

Abstract
Plakoglobin is a protein closely related to β-catenin that links desmosomal cadherins to intermediate filaments. Plakoglobin can also substitute for β-catenin in adherens junctions, providing a connection between E-cadherin and α-catenin. Association of β-catenin with E-cadherin and α-catenin is regulated by phosphorylation of specific tyrosine residues; modification of β-catenin Tyr654 and Tyr142 decreases binding to E-cadherin and α-catenin, respectively. We show here that plakoglobin can also be phosphorylated on tyrosine residues, but unlike β-catenin, this modification is not always associated with disrupted association with junctional components. Protein tyrosine kinases present distinct specificities on β-catenin and plakoglobin, and phosphorylation of β-catenin-equivalent Tyr residues of plakoglobin affects its interaction with components of desmosomes or adherens junctions differently. For instance, Src, which mainly phosphorylates Tyr86 in β-catenin, modifies Tyr643 in plakoglobin, decreasing the interaction with E-cadherin and α-catenin and increasing the interaction with the α-catenin-equivalent protein in desmosomes, desmoplakin. The tyrosine kinase Fer, which modifies β-catenin Tyr142, lessening its association with α-catenin, phosphorylates plakoglobin Tyr549 and exerts the contrary effect: it raises the binding of plakoglobin to α-catenin. These results suggest that tyrosine kinases like Src or Fer modulate desmosomes and adherens junctions differently. Our results also indicate that phosphorylation of Tyr549 and the increased binding of plakoglobin to components of adherens junctions can contribute to the upregulation of the transcriptional activity of the β-catenin-Tcf-4 complex observed in many epithelial tumor cells.
β-Catenin and plakoglobin (also known as γ-catenin) are two closely related proteins essential for the establishment and maintenance of cell-cell contacts among epithelial cells. In adherens junctions, β-catenin links the cytosolic domain of the transmembrane protein E-cadherin to α-catenin, which in turn directly or indirectly associates with the actin cytoskeleton (2, 33, 38). Plakoglobin can substitute for β-catenin in adherens junctions. In addition, plakoglobin is a component of the desmosomes, where it mediates the association of desmosomal cadherins, desmocollin and desmoglein, to desmoplakin and the intermediate filament cytoskeleton (15, 21). This role of plakoglobin is specific and cannot be exerted by β-catenin, even though both proteins are structurally similar. Besides this role, β-catenin also has a signaling activity as a member of the Wnt pathway. When released from E-cadherin and α-catenin, β-catenin can migrate to the nucleus where, through its interaction with the Tcf family of transcription factors, it can activate the transcription of a rapidly increasing number of genes involved in embryonic development and tumorigenesis (5, 37). Probably as a consequence of the importance of this pathway, the translocation of β-catenin to the nucleus is tightly controlled through the activity of a complex implicated in β-catenin degradation. This complex includes the product of the tumor suppressor adenomatous polyposis gene axin and the Thr/Ser protein kinases CKIα and glycogen synthase kinase 3β (27, 37). As a result of the activity of this complex, β-catenin Ser45 is phosphorylated initially by CKIα on Ser45 and sequentially by glycogen synthase kinase 3β on Thr41 and Ser37, Ser33, and Ser29. Upon phosphorylation, β-catenin is polyubiquitylated by β-TrCP ubiquitin ligase and degraded by the proteasome. The activity of the complex is controlled by a signaling pathway triggered by Wnt factors that stabilize cytosolic β-catenin (37). Recently, another parallel mechanism of β-catenin degradation has been described, which depends on the activity of Siah-1 and is activated by p53 (28, 30).
The involvement of plakoglobin in the Wnt pathway has also been a matter of discussion. Kolligs and coauthors observed that plakoglobin can induce transformation of RK3E with a higher efficiency than β-catenin (23). However, data from other laboratories, including ours, indicated that binding of plakoglobin decreases the affinity of Tcf-4 for DNA (31, 44) and prompted the suggestion that the positive effect detected for plakoglobin on Tcf-4-mediated transcription may be indirectly explained by an increased transport of β-catenin to the nucleus (45). To this respect, it has been observed that β-catenin and plakoglobin bind to different sites on Tcf-4 (31). Moreover, other data (reviewed in reference 45) indicate that plakoglobin overexpression is normally associated with tumor suppression, contrary to what happens with β-catenin.
Tyrosine phosphorylation of components of the adhesion complexes regulates the stability of these complexes (11, 26). Thus, stimulation of growth factor receptors or oncogenic Src kinases is implicated in the negative regulation of cell-cell adhesion (4, 16, 29, 40). Increased tyrosine phosphorylation of β-catenin is correlated with the disassembly of adherens junctions and decreased cell adhesion (reviewed in reference 26). Suppression of the association of the epidermal growth factor receptor (EGFR) with β-catenin increases the association with E-cadherin and decreases invasion (6, 40). On the contrary, the activity of several phospho-Tyr (PTyr) phosphatases has been associated with the upregulation of β-catenin-cadherin binding and the prevention of migration (3, 32, 41). Accordingly, inhibition of tyrosine phosphorylation has been reported to result in reassociation of β-catenin to E-cadherin in ras-transformed cells (22, 35).
The phosphorylation of β-catenin residues Tyr654 and Tyr142 specifically decreases the interaction of this protein with E-cadherin and α-catenin, respectively (35, 39). Structural data support these results, since these two Tyr are involved in E-cadherin binding (Tyr654, by establishing an ionic pair with E-cadherin Asp667) (20) and in the stabilization of the β-catenin structure implicated in α-catenin binding (Tyr142) (36). The identification of the Tyr kinases that catalyze these modifications indicates that β-catenin Tyr142 is a good substrate for Fer and Fyn kinases (35), whereas Tyr654 is modified by EGFR-related proteins. Although in vitro assays have not been reported, both the EGFR and its homologue erbB2 interact with β-catenin through the sequence where Tyr654 is located (17, 40, 42) and their activation promotes phosphorylation of Tyr654 (6). Moreover, Src tyrosine kinase phosphorylates β-catenin Tyr654 in vitro with a low level of activity, although it presents a much higher efficiency on Tyr86 (39). The relevance of the phosphorylation of this β-catenin residue is still unknown.
A similar modulation has been proposed for plakoglobin action on desmosomes and adherens junctions. Tyrosine phosphorylation of plakoglobin after EGFR activation has been associated with the loss of interaction with desmoplakin but not with desmoglein (12). Addition of the inhibitor of PTyr phosphatases peroxyvanadate inhibits the interaction of plakoglobin with α-catenin and E-cadherin, an effect that can be reverted by supplementing the system with PTyr phosphatase (18). With this background, we started to study the contribution of the tyrosine kinases mentioned before (EGFR, Src, Fyn, and Fer) in the control of the interactions formed by plakoglobin with other members of desmosomes or adherens junctions. Our results indicate that, although β-catenin and plakoglobin are two closely related proteins, the same protein kinases phosphorylate different residues in both proteins. Moreover, phosphorylation of equivalent residues causes different effects on the interaction of plakoglobin and β-catenin with their cellular partners.
MATERIALS AND METHODS
Generation of plakoglobin mutants.
Human plakoglobin was cloned in the EcoRI site of the pcDNA3.1 plasmid (Invitrogen). Plakoglobin point mutants Y133E, Y133F, Y549E, Y549F, Y643E, and Y643F were obtained by using the QuikChange site-directed mutagenesis kit (Stratagene). A PCR was performed with Pfx polymerase, pcDNA3.1-His-plakoglobin as a template, and oligonucleotide primers containing each mutation. The sense primers used for the generation of Y133E, Y133F, Y549E, Y549F, Y643E, and Y643F were, respectively, 5′-GGCACTGCCACCTTTGCTGCT GCCGTC-3′, 5′-CATCTCATCAACGAACAGGACGATGCC-3′, 5′-CATCTCATCAAC TTTCAGGACGATGCC-3′, 5′-GGCACTGCCACCGAAGCTGCTGCCGTC-3′, 5′-ACACAGCAGCCCGAAACGGATGGTGTG-3′, and 5′-ACACAGCAGCCCTTTACG GATGGTGTG-3′. Modified nucleotides with respect to the plakoglobin sequence (GenBank accession number M23410) are indicated by boldface type. The amplified fragment was treated with DpnI, which digests the parental construct. Finally, the nicked plasmid was transformed and sequenced. The preparation of the plakoglobin fragments Ntail (amino acids [aa] 1 to 113), Arm (aa 111 to 672), Arm 1-6 (aa 111 to 385), Arm 7-12 (aa 380 to 672), and Ctail (aa 667 to 745) has been previously reported (31).
Expression of recombinant proteins.
Mutant plakoglobin cDNAs were inserted in the EcoRI site of the pGEX-6P2 plasmid (Amersham Pharmacia Biotech), expressed in Escherichia coli as glutathione S-transferase (GST) fusion proteins, and purified by affinity chromatography on glutathione-Sepharose (39). Expression and purification of recombinant full-length GST-β-catenin, β-catenin mutants, and the cytosolic domain of human E-cadherin were performed as described previously (35, 39). When required, GST was removed by cleaving with Pre-Scission protease (Amersham Pharmacia Biotech).
Protein binding assays.
Pull-down assays were performed by using purified recombinant proteins fused to a GST tag and extracts from SW-480 cells, as described previously (39). Glutathione-sepharose-bound proteins were analyzed by Western blotting with specific monoclonal antibodies (MAbs) against β-catenin, α-catenin, E-cadherin, plakoglobin, TATA binding protein (TBP), desmoglein (all from Transduction Laboratories), desmoplakin (clones DP1 and 2-2.15; Progen Biotechnik), or Tcf-4 (clone 6H5-3; Upstate Biotechnology). Polyclonal antibody against GST was from Amersham. Reanalysis of the blots with a different antibody was performed after stripping the membranes as described previously (35).
Transient transfections.
Cell lines were routinely grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Transient expression of ectopic proteins was achieved in 50% confluent RWP1, IEC18, IEC18 K-ras, or MDCK cells with the indicated plasmids by using Lipofectamine (Life Technologies) according to the instructions of the manufacturer. When indicated, wild-type or mutant forms of plakoglobin (in pcDNA3.His) were cotransfected with pCMV-Src (13) or pcDNA3-Fer (35). Cells were analyzed 48 h after transfection.
Reporter gene assays.
MDCK or RWP1 cells were cotransfected as described above with wild-type or mutant plakoglobin forms and a plasmid containing three copies of the Tcf-4 binding site upstream of a firefly luciferase reporter gene (plasmid TOP-FLASH) (24). The activity of the product of the Renilla luciferase gene under the control of a constitutive thymidine kinase promoter (Promega) was used as a control. Assays were always performed in triplicate; the averages of the results of three to four independent transfections ± standard deviations are given.
Immunoprecipitation assays.
Cell extracts were prepared from cultured cells resuspended in lysis buffer (50 mM Tris-HCl [pH 7.6], 200 mM NaCl, 5 mM MgCl2, 0.1% Nonidet P-40, 1 mM dithiothreitol, 0.1 mM sodium orthovanadate, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin/ml, and 10 μg of aprotinin/ml) on ice for 30 min. Lysates were cleared at 12,000 × g for 15 min at 4°C. Three hundred micrograms of cell extracts was incubated with 4 μg of antibody/ml for 5 h at 4°C. Precipitated material was removed by centrifugation at 12,000 × g, and the resulting supernatant was incubated for 90 min with 30 μl of protein A-agarose (Sigma). Immunoprecipitates were washed three times with lysis buffer, and bound proteins were directly eluted with electrophoresis sample buffer and either analyzed by Western blotting or used for kinase assays. Desmosomal complexes were solubilized in 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1% digitonin, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin/ml, and 10 μg of aprotinin/ml on ice for 15 min. Lysates were cleared at 16,000 × g for 18 min at 4°C. Alternatively, ectopically expressed proteins were purified with nickel-nitrilotriacetic acid agarose (Qiagen). Two hundred fifty micrograms of cell extracts was incubated in a final volume of 300 μl with 20 μl of a 50% (wt/vol) suspension of nickel-agarose for 1 h at 4°C. Proteins present in the complex were analyzed by Western blotting with specific MAbs.
Immunofluorescence.
Cells, plated on glass coverslips, were fixed with 4% paraformaldehyde for 30 min and permeabilized by incubation with 1% sodium dodecyl sulfate for 10 min. Blocking was carried out for 1 h with phosphate buffered-saline containing 0.1% saponin and 1% bovine serum albumin. Mouse MAb anti-plakoglobin, anti-β-catenin, or anti-α-catenin (Transduction Laboratories) were used to analyze the distribution of these proteins. After washing, binding of primary antibodies was detected with anti-mouse antibodies conjugated to fluorescein-isothiocyanate (Dako, Glostrup, Denmark) or Alexa 488 (Molecular Probes), respectively. Nuclei were counterstained with propidium iodide. Finally, fluorescence was viewed through a TCS-SP2 Leica confocal microscope.
In vitro phosphorylation assays.
Pure recombinant Src and EGFR (cytosolic domain) kinases were purchased from Upstate Biotechnology and Sigma, respectively. Fer kinase was cloned into the BamHI/XbaI sites of pcDNA3-His(C) (Invitrogen), expressed as anti-X-Press and poly-His-tagged protein in RWP-1 cells and purified by nickel-agarose chromatography (35). Fyn kinase was purified from cell extracts by immunoprecipitation with anti-Fyn MAb (Transduction Labs). Phosphorylation assays were performed in a final volume of 30 μl of kinase buffer (25 mM Tris-HCl [pH 6.8], 25 mM MgCl2, 5 mM MnCl2, 0.5 mM EGTA, 1 mM dithiothreitol, 0.25 mM sodium orthovanadate, 0.1 mM ATP) for 1 h at 22°C (Src assays) or 30°C (EGFR, Fer, or Fyn assays). The extent of phosphorylation was determined by analyzing the substrate (plakoglobin) by Western blotting with an anti-PTyr MAb (clone PY20; Transduction Labs).
RESULTS
Both plakoglobin and β-catenin can be divided into three different subdomains (Fig. 1). The central domain is composed of 12 repetitions of 42 amino acids each, known as armadillo repeats, after the β-catenin ortholog in Drosophila melanogaster, armadillo. This region, very similar in both proteins (with a 76% identity) (7), has a basic pI, and its structure has been determined; it forms a super helix composed of 36 small α-helices (3 per each armadillo repeat) (19). Tyr654 lies on the last repeat of this domain, and Tyr142 lies on the limit of this domain and the N-terminal tail. Contrarily to the armadillo repeat domain, the N- and C-terminal tails are mainly acidic (in both the pI is 4.4) and the degree of conservation between β-catenin and plakoglobin is very low (29 and 41%, respectively, for the N- and C-terminal tails). As a consequence of the homologies in these domains, Tyr residues equivalent to β-catenin tyrosines 142 and 654 can be located in plakoglobin: they correspond to tyrosines 133 and 643, respectively. The similarity among the sequences surrounding equivalent Tyr residues in β-catenin and plakoglobin is shown in Fig. 1. On the other hand, β-catenin Tyr86, placed in the N-terminal tail, does not have a correspondent residue in plakoglobin; the equivalent amino acid is Ser74.
FIG. 1.
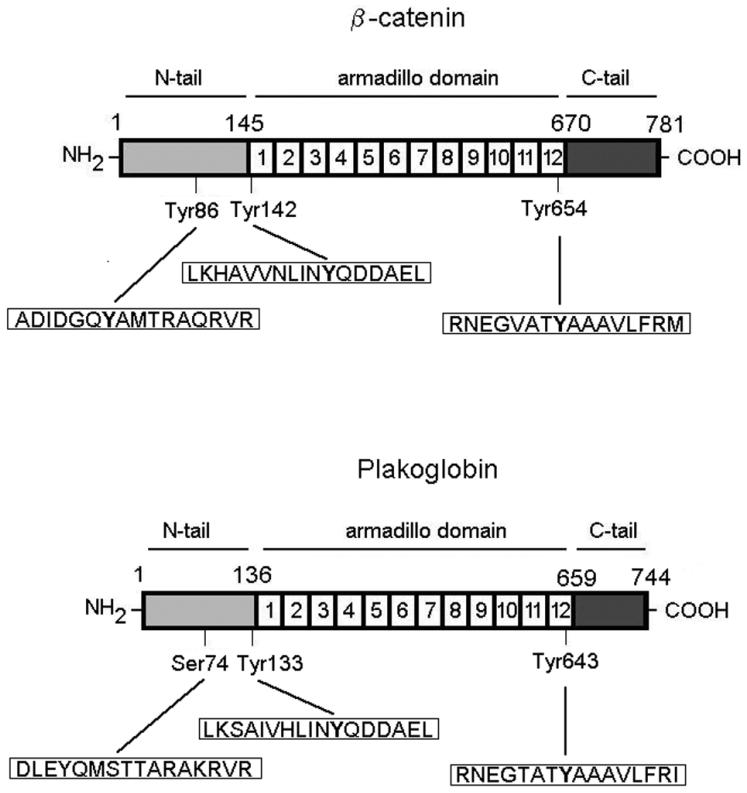
Diagram of β-catenin and plakoglobin. The three different domains that form these two proteins are shown. The amino acid sequences near the three indicated Tyr residues in β-catenin are shown, as are the equivalent sequences in plakoglobin.
We determined whether protein kinases that specifically phosphorylate these three Tyr residues in β-catenin also do it in plakoglobin. As shown in Fig. 2A, recombinant EGFR or purified erbB2 phosphorylates β-catenin. The phosphorylation is not observed when a β-catenin Tyr654→Phe mutant was used as the substrate but was not affected by similar mutations in Tyr86 or Tyr142. These data indicate that EGFR or erbB2 exclusively phosphorylates Tyr654 in β-catenin.
FIG. 2.

EGFR presents different substrate specificities of phosphorylation on β-catenin and plakoglobin. (A) GST-β-catenin fusion proteins (6.7 pmol) were phosphorylated with 0.5 U of recombinant EGFR kinase or RWP1 cell extracts transfected with erbB2. Phosphorylation was analyzed by Western blotting (WB) with anti-PTyr MAb. The membrane was stripped and reprobed for β-catenin as a control, and similar levels of GST-β-catenin were present. (B and C) Five picomoles of GST-plakoglobin deletion mutants (B) or point mutants (C) was phosphorylated with EGFR under the indicated conditions. Samples were analyzed by Western blotting with anti-PTyr MAb and reblotted against GST (B) or plakoglobin (C). (D) Five picomoles of GST or GST-plakoglobin fusion proteins was phosphorylated with EGFR as described above. Pull-down assays were then performed, and the GST proteins were incubated with the indicated amounts of total cell extracts from SW480. The associated proteins were detected with specific MAbs. WT, wild type; +, present; −, absent. The estimated molecular masses of the bands detected with each antibody are indicated.
Similar in vitro assays were performed with recombinant plakoglobin or several fragments as substrates. Plakoglobin was efficiently phosphorylated by EGFR, but different from β-catenin, EGFR did not phosphorylate the plakoglobin armadillo domain, where Tyr643 is located, but modified the C-terminal tail (Fig. 2B). Data from other authors have shown that three Tyr residues in this domain (Tyr693, Tyr724, and Tyr729) are phosphorylated after EGFR stimulation (12). To verify that Tyr643 was not being modified, the same in vitro assays were performed with a Tyr643→Phe mutant. This plakoglobin form incorporated phosphate identically to the wild-type form (Fig. 2C), indicating that Tyr643 is not a substrate of EGFR.
The relevance of plakoglobin phosphorylation by EGFR was also determined. Interaction with desmoplakin was significantly decreased after phosphorylation (Fig. 2D), in agreement with previously published data (12). No changes were observed after phosphorylation in the interaction with desmoglein, E-cadherin, α-catenin, or other cofactors related to the transcriptional activity of this protein (TBP and Tcf-4) (Fig. 2D).
Together, these results indicate that EGFR modifies β-catenin and plakoglobin differently and exerts distinct effects on the interaction of these proteins with some shared partners, such as E-cadherin. These observations prompted us to extend the study to other Tyr kinases, like Src, Fer, or Fyn, which phosphorylate β-catenin.
Src kinase phosphorylates β-catenin mainly on Tyr86, although Tyr654 is also modified with a lower efficiency (39). In plakoglobin, phosphate was incorporated mostly in a fragment comprising armadillo repeats 6 to 12, and to a lesser extent in the C-terminal tail (Fig. 3A). The plakoglobin Tyr643→Phe mutation greatly diminished the amount of PTyr present after Src treatment (Fig. 3B), suggesting that this residue, present in the 12th armadillo repeat, was the main target of this kinase.
FIG. 3.
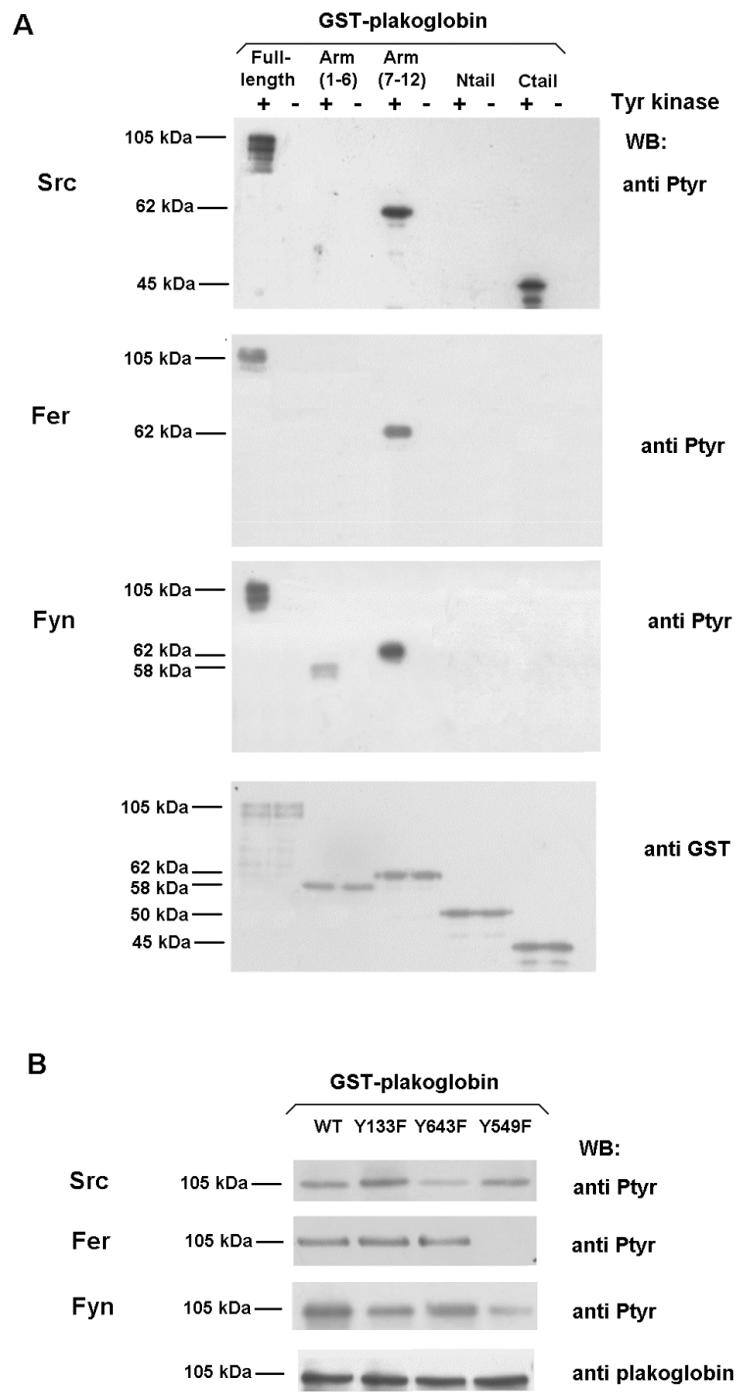
Mapping of residues involved in plakoglobin phosphorylation by Src, Fer, and Fyn kinases. Five picomoles of GST-plakoglobin deletion mutants (A) or the indicated point mutants (B) was phosphorylated with either 0.5 U of recombinant Src (upper panel), Fer kinase purified from transfected RWP1 cells (middle panel), or Fyn kinase immunoprecipitated from transfected RWP1 cells (lower panel) as described in Materials and Methods. Samples were analyzed by Western blotting (WB) with anti-PTyr MAb and reblotted with anti-GST (A) or anti-plakoglobin (B) to ensure that similar levels of GST-plakoglobin forms were present in all cases. +, present; −, absent.
The effect of Fer kinase was also quite different in β-catenin and plakoglobin. Fer and Fyn specifically phosphorylate β-catenin Tyr142, blocking the interaction with α-catenin (35). Fer phosphorylated plakoglobin, but results with armadillo fragments indicated that this kinase was modifying armadillo repeats 7 to 12, comprising aa 381 to 673, but not armadillo repeats 1 to 6, the fragment that contains Tyr133, the equivalent residue to β-catenin Tyr142 (Fig. 3A). Fer was unable to modify this residue, as could be concluded from the experiments performed with plakoglobin Tyr→Phe mutants. Replacement of either Tyr643 or Tyr133 by Phe did not modify the phosphorylation of plakoglobin (Fig. 3B). Thus, in plakoglobin, Fer phosphorylates a residue different from Tyr133 and Tyr643. We investigated the involvement of Tyr549, the only residue located within plakoglobin armadillo repeats 7 to 12 that is not present in β-catenin (Phe561 in β-catenin). Mutation of Tyr549 to Phe confirmed that this Tyr was the target of Fer action: phosphorylation of plakoglobin Tyr549→Phe by this kinase was completely abolished (Fig. 3B).
Similar results were obtained with Fyn kinase. However, although the plakoglobin armadillo fragment 7-12 was again the best substrate for this kinase, the armadillo repeat fragment 1-6 was also modified to a lesser extent. None of the terminal tails were phosphorylated (Fig. 3A). Experiments with Tyr→Phe mutants gave evidence that Fyn mainly phosphorylated Tyr549 and that it phosphorylated Tyr133 with a much lower activity (Fig. 3B).
We investigated the effect of these kinase activities on the interaction of plakoglobin with several partners. Pull-down assays revealed that phosphorylation by Src decreased the binding of plakoglobin with two components of the adherens junctions, α-catenin and E-cadherin, whereas no changes with desmoglein, TBP, or Tcf-4 were detected (Fig. 4A). On the other hand, the affinity of phosphorylated plakoglobin for desmoplakin is higher than that of the unmodified protein (Fig. 4A). This change in affinities was further analyzed by using recombinant proteins: phosphorylation of plakoglobin by Src caused a 2.3-fold decrease in the affinity for E-cadherin (Fig. 4B) and a 4.1-fold rise in the binding to desmoplakin (Fig. 4F). However, neither Src phosphorylation nor the plakoglobin mutant Y643E, designed to mimic the phosphorylation at this residue, had a direct effect on the interaction with α-catenin (Fig. 4C). The apparent contradiction between this result and the decrease in α-catenin binding observed in the pull-down assays can be reconciled if plakoglobin-α-catenin binding is dependent on the presence of E-cadherin, similar to the coordinated interaction of E-cadherin and α-catenin with β-catenin previously reported (10). Indeed, we found that the association of α-catenin and E-cadherin with plakoglobin is interdependent. Previous binding of E-cadherin to plakoglobin significantly facilitated the association of α-catenin (Fig. 4D), although preassociation of α-catenin with plakoglobin did not modify E-cadherin binding to the complex (Fig. 4E). Therefore, the decrease observed in α-catenin binding upon plakoglobin phosphorylation by Src (Fig. 4A) is likely the consequence of reduced E-cadherin binding. No changes were observed in the desmoplakin-plakoglobin binding upon addition of E-cadherin or α-catenin, indicating that, as expected, effects on the affinity for desmoplakin are not dependent on adherens junction proteins (Fig. 4G).
FIG. 4.

Phosphorylation of plakoglobin by Src decreases its interaction with α-catenin and E-cadherin and increases plakoglobin-desmoplakin association. (A) Five picomoles of GST or GST-plakoglobin fusion proteins was phosphorylated with 0.5 U of pp60c-src. Pull-down assays were then performed by incubating the GST proteins with 100 μg of total cell extracts from SW480. The amount of associated proteins was determined by using specific MAbs. (B) Control and Src-phosphorylated plakoglobin (0.35 or 0.7 pmol) were incubated with 1.2 pmol of either GST-cytoE-cadh or GST in a final volume of 200 μl. Plakoglobin (0.03 pmol) was included as a reference (St). The numbers below the lanes indicate the amount of bound protein. (C) GST or GST-plakoglobin fusion proteins (control or phosphorylated by Src) (1.8 pmol) were incubated with 2.4 pmol of α-catenin in a final volume of 200 μl. The amount of bound α-catenin was determined with a specific MAb. (D) Modulation of plakoglobin-α-catenin association by E-cadherin. GST or GST-plakoglobin (0.8 pmol) was incubated with 1.4 pmol of α-catenin. When indicated, binding assays were supplemented with 20 pmol of cytoE-cadh. The amount of associated α-catenin was determined by using a specific MAb. (E) Modulation of plakoglobin-E-cadherin association by α-catenin. GST or GST-plakoglobin (0.8 pmol) was incubated with 1.4 pmol of cytoE-cadh. When indicated, binding assays were supplemented with 20 pmol of α-catenin. The amount of associated cytoE-cadh was determined by using a specific MAb. (F) GST or GST-plakoglobin fusion proteins (5 pmol) were phosphorylated with Src, and pull-down assays were then performed by incubating the GST proteins with 100 or 200 μg of total cell extracts from SW480. The amount of associated desmoplakin was determined by using a specific MAb. (G) GST or GST-plakoglobin (5 pmol) was incubated with 200 μg of total cell extracts from SW480 with the presence of 10 pmol of α-catenin or cytoE-cadh when indicated. The amount of associated desmoplakin was determined by using a specific MAb. WB, Western blotting; +, present; −, absent.
The relevance of Fer phosphorylation was also investigated. Fer affected plakoglobin interaction with desmoplakin and α-catenin. Contrary to what was previously observed for α-catenin-β-catenin interaction, phosphorylation by Fer raised the affinity of plakoglobin for α-catenin (Fig. 5A); using recombinant proteins, this increase was estimated to be 4.4-fold (Fig. 5B). On the other hand, Fer-phosphorylated plakoglobin showed a sevenfold decrease in the affinity for desmoplakin (Fig. 5C). No effect was detected on the interaction of plakoglobin with E-cadherin, desmoglein, TBP, or Tcf-4.
FIG. 5.

Phosphorylation of plakoglobin by Fer and Fyn kinases decreases plakoglobin-desmoplakin interaction and increases plakoglobin-α-catenin association. (A and D) GST or GST-plakoglobin fusion proteins (5 pmol) were phosphorylated with Fer purified from transfected RWP1 cells (A) or Fyn kinase immunoprecipitated from transfected RWP1 cells (D). Pull-down assays were then performed by incubating the GST proteins with 100 μg of total cell extracts from SW480. The amounts of the associated proteins were determined by using specific MAbs. (B and E) GST or GST-plakoglobin (1.2 pmol) (control and Fer phosphorylated [B] or Fyn phosphorylated [E]) was incubated with 0.4 or 0.8 pmol of α-catenin in a final volume of 200 μl. α-Catenin (0.08 pmol) was included as a reference (St). The numbers below the lanes indicate the amounts of bound α-catenin. (C and G) GST or GST-plakoglobin fusion proteins (5 pmol) were phosphorylated with Fer (C) or Fyn kinases (G), and pull-down assays were then performed by incubating the GST proteins with 100 or 200 μg of total cell extracts from SW480. The amount of associated desmoplakin was determined by using a specific MAb. (F) GST or GST-Tcf-4(1-80) (1.2 pmol) was incubated with 0.5 or 1 pmol of plakoglobin incubated or not with Fyn in a final volume of 200 μl. Plakoglobin (0.03 pmol) was included as a reference (St). The numbers below the lanes indicate the amounts of bound plakoglobin. WB, Western blotting; +, present; −, absent.
The effects of Fyn phosphorylation on the association of plakoglobin with α-catenin and desmoplakin were comparable to those obtained with Fer; i.e., an increased binding to α-catenin and a disruption of the interaction with desmoplakin (Fig. 5D). These changes were further confirmed in binding assays with different amounts of recombinant proteins (Fig. 5E and G). Moreover, phosphorylation of plakoglobin by Fyn, and not by Fer, also modified the interaction with the transcriptional factor Tcf-4 (Fig. 5D). We consider that effects on this binding may likely be mediated by the phosphorylation of plakoglobin Tyr133, a residue that is modified by Fyn and not by Fer, albeit with a low efficiency. Even so, Fyn phosphorylation reduced the affinity of plakoglobin for Tcf-4 more than twofold (Fig. 5F). No changes were observed in the interaction with other factors.
All these results indicate that phosphorylation of plakoglobin residues Tyr133, Tyr549, and Tyr643 affects its ability to interact with α-catenin, E-cadherin, desmoplakin, and Tcf-4. To confirm our conclusions, each of these three residues was individually replaced with Glu to mimic the phosphorylation charge. The results obtained with these mutants were consistent with those obtained with the kinases. The plakoglobin Tyr643→Glu mutant showed reduced binding to α-catenin and E-cadherin and a rise in the affinity for desmoplakin. On the contrary, the Tyr549→Glu mutant presented the opposite alterations: an increase in binding to α-catenin and a decreased affinity for desmoplakin without modifications in the association with other factors (Fig. 6). Finally, the Tyr133→Glu mutant showed a complete disruption in the interaction with α-catenin and a parallel increase in desmoplakin binding (Fig. 6). Thus, this mutant has an behavior opposite to what was previously observed upon Fyn phosphorylation of wild-type plakoglobin (Fig. 5D), where the predominant effect is caused by the more efficient modification of Tyr549 (Fig. 3B). A decrease in Tcf-4 binding was also observed in the Tyr133→Glu mutant (Fig. 6).
FIG. 6.
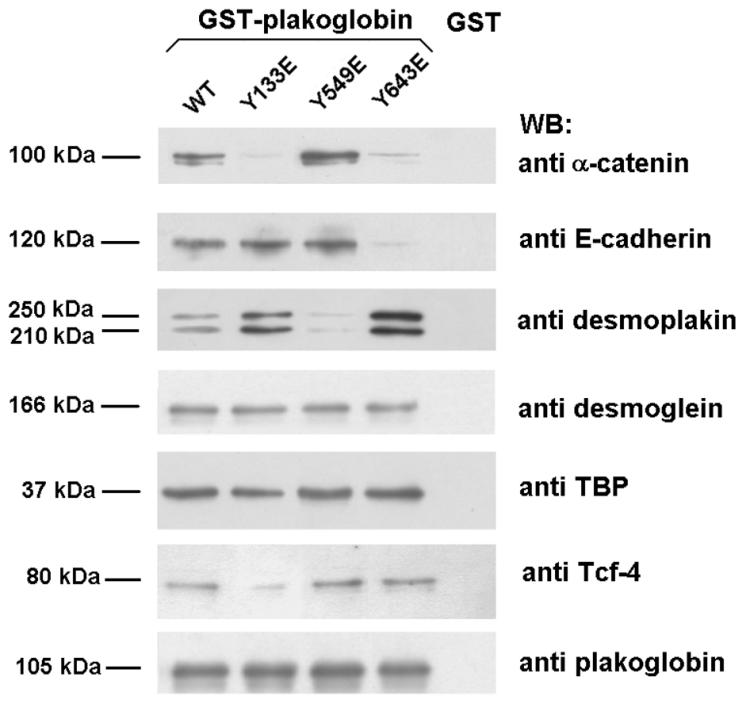
Effect of Tyr-to-Glu point mutants in the association of plakoglobin to its cellular cofactors. Pull-down assays were performed by incubating 8 pmol of GST or GST-plakoglobin fusion proteins with 200 μg of whole-cell extracts from SW480. Protein complexes were affinity purified with glutathione-Sepharose and analyzed by SDS-PAGE and Western blotting (WB). The amounts of the associated proteins were determined by using specific MAbs.
The results shown so far indicate that the effects of phosphorylation of plakoglobin are quite complex, and modification of a specific Tyr residue affects the interaction of several partners. In addition, the action of a specific tyrosine kinase can simultaneously promote the disassembly of the interactions with components of the adherens junctions and the establishment of associations with components of desmosomes. This is what was expected after Src phosphorylation which, upon Tyr643 phosphorylation, reduces binding to α-catenin and E-cadherin and increases the association with desmoplakin. In turn, Fer stimulation, through modification of Tyr549, causes diminished binding of plakoglobin to components of desmosomes (desmoplakin) and increased interaction with adherens junction proteins (α-catenin).
To verify these conclusions, epithelial RWP1 cells were transiently cotransfected with the different Tyr kinases and wild-type plakoglobin (labeled by a poly-His tag). Plakoglobin complexes were purified by Ni2+-agarose, and its association with the different components of adherens junction and desmosome complexes was examined. Overexpression of Src promoted a significant change in the interactions established by plakoglobin; as predicted, higher levels of desmoplakin and lower levels of E-cadherin and α-catenin were purified with this protein (Fig. 7A). On the other hand, transfection of Fer exerted the opposite effect: strengthening the association between plakoglobin and α-catenin while disrupting the interaction with desmoplakin (Fig. 7B). Therefore, our results indicate that these Tyr kinases modulate the interaction of plakoglobin with adherens junctions and desmosome components. Thus, as a consequence of Src phosphorylation, plakoglobin would be mainly associated with desmosomes while Fer activation would promote the preferential linking of plakoglobin to adherens junctions.
FIG. 7.
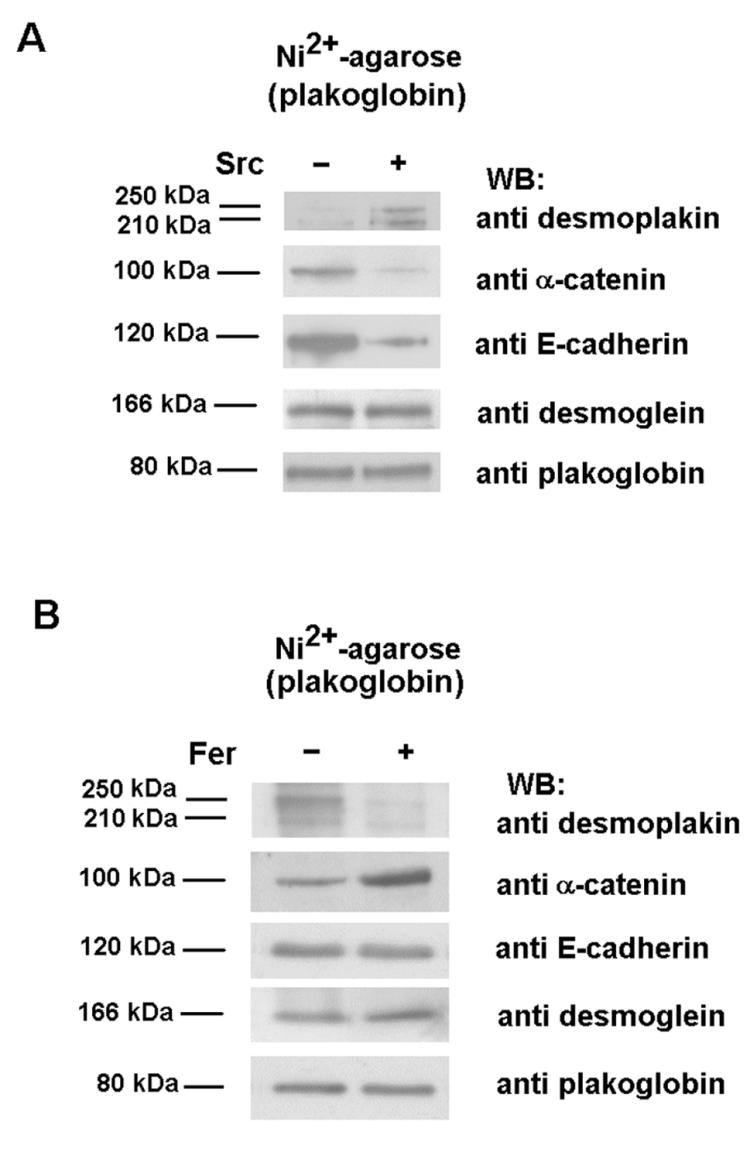
Src and Fyn modify the association of plakoglobin to its cellular partners. RWP1 cells were cotransfected with 5 μg of pcDNA3.1His-plakoglobin and pCMV-Src (A) or pcDNA3.1His-Fer (B), with empty vectors as controls. After 48 h, cell extracts were prepared, His-tagged plakoglobin was purified by chromatography on nickel-agarose, and the associated proteins were analyzed with specific MAbs against α-catenin, E-cadherin, desmoplakin, desmoglein, and plakoglobin. WB, Western blotting; +, present; −, absent.
We also analyzed what happens in conditions in which cell-to-cell contacts are disrupted. Transfection of ras oncogenes to epithelial cells have been shown to disrupt intercellular adhesion (22, 35). In IEC intestinal cells, K-ras transfection stimulates the activity of Fer, Fyn, Src, and EGFR tyrosine kinases and induced the phosphorylation of β-catenin in residues Tyr142 and Tyr654, which was correlated with a downregulated binding to α-catenin and E-cadherin (35). Accordingly, a change in the subcellular distribution of β-catenin was detected after K-ras transfection. Differently than control IEC cells, in which β-catenin was restricted to the cell periphery, IEC K-ras cells presented β-catenin reactivity more dislocated from these regions (Fig. 8). Similar results have been obtained by other groups (22). On the contrary, in IEC cells, plakoglobin showed a more diffuse localization; the protein was observed in the intercellular contacts but also in the cytosol. In IEC K-ras cells, the distribution of this protein seemed to be more localized to the loose contacts established by these cells (Fig. 8). Plakoglobin also showed an increased tyrosine phosphorylation after K-ras transfection (Fig. 9A). Residues Tyr549 and Tyr643 are modified in IEC K-ras cells, since mutation of both amino acids to Phe reduced the phosphorylation of this protein (Fig. 9B). Probably as a consequence of Tyr549 phosphorylation, a higher association of plakoglobin with α-catenin was observed in coimmunoprecipitation experiments, either immunoprecipitation with antiplakoglobin and blotting with anti-α-catenin MAbs or vice versa (Fig. 9A and C). Accordingly, a Tyr549→Phe plakoglobin mutant that cannot be phosphorylated in this residue showed a lower association with α-catenin in IEC K-ras cells than wild-type plakoglobin. These results indicate that plakoglobin Tyr549 phosphorylation and increased binding to α-catenin occur even under conditions with low levels of cell-to-cell interactions.
FIG. 8.
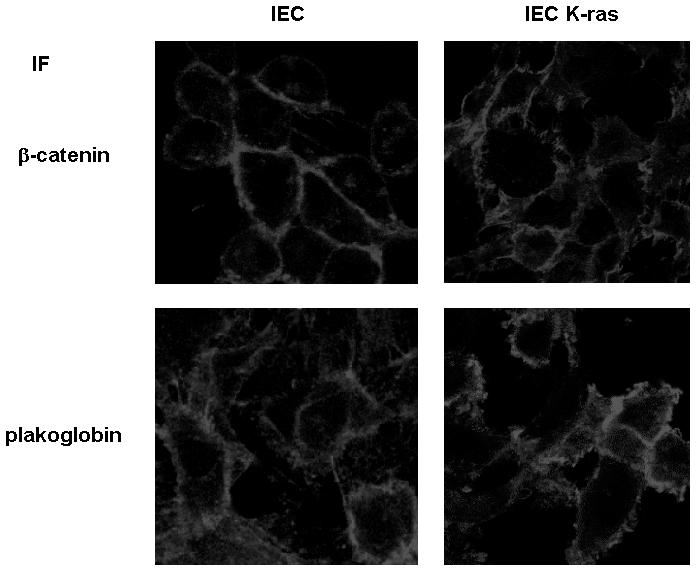
Localization of β-catenin and plakoglobin in IEC and IEC K-ras cells. Cells were grown on glass coverslips as indicated in Materials and Methods and fixed before they reached confluence. The distribution of β-catenin and plakoglobin was analyzed by immunofluorescence with specific MAbs.
FIG. 9.

K-ras transfection of IEC epithelial cells induces plakoglobin phosphorylation and upregulates binding to α-catenin. (A and C) Three hundred micrograms of whole-cell extracts from control and K-ras IEC18 cells were immunoprecipitated (IP) with anti-plakoglobin (A) or anti-α-catenin (B) and followed by immunoblotting with the indicated MAbs. (B and D) IEC18 and IEC18 K-ras cells were transfected with 5 μg of pcDNA3.1His-plakoglobin (wild-type [WT] or the indicated mutants). His-tagged plakoglobin was purified by chromatography on nickel-agarose, and the level of tyrosine phosphorylation (B) or α-catenin association (D) was analyzed by Western blotting (WB) with specific antibodies. The membranes were reprobed for plakoglobin to check that similar levels of expression were obtained.
In addition to its role in the formation of adherens junctions and desmosomes, plakoglobin has also been involved in the regulation of gene transcription (see the introduction). As observed in Fig. 10A, plakoglobin overexpression stimulated the transcriptional activity of β-catenin-Tcf-4 complexes, determined by using the TOP plasmid (24). This effect may be explained by the competition between β-catenin and plakoglobin for binding to several cofactors that has been detected in several systems (45). We checked whether plakoglobin mutants in the phosphorylation sites with altered interactions with E-cadherin and α-catenin behaved differently than the wild-type form on Tcf-4-mediated transcription. IEC K-ras cells were transiently transfected with a Tyr549→Phe mutant which shows decreased binding to α-catenin with respect to the control (Fig. 9D). As shown in Fig. 10A, the stimulatory activity on the TOP activity of this mutant was significantly lower than that of the control. Similar assays were performed with MDCK and RWP-1 cells which show well-formed contacts. Whereas wild-type plakoglobin stimulated TOP activity, plakoglobin mutants with lower affinities for α-catenin (Tyr133→Glu) or E-cadherin and α-catenin (Tyr643→Glu) could not stimulate (and even inhibited) Tcf-4-β-catenin transcriptional activity (Fig. 10A). Binding assays rendered compatible results. Transfection of wild-type plakoglobin to MDCK cells stimulated the amount of β-catenin bound to Tcf-4; on the contrary, the Tyr643→Glu mutant did not modify this association (Fig. 10B). These results indicate that the enhanced transcriptional activity induced by plakoglobin was dependent on its phosphorylation status and suggest that displacement of β-catenin from its interaction with other components of adherens junctions is relevant for the plakoglobin transcriptional activity.
FIG. 10.
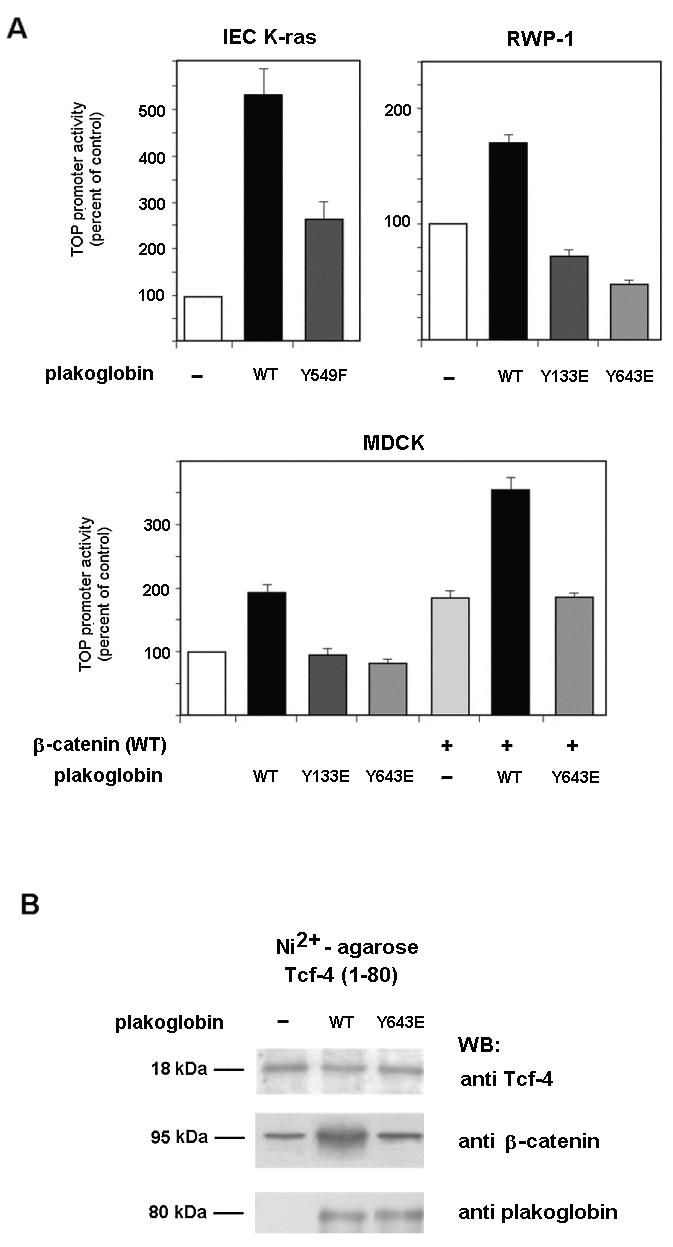
Tyrosine phosphorylation of plakoglobin affects β-catenin-dependent transcription. (A) The indicated cells were cotransfected with plakoglobin plasmid mutants inserted into pcDNA3.1His (150 ng), TOP-FLASH (20 ng), and pTK-Renilla (20 ng) luciferase plasmids in the presence or absence of wild-type (WT) β-catenin. Relative luciferase activity was determined with a dual-luciferase reporter assay system 48 h after transfection and normalized by using the Renilla luciferase activity for each sample. The percentage of activity was calculated by comparing levels of luciferase activity to those obtained after transfection of the pcDNA3.1His plasmid alone. (B) In vivo association between β-catenin and Tcf-4 in MDCK cells overexpressing plakoglobin. Cells were cotransfected with 5 μg of pcDNA3-plakoglobin (wild-type or Tyr643→Glu mutant) or empty vector as a control and 5 μg of pcDNA3.1His-Tcf-4(1-80). His-tagged Tcf-4(1-80) was purified by nickel-agarose chromatography, and associated β-catenin was analyzed by Western blotting (WB) with anti-β-catenin MAb. To verify that the extent of ectopic expression was similar, blots were reanalyzed with anti-Tcf-4. +, present; −, absent.
DISCUSSION
Tyrosine phosphorylation is involved in the regulation of cell-to-cell contacts. It is clearly established that effectors of protein tyrosine phosphatases or kinases modify the formation of adherens junctions and the interactions established among their components (reviewed in reference 26). Phosphorylation of β-catenin is considered to be responsible for destabilizing adhesions. The phosphorylation of specific Tyr residues of β-catenin, Tyr654 and 142, was previously described as being involved in the disruption of the contacts with E-cadherin and α-catenin, respectively (35, 39). Tyr142 is phosphorylated by Fer or Fyn kinases with high efficiency (35). In this work, we show that β-catenin Tyr654 is a good target of EGFR or the related kinase erbB2.
Although not as well studied as β-catenin, plakoglobin has also been reported to undergo tyrosine phosphorylation (12). Plakoglobin, structurally comparable to β-catenin, can substitute for β-catenin in the adherens junctions, interacting simultaneously with E-cadherin and α-catenin. Moreover, plakoglobin is a specific component of the desmosomes, where it mediates the interaction of the desmosome cadherins, desmoglein and desmocollin, with desmoplakin and the intermediate filament cytoskeleton. Other proteins, such as plakophilin, also participate in these junctional complexes (45). Here we describe how the same tyrosine kinases differently phosphorylate equivalent residues in β-catenin and plakoglobin. Thus, EGFR does not phosphorylate the residue equivalent to Tyr654 in β-catenin, i.e., Tyr643 in plakoglobin, but modifies the C-terminal tail of this latter protein. Src works much better on plakoglobin Tyr643 than on β-catenin Tyr654. Lastly, Fer or Fyn cannot phosphorylate plakoglobin Tyr133, the position equivalent to β-catenin Tyr142, with high efficiency. As a matter of fact, Fer cannot modify this residue at all, and Fyn does it with low efficiency (Fig. 3 and data not shown). These two kinases act on a different residue, Tyr 549, a Phe in β-catenin. A summary of the targets of the different kinases is depicted in Fig. 11. At the present moment, we do not know which kinase is responsible for the modification of plakoglobin Tyr133 in vivo nor whether this residue is really modified.
FIG. 11.
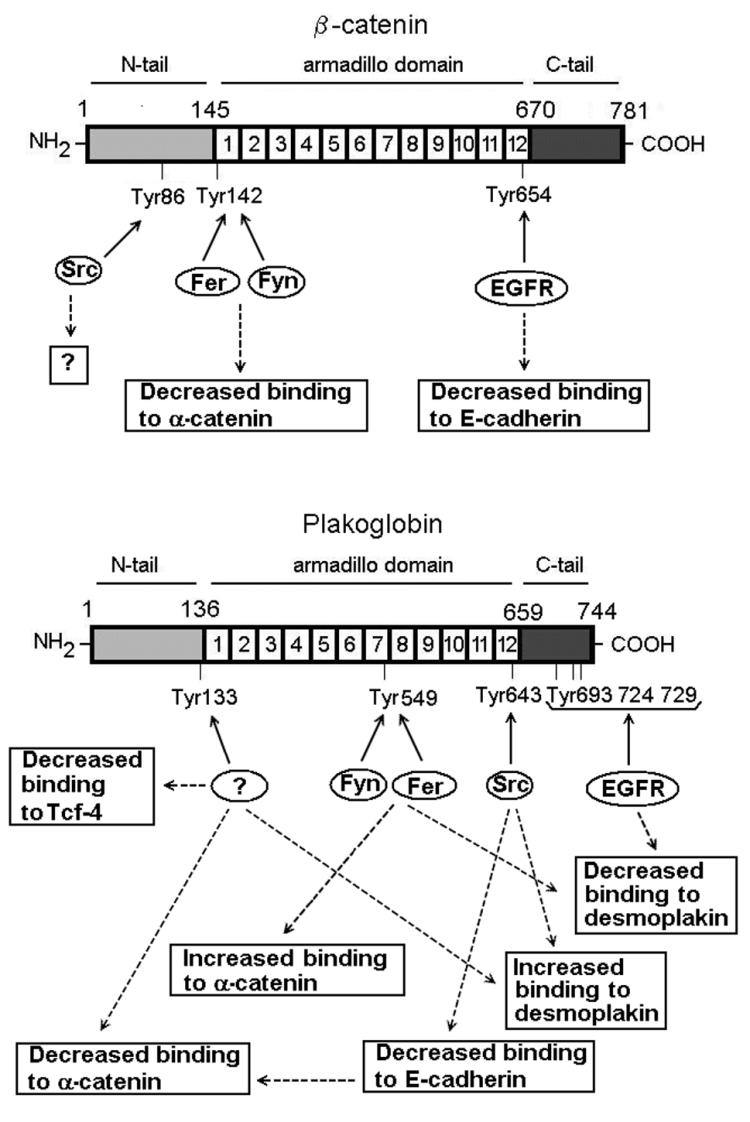
Diagram of β-catenin and plakoglobin tyrosine residues phosphorylated by Src, EGFR, Fer, and Fyn kinases and their corresponding effects. Question marks indicate the points where either an effect of phosphorylation has not been demonstrated (Tyr86 in β-catenin) or the kinase has not been identified (in the case of plakoglobin Tyr133).
As shown in Fig. 11, the consequences of phosphorylation of tyrosine residues in plakoglobin bear similarities with those in β-catenin, although the model seems to be much more complex. Significant differences emerge. For instance, the introduction (through different kinases) of negative charges in Tyr643 and Tyr133 hinders the interaction of plakoglobin with E-cadherin and α-catenin, respectively. However, phosphorylation of plakoglobin Tyr133 exerts an additional effect, blocking the interaction with Tcf-4. Such an inhibition was not detected when β-catenin was phosphorylated in this residue or when it was mutated to Glu (data not shown). This is probably a consequence of the different sites of interaction of Tcf-4 in plakoglobin and β-catenin; as previously described, Tcf-4 associates mainly with armadillo repeats 1 to 6 of plakoglobin (31) while it requires armadillo repeats 3 to 10 of β-catenin (14).
In addition, plakoglobin phosphorylation also modulates the interaction of this protein with other components of the desmosomes. Phosphorylation of Tyr643 increases the association with desmoplakin, a protein that has been shown to interact with the central armadillo domain of plakoglobin (15, 25). Similar effects are observed after modification of Tyr133. Therefore, these two residues seem to act by driving the switch of plakoglobin from adherens junction with desmosome components: phosphorylation of Tyr643 (by Src) or Tyr133 (by a yet unknown kinase) disrupts the interaction of plakoglobin with components of the adherens junctions (E-cadherin and α-catenin, in the case of Tyr643; only α-catenin for Tyr133) and favors in both cases the association with the desmosome protein, desmoplakin. We have shown that this switch takes place when Src is overexpressed in epithelial cells.
Additional sites are modified in plakoglobin compared to β-catenin. EGFR modifies one or more of three tyrosine residues at the C-terminal tail, probably Tyr 693, Tyr724, or Tyr729 (12). As a consequence, the interaction with desmoplakin is dramatically decreased. Fer and Fyn kinases act on Tyr549. Introduction of a negative charge in this residue increases the association with α-catenin and inhibits the interaction with desmoplakin. Therefore, these two kinases act in an opposite fashion than Src: they destabilize the interactions between plakoglobin and the desmosome-associated protein desmoplakin and favor the interaction with adherens junction proteins (α-catenin). Ectopic expression of Fer confirms this conclusion, causing the exchange of proteins bound to plakoglobin.
Our results indicate that tyrosine phosphorylation of plakoglobin is not as unequivocally associated with the loss of cell-to-cell contacts as that of β-catenin. As mentioned, phosphorylation of specific residues of plakoglobin normally plays a dual role, modifying in an opposite fashion the interaction of this protein with components of adherens junctions and desmosomes. These results may explain why some authors have associated increased tyrosine phosphorylation with enhanced cell-to-cell adhesion in some cellular systems such as keratinocytes (8, 9). The elevated ratio of plakoglobin to β-catenin in these cells, resulting in a high presence of plakoglobin in adherens junctions and a distinct distribution of tyrosine kinases (increased levels of Fer or functional equivalent kinases), might explain the results obtained with these cells.
The switch of plakoglobin cellular partners may have another functional significance. Plakoglobin transfection increases the transcriptional activity of β-catenin-Tcf-4 complexes. However, the effect is indirect, since plakoglobin association with Tcf-4, in either the presence or the absence of β-catenin, prevents Tcf-4 binding to DNA (31, 44). Therefore, a maximal activation of β-catenin-Tcf-4-dependent promoters should happen in conditions that prevent plakoglobin translocation to the nucleus, for instance, by an increased binding to α-catenin. Our results are consistent with this conclusion. For instance, increased interaction of plakoglobin with α-catenin is detected under cellular conditions that elevate β-catenin-Tcf-4-dependent transcription, even though cell-to-cell contacts are disrupted. Moreover, the positive correlation of plakoglobin levels with Tcf-4-dependent transcription is not observed with plakoglobin mutants that are unable to interact with components of the adherens junctions and are therefore unable to compete with and release β-catenin from the adhesion complex. Therefore, plakoglobin phosphorylation can contribute to the upregulated β-catenin transcriptional activity detected in epithelial cells after transformation.
Finally, our results suggest that while the general structure of plakoglobin is comparable to that of β-catenin, significant differences exist. For instance, unpublished data from our lab (S. Miravet, J. Piedra, J. Castaño, A. García de Herreros, and M. Duñach, unpublished data), together with already published observations (31, 43), indicate that the N- and C-terminal tails restrict the interaction of different factors with the central plakoglobin armadillo domain, as has also been reported for β-catenin (34). Moreover, analogous to β-catenin, phosphorylation of Tyr residues within the armadillo repeat domain of plakoglobin may prevent the interaction between the tails and the central domain, facilitating the association of different plakoglobin cofactors. Thus, this effect may explain why phosphorylation of Tyr549 facilitates the binding of α-catenin to plakoglobin, an interaction that involves the distant aa 109 to 137 of plakoglobin (1). It was previously reported that β-catenin association with E-cadherin and α-catenin is interdependent (10) and that this effect requires the presence of both terminal tails. According to published results, α-catenin binding to the N-terminal tail of β-catenin results in a looser binding of the C-terminal tail to the armadillo domain, facilitating E-cadherin association with this domain of β-catenin (10). In plakoglobin, a coordinated association of both cofactors was also detected, but it was of the opposite nature. E-cadherin association markedly increases the interaction with α-catenin (Fig. 4D). The effect is stronger in plakoglobin (4.9-fold stimulation) than in β-catenin (2.5-fold). These results suggest that plakoglobin tails are also involved in the control of its interaction with adherens junction proteins. The mechanism is a matter of current research in our laboratory.
In summary, our results indicate that plakoglobin and β-catenin, although structurally and functionally similar, are differently regulated by tyrosine kinases. Our data provide a detailed picture of the exquisite modulation of cell junctions present in epithelial cells and open new perspectives for the explanation of the activity of plakoglobin as an effector of the β-catenin transcriptional activity.
Acknowledgments
We thank D. Garcia-Quintana for critical reading of the manuscript. We also thank H. G. Pálmer for providing the K-ras transfectant clones, A. Mallabiabarrena for help with the confocal microscopy, and A. Ben-Ze'ev, A. Cano, A. Carrera, N. Heisterkamp, and L. Neckers for plasmids.
This work was supported by grants 01/045-00 (from Fundació La Caixa) to A.G.D.H., PM-99-0132 and PM99-0064 (from the Spanish Ministerio de Ciencia y Tecnología) to A.G.D.H. and M.D., respectively, and 2001SGR00410 and 2001SGR00197 (from Direcció General de Recerca). S.M., J.C., and I.R. were recipients of predoctoral fellowships from the Universitat Autònoma de Barcelona, the Spanish Ministerio de Educación y Ciencia, and the Ministerio de Sanidad, respectively.
REFERENCES
- 1.Aberle, H., H. Schwartz, H. Hoschuetzky, and R. Kemler. 1996. Single amino acid substitutions in proteins of the armadillo gene family abolish their binding to α-catenin. J. Biol. Chem. 271:1520-1526. [DOI] [PubMed] [Google Scholar]
- 2.Adams, C., and J. W. Nelson. 1998. Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10:572-577. [DOI] [PubMed] [Google Scholar]
- 3.Balsamo, J., T. Leung, H. Ernst, M. K. Zanin, S. Hoffman, and J. Lilien. 1996. Regulated binding of PTP1B-like phosphatase to N-cadherin: control of cadherin-mediated adhesion by dephosphorylation of beta-catenin. J. Cell Biol. 134:801-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behrens, J., L. Vakaet, R. Friis, E. Winterhager, F. Van Roy, M. Mareel, and W. Birchmeier. 1993. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 120:757-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bienz, M., and H. Clevers. 2000. Linking colorectal cancer to Wnt signaling. Cell 103:311-320. [DOI] [PubMed] [Google Scholar]
- 6.Bonvini, P., W. G. An, A. Rosolen, P. Nguyen, J. Trepel, A. García de Herreros, M. Duñach, and L. M. Neckers. 2001. Geldanamycin abrogates erbB2 association with proteasome-resistent β-catenin in melanoma cells, increases β-catenin-E-cadherin association, and decreases β-catenin-sensitive transcription. Cancer Res. 61:1671-1677. [PubMed] [Google Scholar]
- 7.Butz, S., J. Stappert, H. Weissig, and R. Kemler. 1992. Plakoglobin and β-catenin: distinct but closely related. Science 257:1142-1144. [DOI] [PubMed] [Google Scholar]
- 8.Calautti, E., S. Cabodi, P. Stein, M. Hatzfeld, N. Kedersha, and P. Dotto. 1998. Tyrosine phosphorylation and Src family kinases control keratinocyte cell-cell adhesion. J. Cell Biol. 141:1449-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calautti, E., M. Grossi, C. Mammucari, Y. Aoyama, M. Pirro, Y. Ono, J. Li, and P. Dotto. 2002. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. J. Cell Biol. 156:137-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castaño, J., I. Raurell, J. Piedra, S. Miravet, M. Duñach, and A. García de Herreros. 2002. β-catenin N- and C-tails modulate the coordinated binding of adherens junction proteins to β-catenin. J. Biol. Chem. 277:31541-31550. [DOI] [PubMed] [Google Scholar]
- 11.Daniel, J. M., and A. B. Reynolds. 1997. Tyrosine phosphorylation and cadherin/catenin function. Bioessays 19:883-891. [DOI] [PubMed] [Google Scholar]
- 12.Gaudry, C. A., H. L. Palka, R. L. Dusek, A. C. Huen, M. J. Khandekar, L. G. Hudson, and K. Green. 2001. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J. Biol. Chem. 276:24781-24880. [DOI] [PubMed] [Google Scholar]
- 13.Gómez, S., M. M. Llosas, J. Verdú, S. Roura, J. Lloreta, M. Fabre, and A. García de Herreros. 1999. Independent regulation of adherens and tight junction proteins by tyrosine phosphorylation in Caco-2 cells. Biochim. Biophys. Acta 1452:121-132. [DOI] [PubMed] [Google Scholar]
- 14.Graham, T. A., C. Weaver, F. Mao, D. Kimelman, and W. Xu. 2000. Crystal structure of a β-catenin/Tcf complex. Cell 103:885-896. [DOI] [PubMed] [Google Scholar]
- 15.Green, K. J., and C. Gaudry. 2000. Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 1:208-216. [DOI] [PubMed] [Google Scholar]
- 16.Hazan, R. B., and L. Norton. 1998. The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J. Biol. Chem. 273:9078-9084. [DOI] [PubMed] [Google Scholar]
- 17.Hoschuetzky, H., H. Aberle, and R. Kemler. 1994. β-Catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J. Cell Biol. 127:1375-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu, P., E. J. O'Keefe, and D. S. Rubinstein. 2001. Tyrosine phosphorylation of human keratinocyte β-catenin and plakoglobin reversibly regulates their binding to E-cadherin and α-catenin. J. Investig. Dermatol. 117:1059-1067. [DOI] [PubMed] [Google Scholar]
- 19.Huber, A. H., W. J. Nelson, and W. I. Weis. 1997. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90:871-882. [DOI] [PubMed] [Google Scholar]
- 20.Huber, A. H., and W. I. Weis. 2001. The structure of β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 105:391-402. [DOI] [PubMed] [Google Scholar]
- 21.Jamora, A., and E. Fuchs. 2002. Intercellular adhesion, signaling and the cytoskeleton. Nat. Cell Biol. 4:101-108. [DOI] [PubMed] [Google Scholar]
- 22.Kinch, M. S., G. J. Clark, J. D. Channing, and K. Burridge. 1995. Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia. J. Cell Biol. 130:461-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolligs, F., B. Kolligs, K. Hajra, G. Hu, M. Tani, K. Cho, and E. Fearon. 2000. γ-Catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of β-catenin. Genes Dev. 14:1319-1331. [PMC free article] [PubMed] [Google Scholar]
- 24.Korinek, V., N. Barke, P. Morin, D. van Wichen, R. de Weger, K. Kinzler, B. Vogelstein, and H. Clevers. 1997. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275:1784-1787. [DOI] [PubMed] [Google Scholar]
- 25.Kowalczyk, A. P., E. A. Bornslaeger, J. E. Borgwardt, H. L. Palka, A. S. Dhaliwal, C. M. Corcoran, M. F. Denning, and K. J. Green. 1997. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 139:773-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lilien, J., J. Balsamo, C. Arregui, and G. Xu. 2002. Turn-off, drop-out: functional state switching of cadherins. Dev. Dyn. 224:18-29. [DOI] [PubMed] [Google Scholar]
- 27.Liu, C., Y. Li, M. Semenov, C. Han, G. H. Baeg, Y. Tan, Z. Zhang, X. Lin, and X. He. 2002. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837-847. [DOI] [PubMed] [Google Scholar]
- 28.Liu, J., J. Stevens, C. A. Rote, H. J. Yost, Y. Hu, K. L. Neufeld, R. L. White, and N. Matsunami. 2001. Siah-1 mediates a novel β-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol. Cell 7:927-936. [DOI] [PubMed] [Google Scholar]
- 29.Matsuyoshi, N., M. Hamaguchi, S. Taniguchi, A. Nagafuchi, S. Tsukita, and M. Takeichi. 1992. Cadherin-mediated cell-cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J. Cell Biol. 118:703-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuzawa, S., and J. C. Reed. 2001. Siah-1, SIP and Ebi collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol. Cell. 7:915-926. [DOI] [PubMed] [Google Scholar]
- 31.Miravet, S., J. Piedra, F. Miró, E. Itarte, A. García de Herreros, and M. Duñach. 2002. The transcriptional factor Tcf-4 contains different binding sites for β-catenin and plakoglobin. J. Biol. Chem. 277:1884-1891. [DOI] [PubMed] [Google Scholar]
- 32.Müller, T., A. Choidas, E. Reichmann, and A. Ullrich. 1999. Phosphorylation and free pool of β-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J. Biol. Chem. 274:10173-10183. [DOI] [PubMed] [Google Scholar]
- 33.Nagafuchi, A. 2001. Molecular architecture of adherens junctions. Curr. Opin. Cell Biol. 13:600-603. [DOI] [PubMed] [Google Scholar]
- 34.Piedra, J., D. Martínez, J. Castaño, S. Miravet, M. Duñach, and A. García de Herreros. 2001. Regulation of β-catenin structure and activity by tyrosine phosphorylation. J. Biol. Chem. 276:20436-20443. [DOI] [PubMed] [Google Scholar]
- 35.Piedra, J., S. Miravet, J. Castaño, H. G. Palmer, N. Heisterkamp, A. García de Herreros, and M. Duñach. 2003. p120 catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin interaction. Mol. Cell. Biol. 23:2287-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pokutta, S., and W. I. Weis. 2000. Structure of the dimerization and β-catenin-binding region of α-catenin. Mol. Cell 5:533-543. [DOI] [PubMed] [Google Scholar]
- 37.Polakis, P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837-1851. [PubMed] [Google Scholar]
- 38.Provost, E., and D. L. Rimm. 1999. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr. Opin. Cell Biol. 11:567-572. [DOI] [PubMed] [Google Scholar]
- 39.Roura, S., S. Miravet, J. Piedra, A. García de Herreros, and M. Duñach. 1999. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J. Biol. Chem. 274:36734-36740. [DOI] [PubMed] [Google Scholar]
- 40.Shibata, T., A. Ochiai, Y. Kanai, S. Akimoto, M. Gotoh, N. Yasui, R. Machinami, and S. Hirohashi. 1996. Dominant negative inhibition of the association between β-catenin and c-erbB2 by N-terminally deleted β-catenin suppresses the invasion and metastasis of cancer cells. Oncogene 13:883-889. [PubMed] [Google Scholar]
- 41.Taddei, M. L., P. Chiarugi, P. Cirri, F. Buricchi, T. Fiaschi, E. Giannoni, D. Talini, G. Cozzi, L. Formigli, G. Raugei, and G. Ramponi. 2002. β-Catenin interacts with low-molecular-weight protein tyrosine phosphates leading to cadherin-mediated cell-cell adhesion. Cancer Res. 62:6489-6499. [PubMed] [Google Scholar]
- 42.Takahashi, K., K. Suzuki, and Y. Tsukatani. 1997. Induction of tyrosine phosphorylation and association of β-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence. Oncogene 15:71-78. [DOI] [PubMed] [Google Scholar]
- 43.Wahl, J. K., J. E. Nieset, P. A. Sacco-Bubulya, T. M. Sadler, K. R. Johnson, and M. J. Wheelock. 2000. The amino- and carboxyl-terminal tails of β-cate-nin reduce its affinity for desmoglein 2. J. Cell Sci. 113:1737-1745. [DOI] [PubMed] [Google Scholar]
- 44.Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. Differential mechanisms of LEF/TCF-dependent transcriptional activation by β-catenin and plakoglobin. Mol. Cell. Biol. 20:4238-4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. Plakoglobin and β-catenin: protein interactions, regulation and biological roles. J. Cell Sci. 113:3127-3139. [DOI] [PubMed] [Google Scholar]