

Abstract
Recombination of genes is essential to the evolution of genetic diversity, the segregation of chromosomes during cell division, and certain DNA repair processes. The Holliday junction, a four-arm, four-strand branched DNA crossover structure, is formed as a transient intermediate during genetic recombination and repair processes in the cell. The recognition and subsequent resolution of Holliday junctions into parental or recombined products appear to be critically dependent on their three-dimensional structure. Complementary NMR and time-resolved fluorescence resonance energy transfer experiments on immobilized four-arm DNA junctions reported here indicate that the Holliday junction cannot be viewed as a static structure but rather as an equilibrium mixture of two conformational isomers. Furthermore, the distribution between the two possible crossover isomers was found to depend on the sequence in a manner that was not anticipated on the basis of previous low-resolution experiments.
Genetic recombination is a primary biological process that is critical to the continual evolution of all organisms, and the Holliday junction is a requisite intermediate in nearly all recombination events. Holliday (1) proposed the first and most basic mechanism of genetic recombination: homologous chromatids are nicked at identical sites, then the duplex DNA is partially unwound and the single strands are paired with the complementary strands of the homologs. The structure thus formed, termed the Holliday junction (HJ), may travel (branch-migrate) in either direction along the DNA. Eventually, the structure is cut endonucleolytically (resolved) to give two chromosomes with either a parental (patch) or a recombined (splice) configuration (Fig. 1). Although many postulated mechanisms for recombination exist, they differ only in how recombination is initiated and in how DNA strands are displaced (1–5). Most recombination mechanisms invoke formation of heteroduplex DNA molecules containing branched structures.
Figure 1.

Schematic diagram of the Holliday junction, branch migration, crossover isomerization, and the products formed by HJ resolution (adapted from ref. 12).
It has been reported for more than 40 years that DNA sequence at or near a HJ influences how it is resolved and the corresponding ratio of parental to recombined molecules (6). Results from a variety of experiments suggest that resolution must be predetermined for certain sequences and that not all HJs are identical in structure (e.g., refs. 7–10). To gain an in-depth understanding of recognition and resolution of HJs, it is imperative to characterize their structure and dynamics at the atomic level and to establish how these properties vary with sequence.
A consensus view of HJ structure has emerged from biophysical studies of immobilized four-arm DNA junctions in which the sequence is designed to eliminate the possibility of branch migration (for reviews see refs. 11 and 12). The four helical arms are seen to stack upon each other in a pairwise fashion, forming two duplex domains with an acute interhelical angle (Fig. 2). In this configuration, two contiguous strands run antiparallel to each other and the two crossover strands run in opposite directions. Two alternate arrangements (termed crossover isomers) can be formed for each four-arm junction, distinguished by which two of the four duplex arms stack upon each other and which of the strands cross over from one duplex domain to the other (Fig. 2). The junction at which the four duplex arms meet and the strands cross over is termed the branching point.
Figure 2.

Molecular models of 32 base pair immobilized HJs. (A) Full atom representation of J1 showing the geometrical relationships between the crossover isomers. In the I/II crossover isomer, arm I is stacked on arm II, and arm III on arm IV. The alternate I/IV crossover isomer has arm I stacked on arm IV, and arm II on arm III. Strand 1 is yellow, strand 2 is green, strand 3 is magenta, and strand 4 is cyan. (B) Ribbon diagrams showing the location of the FRET and NMR probes used in these studies. The F1R4 dye-labeled junction for FRET experiments is shown in the two crossover isomers, with fluorescein attached to arm I (F1) in orange and tetramethylrhodamine attached to arm IV (R4) in white. The full molecular structure of the base pair containing the labeled T9 residue for the NMR experiments is also shown, with the 15N-enriched nitrogen atom in red.
In vitro studies of the structure of partially and completely immobilized Holliday junctions by gel mobility and chemical and enzymatic footprinting revealed dominant crossover isomer preferences (11–17). Minor cleavage sites were occasionally observed in restriction enzyme cleavage experiments, and the possibility of this arising from the presence of a small amount of the alternative crossover isomer was discussed (e.g., refs. 13 and 14). Despite the expectation of only limited differences in the thermodynamic stabilities of the two crossover isomers of four-arm junctions, experimental data have been consistently interpreted in terms of HJs existing in one predominant crossover isomer according to the sequence of the four base pairs at the branching point, with, to a first approximation, the sequence of the rest of the molecule having no effect. However, this paradigm has evolved from numerous intrinsically low-resolution experiments.
In this report, we provide convincing direct evidence disproving the conventional wisdom that a given HJ sequence will populate only one of the crossover isomers. Recently developed high-resolution nuclear magnetic resonance (NMR) and time-resolved fluorescence resonance energy transfer (tr-FRET) methods were applied for comparative analysis of three related, immobilized four-arm junctions (Fig. 3): J1, the first immobile HJ designed for biophysical studies (18); J2, an analog of J1 with two base pairs in the junction region swapped; and J2P1, the analog of J2 with a clockwise permutation of the four base pairs at the branching point. These NMR and tr-FRET techniques allow a much more quantitative determination of crossover isomer ratio than was previously possible, which provides new insights on the sequence-dependent properties of Holliday junctions.
Figure 3.

The base sequence of J2 and partial sequences for J1 and J2P1. The branching point residues are in bold. The site of 15N labeling for NMR is denoted by ∗. The four duplex arms of each HJ are labeled I to IV.
MATERIALS AND METHODS
tr-FRET.
Oligonucleotides were synthesized using β-cyanoethyl phosphoramidite chemistry on an Applied Biosystems DNA synthesizer (model 108B) for J1 and J2 and on a Pharmacia LKB gene assembler for J2P1. All strands for J1 and J2 contain a 5′ terminal aminohexyl phosphate linker; however, for J2P1 only those DNA sequences to be dye-labeled were prepared with an amino linker. The donor and acceptor 5′ labeled strands were produced by reacting with succinimidyl ester derivatives (Molecular Probes) of 5-carboxyfluorescein (donor) and 5-carboxytetramethylrhodamine (acceptor) as described previously (19). Donor and donor-acceptor (D-A) labeled four-arm junctions were prepared by combining the appropriate amounts of each of the single strands in a buffer containing 20 mM Tris, 50 mM NaCl, 5 mM MgCl2, and 0.2 mM EDTA at pH 7.5. Fluorescein-labeled DNA strands were added at a 1:3 ratio with respect to the other three strands to ensure that all fluorescein-labeled strands were incorporated into junctions. Samples were then annealed by heating at 363 K for 2 min then slowly cooling to room temperature. The final junction concentrations (fluorescein strand) of the samples were 0.5 μM for J1 and J2, and 5 μM for J2P1. Time-resolved emission profiles were collected using a time-correlated single photon counting apparatus (19). Fluorescein-labeled junctions were excited at 514 nm, and the isotropic emission decay of the fluorescein was monitored at 540 nm. To derive distance distribution information for a specific D-A junction, two time-resolved fluorescence decays were collected, one for the junction containing the D-A pair and one for the corresponding donor-only junction. The decay of fluorescein emission was analyzed using a model which assumes that the distance between any D-A pair can be described by a sum of two continuous, three-dimensional Gaussian distributions (20), each of which is represented by
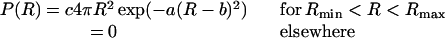 |
1 |
where a and b are adjustable parameters that determine the shape of the distribution and c is a normalizing factor. Rmin and Rmax specify the minimum and maximum possible D-A distances, respectively. Previous experiments determined that the rotation and orientation of the dye molecules in these junctions are such that the orientation factor for energy transfer is close to the free rotation limit and can be ignored in the energy transfer analysis (19). Under these conditions, the complete equation for the intensity decay of the donor in the presence of the acceptor is
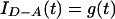 |
2 |
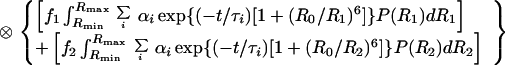 |
where f1 and f2 represent the relative abundances of isomer 1 and 2 in the sample (f1 + f2 = 1), αi and τi represent the intrinsic decay amplitudes and lifetimes of the donor determined from a sum of exponentials fit to single-labeled junctions, P(R1) and P(R2) are the probability distribution describing the D-A distance for each isomer, g(t) is the instrument-response function, and ⊗ denotes the convolution operator. The overall isomeric ratio and the shape of each distance distribution are determined by a least-squares minimization fit of the donor intensity decay to Eq. 2. A full description of the technical details is provided elsewhere (R.S.F., J. C. Van der Schans, and D.P.M., unpublished work).
NMR.
The preparation of samples and the NMR experiments followed methods described previously for J2P1 (21). For 15N-labeled J1 and J2, all seven oligonucleotides were synthesized by Keystone Laboratories (Menlo Park, CA). For the one strand common to both J1 and J2, [1,3-15N]thymidine phosphoramidite (synthesized by the National Stable Isotope Resource, Los Alamos, NM) was used for the thymidine at position 9. Purification by anion-exchange chromatography was carried out using HQ POROS resin in an 8-ml column on a Biocad Sprint (PerCeptives Biosystems, Cambridge, MA). The four strands were combined to a final stoichiometry of 1:1:1:1 at ≈1 mM in each strand. The resulting solution was lyophilized, then redissolved in 420 μl of buffer containing 20 mM Tris⋅d11 (MSD Isotopes) at pH 7.4, 50 mM NaCl, 5 mM MgCl2, 0.2 mM EDTA, 0.003% NaN3, and 10% D2O. The sample was annealed by heating at 363 K for 2 min, then allowing the solution to cool slowly to room temperature.
NMR spectra for J1 and J2 were acquired on a Bruker DMX 750 spectrometer over a temperature range of 274–305 K (data not shown). One-dimensional (1D) 15N-filtered 1H-detected experiments (22) were recorded with a 180° 1H pulse during the 15N evolution period, and a refocusing delay was inserted after the last 15N pulse. Pulse-field gradients were applied for selection of the desired coherence-transfer pathway and water suppression. Two-dimensional 15N-1H heteronuclear multiple quantum correlation spectra (22) were recorded in 18 hr with 512 scans per t1 value and a total of 40 t1 values. Quadrature detection was utilized in the indirect dimension using States-TPPI (23) with a spectral width of 10 ppm. The NMR experiments on J2P1 have been described previously (20). The data were processed on an SGI INDY workstation using felix (versions 2.30 or 95.0, Molecular Simulations). Line broadening of 15 Hz was used for 1D experiments and 10 Hz for 2D experiments, along with deconvolution of the water signal and zero filling to a final matrix of 8,192 × 128 points.
RESULTS
The fluorescence-based approach involves 5′ end-labeling two of the duplex arms of the four-arm junction, one with the fluorophore donor (D) fluorescein and the other with the complementary acceptor (A) tetramethylrhodamine. The distance between donor and acceptor is then determined by fluorescence resonance energy transfer between the dye couple. For a junction labeled on arms I and IV, the D-A distances for each of the two crossover isomers are different (Fig. 2). In the I/IV isomer, the dyes are on opposite ends of a stacking domain, whereas in the alternate I/II crossover isomer, the dyes enclose the acute interhelical angle and are much closer together. Conventionally, the donor decay in a tr-FRET experiment is interpreted by fitting to a discrete donor–acceptor (D-A) distance or a single Gaussian distance distribution (20). To establish isomer distribution, a novel approach for analyzing the experimental data for a system exhibiting distinct structural substates had to be developed. This involved simultaneously fitting two distributions of D-A distances, one for each crossover isomer (R.S.F., J. C. Van der Schans, and D.P.M., unpublished work).
The NMR approach involves selective enrichment of 15N at a single imino nitrogen site in the molecule, combined with isotope-filtered 1H NMR, to allow unimpeded observation of signals from the junction region (21). The previously reported 15N-filtered experiments on J2P1 revealed two peaks for the single imino proton of T9, corresponding to the I/II and I/IV crossover isomers in slow exchange on the chemical shift time scale. The ratio of isomers of J2P1 was found to change with temperature, consistent with one isomer being more stable than the other. To characterize the isomer distribution of J1 and J2, samples were prepared with 15N enrichment at the position corresponding to T9 in J2P1. The isotope-filtered strategy is especially valuable because the NMR spectra of the 15N-labeled junctions can be used to quantitatively assay the crossover isomer distribution.
The results of the tr-FRET bimodal distance distribution analysis and the NMR experiments on J1, J2P1, and J2 are shown in Fig. 4. In the tr-FRET data, peak heights (not the areas under the curves) of each distribution represent the relative population of each crossover isomer. In the NMR data, the relative populations are proportional to the peak heights in the 1D spectra of J1 and J2P1 and to the cross-peak volumes in the 2D spectrum of J2.
Figure 4.

Quantitation of crossover isomer distribution in J1, J2P1, and J2. (A) Normalized D-A distance distributions from time-resolved FRET analysis of J1 (Top), J2P1 (Middle), and J2 (Bottom) with F1R4 labeling. All distances are normalized to R0, the critical transfer distance for the fluorescein-tetramethylrhodamine D-A pair (R0 = 54 Å), which was previously determined from the spectral properties of the donor and acceptor (19). The heights of the two distributions in each panel are proportional to the relative populations of each isomeric species. (B) One-dimensional 15N-filtered 1H-detected NMR spectra of J1 and J2P1 at 280 K and a 2D contour plot of the 15N-1H heteronuclear multiple quantum coherence spectrum of J2 at 285 K. The relative populations of the crossover isomers are proportional to the peak heights in the 1D spectra and to the cross-peak volumes in the 2D spectrum.
For J1, the tr-FRET experiments show that the shorter D-A distance distribution (corresponding to the I/II crossover isomer) is dominant. Similarly, J1 exhibits only a single 15N-filtered 1H resonance at all temperatures examined. The predominance of the I/II crossover isomer is in agreement with the previous hydroxyl radical footprinting experiments on J1 (14). In this junction, the minor species (longer distance distribution) is virtually nonexistent, comprising less than 5% of the total population. Our previous tr-FRET study effectively analyzed this junction in terms of a single distance distribution allowing for a minute contribution from a donor-only species (19). Our present analysis suggests that a small contribution from a second distribution provides an equivalent fit to the experimental data. The relatively broad widths of the distance distributions are a reflection of the flexibility inherent in the four-arm junction structure. This flexibility is believed to be somewhat exaggerated by mobility of the linkers connecting the dyes to the DNA.
The tr-FRET and NMR analyses for J2P1 indicate that this four-arm junction is more conformationally heterogeneous than J1. An examination of the residuals from curve fitting of the tr-FRET data verifies that two Gaussian distance distributions are necessary and sufficient to fit the donor decay profiles (R.S.F., J. C. Van der Schans, and D.P.M., unpublished work). The presence of two distance distributions, corresponding to the I/II and I/IV crossover isomers, is consistent with the two peaks observed in the 15N-filtered NMR spectrum of J2P1 (Fig. 4B). Both methods show that the I/IV isomer is dominant.
The results from the analysis of J2 show that this four-arm junction is also more heterogeneous than J1, although the ratio of the two isomers is different than in J2P1. Two tr-FRET distance distributions and two separate NMR resonances are observed for J2, although the latter could only be detected by 2D 15N-1H heteronuclear multiple quantum coherence experiments (22) because the 1H chemical shifts of the two isomeric species are coincidentally degenerate. As seen in the contour plot of the 2D spectrum in Fig. 4B, the two 15N chemical shifts are resolved for J2. The larger of the two signals is assigned to the I/II crossover isomer on the basis of the results from detailed analysis of the 2D 1H nuclear Overhauser effect spectra (24).
DISCUSSION
The tr-FRET and NMR results clearly show that J1, J2, and J2P1 exist as equilibrium mixtures of crossover isomers, contradicting the commonly held view that a specific HJ sequence populates a specific crossover isomer. From the relative peak heights in the bimodal distance distribution analysis, the ratio of the two crossover isomers (I/II:I/IV) is >19:1 for J1, 1:4 in J2P1, and 1:1 for J2. From the 15N-filtered NMR experiments, the corresponding estimates are >19:1 for J1 (from both 1D and 2D experiments), 1:5 for J2P1 (from the 1D experiment; ref. 21), and 1.5:1 for J2 (from the 2D experiment). An uncertainty of ±10% for the relative populations of the two isomers is estimated for both methods. Thus, the tr-FRET and NMR methods are highly complementary and yield identical results within experimental error, even though they examine the molecule from different vantage points (Fig. 2).
The comparative analysis of J2 and J2P1 provided a most surprising finding. J2P1 corresponds to a one-step cyclic permutation of the base pairs at the junction relative to J2 (Fig. 3). These two four-arm junctions have exactly the same sequence of base pairs at the branching point but differ in terms of the base pairs in the penultimate position. Experiments on synthetic HJ systems using low-resolution techniques have been universal in supporting the belief that the penultimate base pairs have no influence on the crossover isomer preference (11, 13, 14, 16). Hence, the approximately 4-fold difference in the isomer preferences of J2 and J2P1 was not anticipated. This observation demonstrates that the base sequence not only at the branching point, but also at the penultimate position, has a significant role in determining the crossover isomer preferences of four-arm junctions. An effect of the penultimate bases on stacking patterns in three-arm DNA junctions has also been reported (25). Base-stacking interactions are the primary driving force in the formation of DNA structure and, hence, play a dominant role in the pairwise stacking of the duplex arms of four-arm junctions, including the Holliday junction. The energetics of base stacking requires consideration of interactions extending over several base pairs. Consistent with this, our results show that the identity of the penultimate residues influences the relative thermodynamic stabilities of the two crossover isomers.
We have detected substantial differences in the crossover isomer biases of subtlely different, synthetic immobilized four-arm Holliday junctions. But how do these results pertain to natural Holliday junctions? The HJ intermediates in homologous recombination events are formed in regions of sequence homology and, consequently, may have less crossover bias than the nonhomologous four-arm junctions studied in this work. Experiments on double-crossover-branched DNA molecules have shown a dilution of crossover isomer bias as the region of homology increases in size (26). However, the potential for problems associated with conformational strain in these systems has been recognized, and the possibility of small but significant residual biases even in highly homologous regions could not be ruled out (26). Furthermore, significant crossover isomer biases are expected near the ends of homologous regions. The importance of conformational bias in HJs may be even greater in site-specific recombination events in which homology-dependent branch migration may be limited to very small regions. For example, it has been proposed that branch migration of the Holliday intermediate in Flp-mediated recombination of genes may be limited to a central 2- to 4-bp region of symmetry (27).
Crossover isomer bias may be relevant for understanding HJ recognition and processing by junction-resolving enzymes. The resolution of HJs can conceivably occur from either unfolded symmetric structures or directly from the folded HJs. If resolution occurs from the folded HJ, the choice of crossover isomer will determine whether parental (patch) or recombined (splice) products are formed (Fig. 1). Sequence-dependent variation in crossover isomer preference, as we have observed, might then exert a significant effect on the observed ratio of parental versus recombined products.
Our studies provide new, direct evidence for the coexistence of the two crossover isomers and the sequence-dependence of the crossover isomer preference in four-arm DNA junctions. They also contribute to an increasing appreciation of the subleties of Holliday junction structure and the potential roles of its structural properties in determining the outcome of genetic recombination processes.
Acknowledgments
We thank Dr. Jens Madsen for his assistance with the NMR experiments, Dr. Peggy S. Eis for assistance with the tr-FRET measurements of J2, J. C. Van der Schans for help with oligonucleotide synthesis, Drs. Brigitte Lavoie, George Chaconas, Anthony Pelletier, and Joel Gottesfeld for helpful discussions, and Dr. Fred Heffron for his encouragement and enthusiasm in the initial stages of our work on Holliday junctions. This work was supported by a grant from the National Science Foundation (MCB-9317369) to W.J.C. and D.P.M. and a fellowship from the National Institutes of Health (F32GM16849) to S.M.M. W.J.C. is a recipient of an American Cancer Society Faculty Research Award (FRA-436). The NMR spectrometer was purchased with partial funding from the National Institutes of Health (S10RR08975).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: HJ, Holliday junction; tr-FRET, time-resolved fluorescence resonance energy transfer; NMR, nuclear magnetic resonance.
References
- 1.Holliday R. Genet Res. 1964;5:282–304. [Google Scholar]
- 2.Meselson M S, Radding C M. Proc Natl Acad Sci USA. 1975;72:358–361. doi: 10.1073/pnas.72.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szostak J W, Orr-Weaver T L, Rothstein R, Stahl F W. Cell. 1983;33:24–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 4.Radding C. Annu Rev Genet. 1982;16:405–437. doi: 10.1146/annurev.ge.16.120182.002201. [DOI] [PubMed] [Google Scholar]
- 5.Smith G R. Cell. 1983;34:709–710. doi: 10.1016/0092-8674(83)90525-1. [DOI] [PubMed] [Google Scholar]
- 6.Whithouse H L K. Genetic Recombination: Understanding the Mechanism. New York: Wiley; 1982. [Google Scholar]
- 7.Hicks J B, Strathern J N, Klar A J S. Nature (London) 1979;282:478–481. doi: 10.1038/282478a0. [DOI] [PubMed] [Google Scholar]
- 8.Haber J E, Mascioli D W, Rogers D T. Cell. 1980;20:519–528. doi: 10.1016/0092-8674(80)90638-8. [DOI] [PubMed] [Google Scholar]
- 9.Rothstein R. Cold Spring Harbor Symp Quant Biol. 1984;49:629–637. doi: 10.1101/sqb.1984.049.01.071. [DOI] [PubMed] [Google Scholar]
- 10.Hastings P J. In: Meiosis. Moens P B, editor. Orlando, FL: Academic; 1987. pp. 107–156. [Google Scholar]
- 11.Lilley D M J, Clegg R M. Q Rev Biophys. 1993;26:1311–1375. doi: 10.1017/s0033583500004054. [DOI] [PubMed] [Google Scholar]
- 12.Seeman N C, Kallenbach N R. Annu Rev Biophys Biomol Struct. 1994;23:53–86. doi: 10.1146/annurev.bb.23.060194.000413. [DOI] [PubMed] [Google Scholar]
- 13.Duckett D R, Murchie A I H, Diekmann S, von Kitzing E, Kemper B, Lilley D M. Cell. 1988;55:79–89. doi: 10.1016/0092-8674(88)90011-6. [DOI] [PubMed] [Google Scholar]
- 14.Churchill M E A, Tullius T D, Kallenbach N R, Seeman N C. Proc Natl Acad Sci USA. 1988;85:4653–4656. doi: 10.1073/pnas.85.13.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller J E, Kemper B, Cunningham R P, Kallenbach N R, Seeman N C. Proc Natl Acad Sci USA. 1988;85:9441–9445. doi: 10.1073/pnas.85.24.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J-H, Churchill M E A, Tullius T D, Kallenbach N R, Seeman N C. Biochemistry. 1988;27:6032–6038. doi: 10.1021/bi00416a031. [DOI] [PubMed] [Google Scholar]
- 17.Guo Q, Lu M, Kallenbach N R. Biopolymers. 1991;31:359–372. doi: 10.1002/bip.360310402. [DOI] [PubMed] [Google Scholar]
- 18.Seeman N, Kallenbach N R. Biophys J. 1983;44:201–209. doi: 10.1016/S0006-3495(83)84292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eis P S, Millar D P. Biochemistry. 1993;32:13852–13860. doi: 10.1021/bi00213a014. [DOI] [PubMed] [Google Scholar]
- 20.Haas E, Wilchek M, Katchalski-Katzir E, Steinberg I Z. Proc Natl Acad Sci USA. 1975;72:1807–1811. doi: 10.1073/pnas.72.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlström G, Chazin W J. Biochemistry. 1996;35:3534–3544. doi: 10.1021/bi952571n. [DOI] [PubMed] [Google Scholar]
- 22.Griffey R H, Poulter C D, Bax A, Hawkins B L, Yamaizumi Z, Nishimura S. Proc Natl Acad Sci USA. 1983;80:5895–5897. doi: 10.1073/pnas.80.19.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marion D, Ikura M, Tschudin R, Bax A. J Magn Res. 1989;85:393–399. [Google Scholar]
- 24.Chen S-M, Chazin W J. Biochemistry. 1994;33:11453–11459. doi: 10.1021/bi00204a007. [DOI] [PubMed] [Google Scholar]
- 25.Lu M, Guo Q, Kallenbach N R. Biochemistry. 1991;30:5815–5820. doi: 10.1021/bi00238a001. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Seeman N C. J Mol Biol. 1994;238:658–668. doi: 10.1006/jmbi.1994.1327. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Lee J, Jayaram M. J Biol Chem. 1995;270:19086–19092. doi: 10.1074/jbc.270.32.19086. [DOI] [PubMed] [Google Scholar]