give stabilizing and destabilizing effects of a particular amino acid residue in terms of Gibbs free enthalpy ΔΔG. For a given position, the affinities of 19 nonapeptide sublibraries can vary over two orders of magnitude, indicating the selectivity spectrum of TAP.
Figure 2.

(A) Stabilizing factors for the side chains of all possible nonapeptide sublibraries. Affinities of 9 × 19 = 171 peptide sublibraries X8O (differing in O, which represents 19 defined amino acid residues in positions 1 to 9) were determined as molar excess needed to achieve 50% inhibition of specific binding of the reporter peptide *RRYQKSTEL*. These IC50 values were standardized for the total random library X9 as a reference (IC50/IC50;ref.; Left). The difference of free enthalpies ΔΔG are given (Right) to allow for easy inspection of stabilizing and destabilizing effects. Error bars indicate SD (n = 10). Amino acids are given in signal-letter code. (B) Variance of the stabilizing factors at different positions (see Material and Methods).
Most interestingly, the effects from the peptide side chains were not equally distributed over the entire peptide (Fig. 2B). An N-terminal region from positions 1 to 3 as well as the C-terminal residue were found to be critical, whereas positions 4 to 8 appeared to be nearly nonselective, as if there were almost no physical interaction of the peptide side chains with TAP at these positions. Aromatic, hydrophobic, and positively charged amino acid residues—in particular, Phe, Tyr, Arg, and Leu—were preferred at the C terminus, whereas the negatively charged residues Asp and Glu as well as Asn and Ser had a strongly destabilizing effect. Sequence positions 1 to 3 showed nearly the same variance in the observed IC50 values, albeit with no prevailing pattern for side chains. Negatively charged residues were disfavored at positions 1 and 3, while positively charged residues and, in particular, Arg were stabilizing at the first three positions. Also, aromatic and hydrophobic residues were favored, in particular at position 2 and 3. The most pronounced effect found for any residue was the strongly disfavored Pro at position 2.
Although our approach was not designed to identify the best binding sequence, which might also depend on effects from neighboring amino acids, we could predict high- and low-affinity peptides. For the two extremes tested, we found KD = 137 nM (IC50/IC50;ref. = 0.94; Table 2) for NRYMPRIRY and no detectable affinity (KD > 1 mM) for EPGNTWDED, as compared with the averaged KD = 2.4 μM (IC50/IC50;ref. = 17) for the randomized peptide library X9. This not only demonstrates the resolution achievable with the combinatorial concept but further illustrates the range of TAP selectivity.
Table 2.
Affinity constants of selected peptides
| Peptide | KD, μM |
|---|---|
| RRYQKSTEL | 0.146 |
| RRYNASTEL | 0.473 |
| rrynastel (inverse peptide) | >1,000 |
| letsanyrr (retro-inverse peptide) | >1,000 |
| Acetyl-RRYNASTEL | 82.3 |
| RRYNASTEL-amide | 101.6 |
| Acetyl-RRYNASTEL-amide | >1,000 |
| GRYNASTEL | 3.74 |
| Methyl-GRYNASTEL | 100.0 |
| NRYMPRIRY | 0.137 |
| EPGNTWDED | >1,000 |
Amino acid residues are given in single-letter code. d amino acids are indicated by small letters.
Major Contribution to Binding Is from the Peptide Backbone.
The broad-length selectivity of TAP as well as the lack of dominant anchoring residues as compared with MHC class I molecules led us to speculate that the mechanism of peptide binding must be different from that for class I molecules. We thus interpreted the destabilizing effect of Pro at position 2 as a hint for the relevance of the peptide backbone. Involvement of the peptide backbone has already been implied during photo-crosslinking experiments when peptide affinity was lost with N- and C-terminally blocked peptides (17). We can confirm this effect by using the peptide RRYNASTEL (Table 2), and for the N terminus, we can attribute it to disruption of hydrogen bonds and not to a missing positive charge, because N-terminal methylation of the peptide decreases the affinity for TAP 30-fold.
To investigate our backbone hypothesis further, we synthesized a peptide with the same sequence as our reporter peptide, containing only d amino acids (inverse peptide), as well as an all-d peptide with inverted sequence (retro inverse). Both peptides showed no detectable binding to TAP (KD > 1 mM, Table 2). In the inverse peptide, the backbone can adopt a conformation identical to that of the all-l peptide, only the side chains protrude in different directions, possibly causing steric hindrance (Fig. 3). In contrast, the result from the retro-inverse peptide is more interesting: conformations are possible, where the side chains of the all-l and the retro-inverse peptide have the same relative positions, but C and N atoms of the backbone are reverted, disrupting hypothetical hydrogen bonds to the backbone. For a more detailed analysis of this phenomenon we synthesized two sets of nonapeptides (a defined epitope and randomized library) containing only one d-amino acid position. Strikingly, the destabilizing effect from d-residues is not equally distributed over the peptide (Fig. 4): an N-terminal region and the very C terminus were most strongly affected, with the N-terminal effect more pronounced. In between and particularly at position 6, peptide binding was hardly influenced by this modification of the backbone. It should be noted that the effects of exchanging a residue to its d-equivalent were much stronger than those observed with any of the X8O sublibraries containing only l amino acids. Interestingly, the regional distribution matches that of side chain effects.
Figure 3.

All-l, inverse-, and retro-inverse peptide. An all-l amino acid dipeptide is depicted in extended conformation with all-d peptides (having the same or a reverted amino acid sequence) to compare relative positions of side chains and backbone atoms.
Figure 4.

Affinities for peptides derived from the nonapeptide RRYNASTEL (A), the randomized nonapeptide library (X9; B), the 15-mer peptide REIRRYNASTELLIR (C), or the randomized undecapeptide libraries (X12; D) containing one d-amino acid position. Affinities were determined as IC50 values standardized for the corresponding all-l peptides or all-l peptide libraries (X9 or X12). Error bars indicate SD (n = 10).
Human TAP preferentially recognizes peptides 8–16 residues in length (15). To generalize the binding motif for peptides of different lengths, we used longer peptides with one d position. For synthetic reasons, we chose a randomized dodecamer peptide library and the pentadecamer sequence REIRRYNASTELLIR with known high affinity to TAP (16). Again, d amino acids showed the strongest effects at positions 1 to 3. Also, the effects we found for the C termini were present but not so pronounced for the two libraries tested. Because of the random situation, a disfavored backbone conformation could be compensated by the overall pool of sequences or negatively charged residues could replace the charged C terminus. For the longer peptides, however, the region with virtually no influence from a d amino acid extended over up to 10 residues.
DISCUSSION
Studies comparing the transport efficiencies of TAP for different peptides have been hampered by the fact that peptides without an ER-based retention (N-glycosylation or MHC class I binding) were undetectable, possibly because of a so far uncharacterized ATP-dependent peptide export from the ER (6). We have applied a combinatorial approach, including complex peptide libraries, in a direct, bimolecular assay system. Selectivity and mechanism of peptide binding to the human TAP complex were explored independently of sequence context and free from the possible bias of peptide retention and export kinetics. We conclude that human TAP exhibits a marked selectivity with an IC50/IC50;ref. value of 17 for the total random nonapeptide library, as opposed to a hypothetical value of 1 for a totally nonselective system and the value of about 200 observed for the highly selective MHC class I molecules H-2Kb in a similar approach with a random octapeptide library (23). This is further emphasized by the difference of affinity for two peptides predicted from our results to have high and low affinity, covering more than three orders of magnitude. In addition, we found differences for individual amino acid substitutions at a single position of up to 80-fold, whereas in retention-based assays typically only differences of 2- to 3-fold were found. Our results are thus in contrast to experiments that rely on glycosylation for peptide retention in the ER and that state human TAP to be rather nonselective. We have three possible explanations for this. (i) In previous studies peptides were mostly derived from MHC class I-binding motifs and thus are likely to have high affinity for TAP; as a consequence, effects of particular residues in an environment of other preferred residues might be underestimated. (ii) Typically, the peptide concentrations used in these studies (600 nM) were significantly above the expected KM value for most of the MHC class I-motif derived peptides. Thus, there is no linear correlation between KM and observed transport rate at these concentrations, leading to compensation of differences in KM at the transport rate level. (iii) Assays are typically run over 10–20 min with up to 5% of input peptide glycosylated, possibly leading to depletion of activated sugars for glycosylation and consequently further equalizing KM differences. All three factors might contribute to an underestimation of the differences in peptide affinities. Thus, the direct bimolecular assay used in this study resulted in much higher resolution, and we can conclude that peptide translocation by TAP is indeed a process of marked selectivity.
Apart from the more drastic differences with respect to absolute values, we found good correlation of the relative order of side chain affinities with the data available for human TAP. The C terminus has been studied most extensively in other assay systems, with preferred hydrophobic and positively charged residues, moderate affinity of polar residues, and Asp as well as Gly clearly disfavored. This is identical to the pattern we found, and, interestingly, it also matches with Phe, Tyr, Val, Ile, and Leu as the anchor positions for the human MHC class I alleles examined so far. Furthermore, it has been speculated that the C-terminal position is the only position with any preference for human TAP (11). Indeed, the C terminus in our system also proved to have the most pronounced effect, with the important exception of Pro at position 2. Positions 1 to 3 are also important, although on average they displayed only 60% of the effect of position 9; but, taken together, the major contribution of the side chains is from these positions. We could not define a clear pattern for classification of side chain effects at positions 1 to 3, nor could we find strong correlation with MHC class I anchors (24) at these positions. In fact, Pro at position 2 as favored by most HLA-B alleles had the strongest destabilizing effect of a side chain seen in our system. Nevertheless, it came as a surprise that even with our high-resolution method the peptide positions 4 to 8 were irrelevant, with stabilizing factors averaging 1- to 2-fold. Interestingly, it was recently shown that the T cell receptor (TCR) interacts mainly with residues 5 to 8 of a MHC class I-associated nonapeptide (25). Thus, antigen recognition by the TCR is in a peptide region where TAP exerts minimal selection.
The most striking result is the relevance of interaction with the peptide backbone for binding to TAP. Again, we found the contact regions not to be distributed equally over the peptide but concentrated at positions 1 to 3 and 9, with almost no effect from positions 4 to 8. Together with the strict requirement for free N and C termini, this leads us to propose a model of peptide binding to TAP in which the peptide is fixed at an N-terminal region (positions 1 to 3) and the C-terminal residue, primarily via the peptide backbone, but with a marked contribution from the side chains at these positions. Thus, the destabilization observed with Pro-containing peptides in different assay systems (26, 27) is due to disruption of this interaction with the peptide backbone. Residues 4 to 8 of a nonamer peptide could span a cavity with virtually no contact with TAP, allowing for longer peptides to be adopted, whereas peptides of less than eight amino acids are too short to span the distance of interaction sites. This model would explain not only the discrete lower limit and more flexible upper limit for peptide length, but also findings that TAP tolerates peptides with bulky labels at the side chains (16).
Peptide translocation across the ER membrane by TAP has to fulfill two important requirements in terms of antigen processing: (i) transport has to be highly effective; thus, at low given peptide concentrations in the cytosol, TAP needs to have high peptide affinities, and (ii) one TAP allele has to supply several hundreds of different MHC class I molecules with peptides for presentation to a nearly unlimited number of different cytotoxic T cells. Therefore, TAP should not put a further restriction on the pool of peptides available at the MHC class I–TCR contact. We believe that TAP achieves high affinity without restrictions in selectivity by fixing the peptide backbone at positions 1 to 3 and at the C-terminal residue. Fixation at the peptide termini allows for a size-selection mechanism that is optimized in a coevolution to match the length required by MHC class I molecules and the length distribution provided by the proteasome complex (28, 29). Additional binding energy is gained from interaction with C-terminal residues in a way that does not further restrict the presentable pool of peptides. An important exception is Pro, at position 2, implementing a negative effect on the backbone fixation. Because Pro at position 2 is a strict prerequisite for several HLA-B molecules (24), an N-terminal exopeptidase activity in the ER lumen for loading onto these molecules was suggested (30, 31). This would also explain that peptide length does not strictly match with the upper limit for proteasome and MHC, possibly extending the range of available antigens by combining the pools of unprocessed and processed peptides. TAP might thus have evolved a mechanism for binding of peptides that serves the means of high peptide affinity for efficient transport and supplying peptides optimal for binding to different MHC I alleles without restricting T cell response.
Acknowledgments
We thank Drs. Wolfgang Baumeister and Robert Huber for critically reading the manuscript. This work is supported by the Deutsche Forschungsgemeinschaft.
ABBREVIATIONS
- MHC
major histocompatibility complex
- ER
endoplasmic reticulum
- DMF
dimethylformamide
References
- 1.Spies T, DeMars R. Nature (London) 1991;351:323–324. doi: 10.1038/351323a0. [DOI] [PubMed] [Google Scholar]
- 2.Powis S J, Townsend A R M, Deverson E V, Bastin J, Butcher G W, Howard J C. Nature (London) 1991;354:528–531. doi: 10.1038/354528a0. [DOI] [PubMed] [Google Scholar]
- 3.Attaya M, Jameson S, Martinez C K, Hermel E, Aldrich C, Forman J, Fischer Lindahl K, Bavan M J, Monaco J J. Nature (London) 1992;355:647–649. doi: 10.1038/355647a0. [DOI] [PubMed] [Google Scholar]
- 4.Neefjes J J, Momburg F, Hämmerling G J. Science. 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- 5.Androlewicz M J, Anderson K S, Cresswell P. Proc Natl Acad Sci USA. 1993;90:9130–9134. doi: 10.1073/pnas.90.19.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shepherd J C, Schumacher T N, Ashton-Rickardt P G, Imaeda S, Ploegh H L, Janeway C A J, Tonegawa S. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 7.Meyer T H, van Endert P M, Uebel S, Ehring B, Tampé R. FEBS Lett. 1994;351:443–447. doi: 10.1016/0014-5793(94)00908-2. [DOI] [PubMed] [Google Scholar]
- 8.Momburg F, Roelse J, Hämmerling G J, Neefjes J J. J Exp Med. 1994;179:1613–1623. doi: 10.1084/jem.179.5.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koopmann J O, Post M, Neefjes J J, Hämmerling G J, Momburg F. Eur J Immunol. 1996;26:1720–1728. doi: 10.1002/eji.1830260809. [DOI] [PubMed] [Google Scholar]
- 10.Heemels M T, Schumacher T N M, Wonigeit K, Ploegh H L. Science. 1993;262:2059–2063. doi: 10.1126/science.8266106. [DOI] [PubMed] [Google Scholar]
- 11.Momburg F, Roelse J, Howard J C, Butcher G W, Hämmerling G J, Neefjes J J. Nature (London) 1994;367:648–651. doi: 10.1038/367648a0. [DOI] [PubMed] [Google Scholar]
- 12.Heemels M T, Ploegh H L. Immunity. 1994;1:775–784. doi: 10.1016/s1074-7613(94)80019-7. [DOI] [PubMed] [Google Scholar]
- 13.Schumacher T N, Kantesaria D V, Heemels M T, Ashton-Rickardt P G, Shepherd J C, Früh K, Yang Y, Peterson P A, Tonegawa S, Ploegh H L. J Exp Med. 1994;179:533–540. doi: 10.1084/jem.179.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obst R, Armandola E A, Nijenhuis M, Momburg F, Hämmerling G J. Eur J Immunol. 1995;25:2170–2176. doi: 10.1002/eji.1830250808. [DOI] [PubMed] [Google Scholar]
- 15.van Endert P M, Tampé R, Meyer T H, Tisch R, Bach J F, McDevitt H O. Immunity. 1994;1:491–500. doi: 10.1016/1074-7613(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 16.Uebel S, Meyer T H, Kraas W, Kienle S, Jung G, Wiesmüller K H, Tampé R. J Biol Chem. 1995;270:18512–18516. doi: 10.1074/jbc.270.31.18512. [DOI] [PubMed] [Google Scholar]
- 17.Androlewicz M J, Cresswell P. Immunity. 1994;1:7–14. doi: 10.1016/1074-7613(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 18.van Endert P M, Riganelli D, Greco G, Fleischhauer K, Sidney J, Sette A, Bach J F. J Exp Med. 1995;182:1883–1895. doi: 10.1084/jem.182.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Androlewicz M J, Cresswell P. Immunity. 1996;5:1–5. doi: 10.1016/s1074-7613(00)80304-0. [DOI] [PubMed] [Google Scholar]
- 20.Stevanovic S, Jung G. Anal Biochem. 1993;212:212–220. doi: 10.1006/abio.1993.1314. [DOI] [PubMed] [Google Scholar]
- 21.Metzger J W, Kempter C, Wiesmüller K H, Jung G. Anal Biochem. 1994;218:261–277. doi: 10.1006/abio.1994.1266. [DOI] [PubMed] [Google Scholar]
- 22.Wiesmüller K-H, Feiertag S, Fleckenstein B, Kienle S, Stoll D, Hermann M, Jung G. In: Peptide and Cyclopeptide Libraries: Automated Synthesis, Analysis and Receptor Binding Assays. Jung G, editor. Weinheim, Germany: Verlag Chemie; 1996. pp. 203–246. [Google Scholar]
- 23.Udaka K, Wiesmüller H-H, Kienle S, Jung G, Walden P. J Exp Med. 1995;181:2097–2108. doi: 10.1084/jem.181.6.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rammensee H-G, Friede T, Stevanovic S. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 25.Garboczi D N, Ghosh P, Utz U, Fan Q R, Biddison W E, Wiley D C. Nature (London) 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 26.Neefjes J, Gottfried E, Roelse J, Grommé M, Obst R, Hämmerling G J, Momburg F. Eur J Immunol. 1995;25:1133–1136. doi: 10.1002/eji.1830250444. [DOI] [PubMed] [Google Scholar]
- 27.Neisig A, Roelse J, Sijts A J A, Ossendorp F, Feltkamp M C W, Kast W M, Melief C J M, Neefjes J J. J Immunol. 1995;154:1273–1279. [PubMed] [Google Scholar]
- 28.Wenzel T, Eckerskorn C, Lottspeich F, Baumeister W. FEBS Lett. 1994;349:205–209. doi: 10.1016/0014-5793(94)00665-2. [DOI] [PubMed] [Google Scholar]
- 29.Ehring B, Meyer T H, Eckerskorn C, Lottspeich F, Tampé R. Eur J Biochem. 1996;235:404–415. doi: 10.1111/j.1432-1033.1996.00404.x. [DOI] [PubMed] [Google Scholar]
- 30.Falk K, Rötzschke O, Rammensee H-G. Nature (London) 1990;348:248–251. doi: 10.1038/348248a0. [DOI] [PubMed] [Google Scholar]
- 31.Roelse J, Grommé M, Momburg F, Hämmerling G, Neefjes J. J Exp Med. 1994;180:1591–1597. doi: 10.1084/jem.180.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]