

Abstract
Adoptive immunotherapy — the isolation of antigen-specific cells, their ex vivo expansion and activation, and subsequent autologous administration — is a promising approach to inducing antitumour immune responses. The molecular identification of tumour antigens and the ability to monitor the persistence and transport of transferred cells has provided new insights into the mechanisms of tumour immunotherapy. Recent studies have shown the effectiveness of cell-transfer therapies for the treatment of patients with selected metastatic cancers. These studies provide a blueprint for the wider application of adoptive-cell-transfer therapy, and emphasize the requirement for in vivo persistence of the cells for therapeutic efficacy.
The magnitude and specificity of immune responses to pathogenic organisms have inspired a search for ways to arm the immune system and aim it at a patient’s own metastatic cancer. Knowledge of immune regulation and effector-cell function have grown rapidly in the past decade, and with this knowledge has come incremental gains in the ability to manipulate host immunity and specifically target tumour cells for immune destruction. A convergence of information from the fields of molecular biology and cellular immunology has opened new opportunities for clinical trials using potent immunomodulators and molecularly defined immunogens. The clinical practice of immunotherapy for the treatment of patients with cancer is capitalizing on these advances, and recent efforts in adoptive-cell-transfer (ACT) therapy provide an excellent example of this progress.
A key advance in immunology in the past decade has been the elucidation of the antigenic basis of tumour-cell recognition and destruction. The ultimate effector cell that mediates the rejection of solid tumours in pre-clinical animal models is the CYTOTOXIC T LYMPHOCYTE (CTL). Each CTL expresses a clonotypically unique T-cell-antigen receptor (TCR) that confers specificity for a particular target antigen. The antigens recognized by CTLs consist of peptide fragments, which are bound within the major clefts of MHC-CLASS-I MOLECULES on the cell surface. Cells are exposed to immune-system scrutiny by loading peptide fragments of newly synthesized cellular proteins onto MHC-class-I molecules, which are then transported to the cell surface1.
As in normal cells, the surfaces of tumour cells contain MHC–peptide antigens that reflect their expressed ‘proteome’. T-cell lines can be generated that specifically recognize the HLA-RESTRICTED ANTIGENS that are expressed by tumour cells. These T cells have been used in combination with molecular cloning techniques to identify many genes that encode tumour antigens, and the peptide epitopes derived from them2. Tumour antigens have been conceptually grouped into categories based on the genes that encode them. Some tumour antigens, such as an epitope from a mutated β-catenin gene3, arise de novo and are unique to individual cancer cells. Other tumour antigens, such as NY-ESO-1(REF. 4), are derived from the aberrant expression of non-mutated genes, the products of which are normally expressed only in testes or fetal tissues. Other tumour antigens, such as MART-1 or gp100 (REFS. 5,6) are derived from non-mutated, cell-lineage-specific proteins. A similar diversity of tumour antigens is recognized by MHC-CLASS-II-restricted CD4+ T CELLS2.
Summary
Adoptive-cell-transfer (ACT) therapy for patients with cancer relies on the ex vivo generation of highly active, tumour-specific lymphocytes, and their administration in large numbers to the autologous host.
Preclinical models have identified characteristics of lymphocyte cultures that are required for successful ACT therapy. The most important characteristic is the presence of high affinity, tumour-antigen-specific CD8+ T cells. There is generally a direct correlation between treatment efficacy and the number of transferred, tumour-specific cells.
Preclinical models have also identified ways to manipulate the host immune environment that increase ACT therapeutic efficacy. These include immunosuppression before cell administration and concurrent interleukin 2 administration with the transferred T cells.
ACT therapy directed at viral antigens has been effective for elimination of Epstein–Barr virus (EBV)-induced post-transplant lymphoproliferative disease. EBV-specific lymphocyte cultures suitable for ACT therapy were generated by repetitive in vitro stimulation using EBV-transformed lymphoblastoid lines.
Lymphocyte cultures that were selected for reactivity against melanoma antigens, including melanocyte-differentiation antigens, mediated cancer regression in some patients with metastatic melanoma. Melanoma-reactive cultures that were suitable for ACT therapy were generated from tumour-infiltrating lymphocytes that were rapidly expanded with anti-CD3 antibody.
The generation of tumour-antigen-specific lymphocyte cultures is evolving rapidly, spurred on by the identification of tumour antigens and the T-cell receptors that recognize them.
Further improvements to ACT therapy will depend on a deeper understanding of basic immunological processes, including the role of CD4+ T cells in the antitumour inflammatory response, the ability of lymphocytes to persist in vivo and travel to tumours, and the mechanisms of ACT augmentation by previous host immunosuppression.
The productive engagement of a TCR by cognate MHC/antigen complexes on the target-cell surface triggers the CTL’s effector functions and can result in the destruction of the target cell. CTLs induce target-cell destruction through the release of inflammatory cytokines (such as tumour-necrosis factor-α and interferon gamma (IFN-γ)), the induction of apoptosis-inducing proteins (including FAS ligand (FASL) and TNF-related apoptosis-inducing ligand (TRAIL)) and cytotoxic degranulation, which leads to perforin-mediated lysis7,8. However, the existence of tumour-reactive CD8+ T CELLS in the peripheral circulation of a patient or an experimental animal is not sufficient to cause the rejection of an established tumour. Clinical trials using peptide antigens for vaccination succeeded in routinely generating tumour-reactive CTLs in patients9,10, but vaccination alone only sporadically induced tumour regression in patients with metastatic disease. Even in transgenic mice, which were engineered to enable every T cell to express a tumour-reactive TCR, tumours still grew progressively11,12.
The lack of inflammatory rejection of tumours by immunized patients and TCR transgenic mice is not well understood at the cellular and molecular level. Many mechanisms could account for the failure of antigen-specific CD8+ T cells to eliminate antigen-expressing tumour cells in vivo (FIG. 1). For instance, the tumour-antigen-specific T cells themselves could be functionally deficient, rendered ANERGIC, or unable to fully differentiate in the tumour environment13. The tumour environment could lack a ‘danger signal’ or other innate immune stimulation, preventing a general inflammatory reaction from evolving14. Alternatively, active immune regulatory mechanisms such as CD4+CD25+ T cells might impede any endogenous immune reaction to cancer cells15,16. Whatever the mechanism, without an inflammatory immune response, the CD8+ T cells of the adaptive immune system are rendered ineffective. As a tumour grows and metastasizes, additional systemic immunosuppression could develop, and antigen-escape variants of the tumour could arise17.
Figure 1. Mechanisms that limit immune responses.
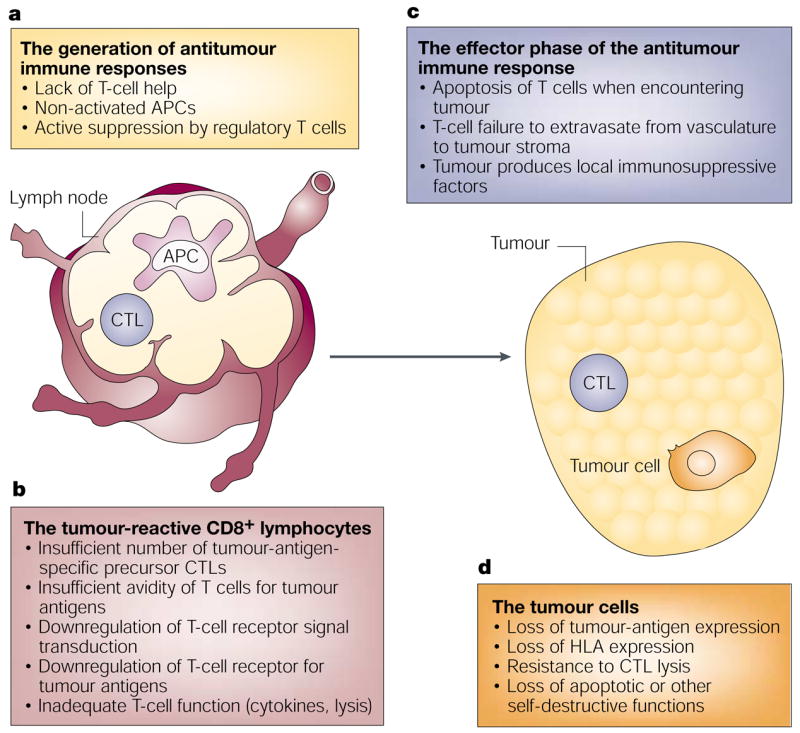
Many mechanisms could account for the limited effectiveness of endogenous or vaccine-induced immune responses to tumours. a | Inadequacies of the afferent stages of an antitumour immune response might include a lack of helper T cells, non-activated antigen-presenting cells (APCs), or active suppression by CD4+CD25+ regulatory T cells. b | The CD8+ cytotoxic T lymphocytes (CTLs) could be insufficient in number, or deficient in T-cell-receptor avidity or receptor signalling, or express sub-optimal function (low lysis or T-helper-2-cell polarized cytokine release). c | The efferent phase of the immune response might be blocked by mechanisms including failure of T cells to traffic to tumour sites, production of immunosuppressive factors by the tumour, or CTL apoptosis on encountering tumour cells. d | Finally, the tumour cells might acquire resistance to CTL attack through loss of tumour-antigen expression, loss of human-leukocyte antigen (HLA) expression, acquisition of resistance to CTL lysis, or loss of capacity for apoptosis.
This bleak picture of tumour immune evasion has engendered many strategies for immunotherapeutic intervention. ACT therapy is, however, one of the most promising immunotherapy approaches. Several strategies are simultaneously used to allow ACT to overcome tumour-escape mechanisms and enable a successful immune attack against metastatic disease (BOX 1). With this approach, an investigator first selects effector T cells with optimal characteristics, such as highly avid tumour antigen specificity. The selected lymphocytes are then expanded and activated ex vivo, circumventing in vivo host immune-regulatory mechanisms and potentially suppressive tumour influences. Finally, the patient receives systemic immunosuppressive chemotherapy before ACT, avoiding any toxic effects to the relevant effector cells and generating a host environment that is conducive to the function of the transferred cells. These components of ACT therapy have been shown to generate an inflammatory response that is capable of destroying growing tumours. In practice, this strategy has led to substantial tumour regressions in patients with melanoma or lymphoma18,19, and promises to provide treatment options for other refractory-tumour types.
Box 1 | Adoptive-cell-transfer therapy for patients with cancer.
Adoptive-cell-transfer therapy provides three opportunities for immune manipulation, which are not readily achieved with other immunotherapeutic approaches. First, highly active, tumour-reactive lymphocyte cultures with optimal characteristics can be selected. For patients with melanoma, the generation of tumour-infiltrating lymphocyte (TIL) cultures was optimized to produce highly avid, tumour-antigen-reactive cells that secreted high levels of interferon-γ (IFN-γ). This was accomplished by establishing several, independent TIL lines and assaying each line for recognition of a panel of melanoma cells. The most active TIL lines were identified by enzyme-linked immunosorbent assay (ELISA) and were selected for further expansion (see a). Second, lymphocyte cultures can be rapidly expanded ex vivo, circumventing normal immune-regulatory mechanisms and obviating a potentially suppressive tumour environment. For treatment of patients with melanoma, the highly selected TIL cultures were rapidly expanded using anti-CD3 antibody, exogenously supplied interleukin 2 and irradiated allogeneic peripheral blood mononuclear ‘feeder’cells (see b). Third, a patient can receive systemic immunosuppression without compromising the antitumour lymphocytes that are being cultured ex vivo. Patients with melanoma received a non-myeloablative but lymphodepleting chemotherapy regimen (see c). These three immune manipulations can be theoretically applied to treat patients with any tumour histology, and they have been successfully combined for the treatment of patients with metastatic melanoma.

ACT — preclinical studies
Our understanding of tumour immunology has been greatly influenced by ACT studies in animal models. These ACT studies provided the earliest evidence that the immune system could sustain a therapeutic effect against established tumours. Early ACT studies involved splenocytes from immunized mice that were used to treat transplanted SYNGENEIC TUMOURS — more recently these studies have been refined, using TCR-transgenic mouse models. The early studies concentrated on defining the characteristics of the transferred T cell that influenced treatment effectiveness. CD8+ T cells were found to be absolutely required for antitumour effects in many models20,21, and CD4+ T cells were also required for effective treatment of some tumours22–24. In addition, the number of cells transferred was directly correlated with treatment efficacy. Adoptively transferred CD8+ T cells could eliminate only tumour cells that expressed their cognate antigen; recently, a correlation between the antigen avidity of the transferred T cells and efficacy in ACT therapy has been shown25. Some functional requirements of the cells for effective ACT were elucidated in animal models, including a correlation between IFN-γ production by T-cell cultures and therapeutic efficacy26,27. The definition of cellular characteristics that are required for therapeutic efficacy in mouse models has led to the examination of similar cellular traits in clinical trials.
Characterization of the transferred T cells has been complemented by analysis of systemic treatments that could improve ACT therapy in mouse models. For example, administration of T-cell growth factors, such as interleukin-2 (IL-2), has greatly improved the persistence and effectiveness of transferred cells28–31. Vaccination with tumour antigens after cell transfer can also increase the persistence of the antigen-specific T cells and improve their effectiveness in mouse tumour-therapy models32,33. This provides a rationale for pursuing vaccination strategies along with ACT therapy in the clinic. Similarly, previous immune suppression markedly improved the efficacy of transferred cells in several mouse models of tumour therapy34–37, and this strategy has also been tested clinically18. The relative contributions of these or other mechanisms of host immune manipulation to ACT therapy remain to be fully elucidated in both murine and human systems.
ACT therapy for patients
Preclinical studies have clearly shown the potential for ACT therapy, but a direct translation of the preclinical findings to the clinical treatment of cancer has been hampered as tumour-antigen-specific effector cells are difficult to isolate.
Epstein–Barr-virus-expressing tumours
This problem has been successfully addressed in studies of patients with Epstein–Barr virus (EBV)-induced lymphoma. Most healthy adults have a persistent but cryptic EBV infection that is held in check by antiviral immune mechanisms. By contrast, the suppressed immune systems of patients who have received bone-marrow or solid-organ transplants are not able to mount an effe tive response against activated virus. Immunosuppressed patients who have received a transplant are at an increased risk of developing EBV-induced lymphoproliferative disease and lymphoma. ACT therapy can effectively treat EBV-transformed B-cell-lineage (EBV-B)-induced lymphoproliferation and restore anti-EBV immunity19,38–40, and the treatment of patients with EBV-related lymphoma using viral-antigen-specific T cells has become a paradigm for ACT therapy.
The constitutive production of viral antigens by EBV-induced tumours makes them highly susceptible to immune attack. In patients who received bone-marrow transplants, infusions of small numbers of unselected peripheral blood lymphocytes (PBLs) from the bone-marrow donor could mediate antiviral and anti-lymphoma effects41. However, donor lymphocyte infusions could also initiate or exacerbate GRAFT-VERSUS-HOST DISEASE, and more specific cellular therapies were sought. Lymphocyte cultures highly enriched for EBV-antigen-specific T cells were generated by repetitive in vitro stimulation of PBLs with EBV-B lymphoblastic cell lines19. These EBV-B cells efficiently presented viral antigens, and provided other CO-STIMULATION necessary to activate and expand T cells in vitro. Starting with PBLs, several rounds of EBV-B stimulation generated cultured T cells that were ideal for ACT therapy. These cultured cells were highly tumour reactive, phenotypically heterogeneous (containing both CD8+ and CD4+ T cells) and maintained their proliferative potential. Most importantly, these cultures were effective in ACT therapy for inhibiting EBV-induced lymphoproliferation. This method of repetitive in vitro restimulation to increase the fraction of tumour-specific cells generates effector lymphocyte populations, and these could be used in ACT therapy of other tumour types.
Non-viral-antigen-expressing tumours
For patients with tumours that do not express viral antigens, the development of ACT therapies has had limited success because the generation of tumour-antigen-specific cells has been problematic. Many clinical trials were undertaken using non-specific, activated lymphocytes to treat patients with metastatic cancer. For instance, the generation and transfer of LYMPHOKINE-ACTIVATED KILLER CELLS (LAKs) has been extensively investigated in preclinical and clinical models42–44. Others have tried non-specific PBLs that were activated in vitro with anti- CD3 antibodies45–47. Some of these trials with non-specific cells resulted in sporadic or inconsistent tumour responses. In other trials, the concurrent use of IL-2 or chemotherapy with the transfer of non-specific cells confounded the interpretation of response rates. A prospective randomized trial did not show a statistically significant benefit of LAK ACT therapy in patients with metastatic renal-cell carcinoma or melanoma compared with IL-2 therapy alone48. Overall, the low response rates reported in these clinical studies, together with data from many animal treatment models, indicate that tumour-antigen-specific lymphocytes are crucial for successful ACT therapy in patients with solid malignancies.
Generating antigen-specific lymphocytes
Several ideas for improving the generation and selection of antigen specific lymphocyte cultures for ACT therapy have been tested clinically. One approach involved vaccinating patients with their autologous tumour cells, harvesting the tumour-vaccine-draining lymph node, and stimulating the recovered lymphocytes for ACT in vitro49–52. Some tumour specificity in the resulting lymphocyte cultures was shown in vitro, and these studies have led to improved methods for activating and culturing lymphocytes to clinically relevant numbers. However, the limited clinical benefit that is seen in most of these clinical trials emphasizes the need for increased specificity of tumour-antigen-reactive cells for successful ACT therapy. Additional methods for expanding tumour-antigen-specific lymphocytes from the peripheral blood of immunized patients need to be explored.
The molecular identification of tumour antigens has enabled the investigation of new approaches to generate tumour-specific cultures for ACT. One approach involves the use of dendritic cells that have been pulsed with peptide epitopes from melanoma antigens to stimulate PBLs isolated from patients with melanoma. This approach is similar to that used to generate EBV-specific cultures. In a pilot trial with eight patients, ACT with these cells was found to be safe and well tolerated, modest T-cell persistence was observed, and one objective partial response to treatment was reported53. Although this approach is promising, it has been logistically difficult to generate sufficient numbers of dendritic cells to drive large, clinical-scale expansions of lymphocyte cultures. Some investigators have suggested the replacement of dendritic cells with artificial antigen-presenting cells54–57 for T-cell expansion, and this approach might hold promise for future clinical studies.
The ultimate example of antigen specificity is embodied in the application of cloned T cells to ACT therapy. The adoptive transfer of T-cell clones was effective in preventing complications from cytomegalovirus (CMV) infection in some bone-marrow-transplant recipients58. CMV infection, like EBV reactivation, constitutes an important medical problem for immunosuppressed transplant patients. CMV-specific lymphocyte cultures were generated from the PBLs of bone-marrow donors through in vitro stimulation with CMV-infected fibroblasts. Individual virus-specific clones were then isolated and expanded in vitro and transferred to the marrow recipient for CMV prophylaxis. These CMV-specific CD8+ T-cell clones were shown to be successful in preventing acute infections in the post-transplant period. Interestingly, transferred clones did not persist in all patients, and the endogenous reconstitution of antiviral CD4+ immunity was correlated with CD8+ T-cell persistence.
In contrast to the clinical efficacy that is seen in trials with anti-CMV clones for viral prophylaxis, cloned melanoma-reactive T cells were ineffective at mediating marked tumour regression. Objective tumour regressions (defined as greater than a 50% decrease in the sum of the products of perpendicular diameters of all lesions with no growth in any lesion) were not seen in immunocompetent or immunosuppressed patients when clones were transferred with or without IL-2 administration59–61. In our studies, transferred tumour-reactive clones disappeared rapidly from the circulation of patients, whether monitored by sensitive fluorescence-cytometry or polymerase-chain-reaction methods (BOX 2). In a study by Yee et al., extended administration of low-dose IL-2 enhanced the clonal-lymphocyte survival, but was not sufficient to induce objective tumour responses. Clones that reacted against HIV antigens also failed to persist in vivo, when tested for their ability to treat patients with HIV infection62,63. An important conclusion from these studies is that highly avid recognition of tumour antigens by the transferred lymphocyte culture is necessary, but is not sufficient, for treatment efficacy. Several explanations could account for the lack of clonal persistence and therapeutic efficacy in these ACT trials. These include a requirement for antigen-specific CD4+ T cells64; proliferative exhaustion or terminal differentiation of clones during expansion62; aberrant transport or reduced survival of ineffective cells65; or selection by clones of antigen-loss tumour variants in vivo61.
Box 2 | Assessing T-cell persistence after transfer.
Monitoring the persistence and localization of adoptively transferred T cells is crucial for evaluating and improving adoptive-cell-transfer (ACT) therapies, and ultimately for understanding the interaction between the immune system and growing tumours. Several new approaches are complementing well-established techniques to reveal the fate and in vivo function of adoptively transferred T cells:
Gene marking. Exogenous DNA can be stably integrated into the genome of T cells before adoptive transfer by retroviral transduction or other means. The ‘marked’ cells can be quantified by polymerase-chain-reaction (PCR) analysis of the inserted gene, or re-isolated after transfer by functional selection for the product of the transferred gene (for example, G418 resistance conferred by the neomycin-transferase gene).
Reverse-transcriptase PCR (RT-PCR) approaches. Each T cell expresses a single T-cell antigen receptor (TCR), generated by several clonotypic somatic recombination events at both the TCRβ and TCRα gene loci. A mature TCRβ-chain messenger RNA (mRNA) is depicted with the linearly arrayed constant (C), junctional (J), diversity (D) and variable (V) regions followed by a poly-A tail. Judicious choice of PCR primers allows the tracking of individual clones within a bulk population of T cells or within a tissue that consists of heterogeneous cell populations. A general survey of the T-cell repertoire is possible using a complete family of TCRβ-chain-variable (TCRBV) forward primers and a single TCR-C-region reverse primer (BV specific). Clone-specific primers can be designed based on the clonotypically unique VDJ junctional sequences generated by somatic recombination during T-cell ontogeny. In combination with the expressed TCRBV-specific primer, clonotype-specific PCR can be accomplished. Sequencing of many cDNA clones obtained by random amplification of cDNA ends (RACE) using a TCRBV primer can provide a quantitative assessment of the frequency of individual T cells.
TCR-fluorescence cytometry (FACS) approaches. A panel of fluorescently labelled antibodies that recognize structural components of individual TCRBV regions can be used to survey skewing of the TCR repertoire. Alternatively, fluorescently labelled HLA–peptide-antigen complexes can be multimerized to increase their TCR-binding avidity. These reagents (HLA–antigen tetramers) can be used to label T cells for quantitative, antigen-specific fluorescence cytometry.
Functional assays. Established methods that measure T-cell function, such as cytokine release, lysis and proliferation assays, are now complemented by methods that count the individual cells that contribute to these bulk effects. These methods include FACS-based methods that use anti-cytokine antibodies to measure intracellular cytokine release, and enzyme-linked immunosorbent spot (ELISpot) assays, which use cytokine-specific monoclonal antibodies to detect and quantify cytokine production by individual T cells.

A highly effective method for generating tumour-specific lymphocyte cultures from patients with melanoma was indicated by preclinical studies in mice. Tumour-infiltrating lymphocytes (TILs) from transplantable mouse sarcomas cultured in high levels of IL-2 showed specific lytic activity towards the cognate tumour cells in vitro. These lymphocytes also mediated tumour regression when transferred into tumour-bearing mice31. Some human tumours, including melanoma66–69, renal-cell carcinoma70 and glioma71, could also generate TILs that were suitable for use in ACT therapy. Melanoma lesions seem particularly suited to this approach, as patients with melanoma can be naturally immunized against antigens expressed by their own tumours72, and melanoma lesions seem to be a repository for tumour-reactive T cells. The use of TILs in the treatment of patients with cancer histologies other than melanoma has not been effective, probably because TILs from other tumour types rarely produce CD8+ T cells that recognize the autologous tumour73–77.
TIL cultures generated from melanoma lesions were often highly lytic — especially when tested against their autologous tumour78–80 — and most TILs contained both CD8+ and CD4+ T cells. Recent refinement in methods for growing TIL cultures suitable for ACT therapy resulted in the generation of autologous tumour-reactive lymphocytes from 78% of patients81. In clinical trials at our institution, 34% of immunocompetent patients with melanoma who were treated with bulk TIL administration and high-dose IL-2 therapy achieved objective clinical responses82. Most of the responses, however, were transient, and limited persistence of the transferred cells was seen83.
ACT therapy following lymphodepletion
Several lines of evidence indicate that the host immune environment can markedly impact the efficacy of ACT therapy. Evidence from preclinical models indicated that host immunosuppression before ACT was highly effective for increasing the impact of the transferred cells. The mechanisms of enhancement of ACT therapy by immune suppression have not been unambiguously determined, but several hypotheses have been investigated. In 1982, North described a CD4+ T-lymphocyte population that suppressed the antitumour effects of ACT therapy37. More recently, Sakaguchi et al. showed that the elimination of CD4+CD25+ regulatory T cells can greatly improve adoptive immunotherapy16. Alternatively, lymphocyte depletion might create ‘space’ in the lymphocyte compartment84, and provide homeostatic lymphocyte-survival and proliferative signals such as IL-7 (REF. 85). Others have suggested that the success of ACT therapy for the treatment of EBV-related lymphoma and for prophylaxis of CMV infection is due, in part, to the immunosuppressed status of the patients receiving treatment. These examples constitute a strong rationale for the use of clinical trials to investigate the combination of lymphodepletion and ACT therapy.
We recently investigated the addition of a non-myeloablative, but lymphodepleting conditioning regimen to ACT therapy for the treatment of patients with metastatic melanoma18 (BOX 1). Patients had received a lymphodepleting chemotherapy regimen consisting of cyclophosphamide and fludarabine before lymphocyte administration. This conditioning regimen caused transient myelosuppression and the virtual elimination of circulating lymphocytes for about a week. Patients typically recovered endogenous marrow function and reconstituted their lymphocyte compartment to normal levels within 2–3 weeks of chemotherapy60. Some patients experienced delayed recovery of CD4+ T-cell counts and extended immunosuppression. Highly selected, highly expanded, tumour-reactive TIL cultures were administered to the lymphodepleted patients, who also received IL-2 therapy. This treatment resulted in the substantial regression of metastatic melanoma deposits in some treated patients.
A summary of this treatment approach for the first 13 HLA-A2 patients treated is shown in TABLE 1. All 13 patients involved in the study had progressive metastatic melanoma before ACT therapy, despite having undergone several previous treatments that included surgery, experimental immunotherapy, high-dose IL-2 therapy and (for many) aggressive chemotherapy. Six patients showed a greater than 50% reduction at all tumour sites, and four others showed mixed responses with substantial shrinkage of some lesions, but growth at other sites. Significant levels of tumour regression occurred in metastatic deposits in the lungs, liver, cutaneous and subcutaneous tissues, and lymph nodes (FIG. 2). The tumour regressions observed in some patients responding to ACT therapy were accompanied by striking immunological findings. TIL transfer following non-myeloablative conditioning led to the in vivo proliferation of the transferred cells in some patients. These cells repopulated the patients’ immune systems and persisted at high levels in the peripheral blood. Two patients experienced stable engraftment of self-antigen-reactive T-cell clones, which made up over 70% of circulating CD8+ T cells, for many months after TIL administration. This engraftment and long-term persistence of tumour-specific lymphocytes in patients with metastatic cancer represents the achievement of one of the main goals of the immunotherapy of patients with cancer.
Table 1.
Clinical outcome of chemotherapy and adoptive cell transfer for 13 HLA-A2+ patients18.
| Patient | Age/sex | Sites of evaluable metastases | Cells (×1010) | IL-2 (doses) | Antigen specificity | Response (duration in months) | Autoimmunity |
|---|---|---|---|---|---|---|---|
| 1 | 18/M | Axillary, mesenteric, pelvic lymph nodes | 2.3 | 9 | Autologous | PR (29) | None |
| 2 | 30/F | Cutaneous, subcutaneous | 3.5 | 8 | MART-1, gp100 | PR (8) | Vitiligo |
| 4 | 57/F | Cutaneous, subcutaneous | 3.4 | 9 | Autologous, gp100 | PR (2) | None |
| 6 | 37/F | Lung, intraperitoneal, subcutaneous | 9.2 | 6 | Autologous | PR (17+) | None |
| 9 | 57/M | Cutaneous, subcutaneous | 9.6 | 10 | Autologous, MART-1 | PR (11) | Vitiligo |
| 10 | 55/M | Lymph nodes, cutaneous, subcutaneous | 10.7 | 12 | MART-1 | PR (14) | Uveitis |
| 5 | 53/M | Brain, lung, lymph nodes | 3.0 | 7 | Autologous, gp100 | NR/m | None |
| 7 | 44/M | Lymph nodes, subcutaneous | 12.3 | 7 | Autologous, MART-1 | NR/m | Vitiligo |
| 11 | 29/M | Liver, pericardial, subcutaneous | 13.0 | 12 | MART-1 | NR/m | Vitiligo |
| 12 | 37/F | Liver, lung, gallbladder, lymph nodes | 13.7 | 11 | Autologous, MART-1 | NR/m | None |
| 3 | 43/F | Brain, cutaneous, liver, lung | 4.0 | 5 | Autologous, gp100 | NR | None |
| 8 | 48/M | Subcutaneous | 9.5 | 12 | Gp100 | NR | None |
| 13 | 41/F | Subcutaneous | 7.7 | 7 | MART-1 | NR | None |
| Total | |||||||
| 13 | 6 PR, 4 NR/m, 3NR | 5+/8− | |||||
All patients enrolled in this clinical trial had refractory metastatic melanoma after high-dose interleukin (IL)-2 therapy. Patients were treated before adoptive-cell-transfer (ACT) therapy with chemotherapy consisting of 2 days of cyclophosphamide at 60 mg per kg of body weight followed by 5 days of fludarabine at 25 mg/m2. Cells were infused on the first day following chemotherapy by intravenous administration over 30–60 minutes, and patients received high-dose IL-2 therapy consisting of 720,000 IU/kg by bolus intravenous infusion every 8 hours to tolerance. Some patients with mixed or minor responses received an additional course of ACT therapy. Antigen specificity was determined by cytokine-release assays. Autologous, recognition of autologous tumour cells mediated by undetermined antigens; gp100, recognition of the HLA-A2-restricted gp100 peptide; MART-1, recognition of the HLA-A2-restricted ‘melanoma antigen recognized by T cells’ (MART)-1 peptide; NR, no objective response; NR/m, partial response of some lesions with progression at other sites; PR, partial response (greater than 50% reduction in the sum of the cross-sectional area of all lesions with no progressive lesion and no new lesion).
Figure 2. Tumour regression by adoptive-cell-transfer therapy.
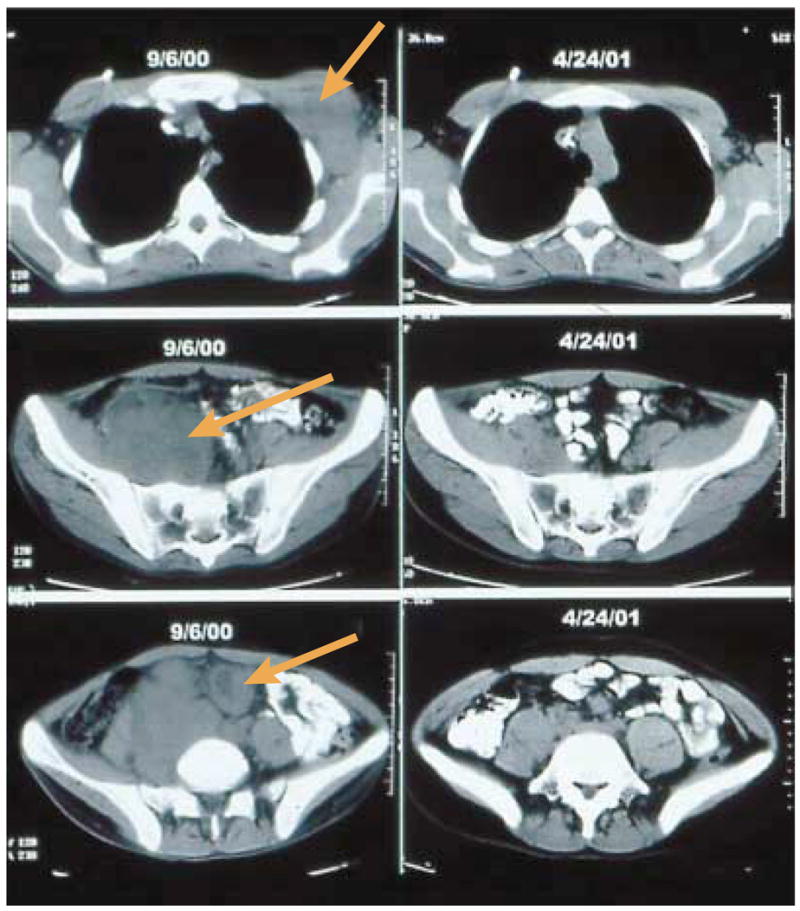
Activated T cells can mediate the regression of a large excess of metastatic melanoma. Computed tomography scans of the trunk and pelvis of one patient document the regression of bulky metastases (arrows) in axillary (top), pelvic (middle) and mesenteric (bottom) lymph nodes, mediated by adoptive-cell-transfer therapy following non-myeloablative but lymphodepleting chemotherapy. Tumour deposits were present before treatment and substantially shrank or completely resolved when evaluated 8 months later.
Interestingly, five of the patients also experienced the autoimmune destruction of normal melanocytes. Vitiligo (skin depigmentation) was seen in four patients, and has been previously described in melanoma patients receiving an immune-based therapy86,87. Autoimmune inflammation of the eye, uveitis, was effectively treated with local steroids in one additional patient treated in this trial. The concordant onset of melanoma regression and autoimmune melanocyte destruction raises interesting parallels between autoimmunity and tumour therapy. Others have reported a link between these immune phenomena in mouse models and in human patient populations. Successful, antigen-directed immunotherapy of the B16 murine melanoma also led to the onset of normal melanocyte destruction88,89. Furthermore, non-specific suppression of immune regulation through CTLA-4 blockade in patients with melanoma led to tumour regression in some patients90 that was also associated with autoimmune toxicity. These results indicate that autoimmunity and tumour rejection share some underlying mechanisms or require immune responses of a similar magnitude. These results also indicate that ACT therapy is capable of unleashing a potent tissue-destructive capacity, and the challenge for the future is to learn to direct these responses to antigens that are unique to tumours or that are expressed only on non-essential normal tissues.
Improving ACT therapy
Recent clinical success with ACT therapy is motivating additional investigation of this treatment modality. Substantial improvements in clinical outcomes for patients can be obtained by improvements in generating T cells for transfer and in manipulating the host immune environment to enhance in vivo antitumour activities. The generation of active, tumour-specific lymphocyte cultures with the characteristics necessary for in vivo effectiveness remains a considerable obstacle to the application of ACT therapy. The molecular characterization of tumour antigens has created the possibility of using these defined tumour antigens to generate tumour-specific lymphocyte cultures for patient treatment by repetitive in vitro stimulations. Another approach involves the genetic engineering of T cells to express a defined antigen specificity. Several investigators have reported that the transfer to PBLs of a gene encoding a tumour-antigen-specific T-cell receptor conferred specific recognition of tumour cells to the T cells87,91,92. Clinical trials with these genetically enginereed lymphocytes will soon begin to assess the utility of this approach.
Concurrently, a growing appreciation of lymphocyte characteristics other than antigen specificity that can impact lymphocyte behaviour in vivo is influencing the practice of ACT therapy. For instance, the discovery that the maturation state of CD8+ T lymphocytes determines their in vivo transport and persistence during an immune response93,94 indicates new avenues for manipulating T-cell behaviour. These might include the alteration of in vitro T-cell cultures to maintain a central memory phenotype — for instance by altering the cytokine milieu during T-cell growth —or genetically engineering lymphocytes to constitutively express their own growth factors that would enhance survival after cytokine withdrawal in vivo95. A more detailed understanding of these and other factors influencing CD8+ T-cell persistence in vivo could improve ACT therapy.
Rapid progress in understanding the role of the host immune environment on tumour therapy is also likely to impact on the practice of cell-based therapies. For instance, the dual role of CD4+ T cells in tumour immunity has important clinical implications. The recent reports describing a requirement of CD4+ T cells for persistence of CD8+ T cells after an immune response96,97 underscores a potential role of CD4+ T cells in TIL cultures for ACT therapy. By contrast, the demonstration that CD4+CD25+ T cells suppress autoimmunity and might be potent inhibitors of antitumour effects in mice indicates a rationale for additional investigation of lymphodepleting conditioning for ACT therapy. These opposing effects of CD4+ T cells highlight the need for additional investigation of CD4+ T-cell-mediated antitumour immunity. A better understanding of the homeostatic regulation of T-cell number and activation, and the role of concurrent vaccination after T-cell transfer has similar potential for improving ACT therapy. Each of these manipulations of the host immune environment is being translated for clinical investigation in ACT clinical protocols. These technologies can be combined with emerging strategies and new biological therapeutics for systemic immune stimulation to improve and enhance the scope and efficacy of ACT therapy.
Supplementary Material
- CYTOTOXIC T LYMPHOCYTES (CTLs)
T lymphocytes that exert a cytolytic function following engagement by their T-cell antigen receptor on target cells. CTLs express the co-receptor CD8 and recognize antigenic peptides (or CTL epitopes) that are presented by HLA-class-I molecules.
- MHC-CLASS-I MOLECULES
Highly polymorphic glycoproteins that are expressed by every nucleated cell of vertebrates, and that are encoded by the gene cluster ‘major histocompatibility complex’ (MHC). The human MHC molecules are termed HLA (human leukocyte antigen) molecules. MHC-class-I molecules mainly present peptides from intracellular proteins to cytotoxic T cells.
- HLA-RESTRICTED ANTIGENS
T-cell receptors recognize antigen peptides on the surface of antigen-presenting cells in the context of an HLA molecule. Each HLA allele can bind only a fraction of the potential peptide pool, and this ‘restricts’ the peptide repertoire. So, the HLA-restricted antigenic peptides provide a unique immunological ‘signature’ that allows immune discrimination of tumour cells, but not normal cells.
- MHC-CLASS-II MOLECULES
Peptide receptors, similar to class-I molecules in structure and function, but expressed by a small set of professional antigen-presenting immune cells. They mainly present peptides from extracellular proteins to T-helper cells.
- CD4+ T CELLS
T cells bearing the CD4 surface glycoprotein, which recognizes MHC-class-II molecules. These cells provide helper function to CD8+ T cells through the release of cytokines and the activation of professional antigen-presenting cells.
- CD8+ T CELLS
T cells bearing the CD8 cell-surface glycoprotein, which recognizes MHC-class-I molecules on target cells. CD8+ T cells are usually cytotoxic T cells.
- ANERGIC T CELLS
T cells that are unable to undergo proliferation, secretion of inflammatory cytokines or other functions in response to antigens.
- SYNGENEIC TUMOURS
Tumours derived from mice with an identical genetic background; specifically, the same inbred line.
- GRAFT-VERSUS-HOST DISEASE
A toxic reaction that is mediated by donor-derived T lymphocytes within the graft towards the recipient’s organs. The attack is usually directed toward the skin, gut, liver and haematopoietic cells.
- CO-STIMULATION
Optimal stimulation of T-cell proliferation requires two signals. Signal one is transduced through T-cell-antigen receptors. Signal two is generically referred to as co-stimulation; several receptors on T cells have been reported to mediate co-stimulation, including CD28.
- LYMPHOKINE-ACTIVATED KILLER CELLS (LAKs)
Lymphocytes that have been cultured in high concentrations of cytokines (including interleukin 2). They show characteristic functional and phenotypic properties, such as in vitro lysis of tumour cells, in the absence of MHC restriction.
- CD3
A multimeric component of the T-cell-receptor signalling apparatus. Direct activation of T cells by antibody cross-linking of CD3 complexes on the cell surface bypasses the requirement for T-cell receptor engagement of HLA-restricted antigens.
Footnotes
DATABASES
Cancer.gov: http://cancer.gov/melanoma
LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/
β-catenin | CD4 | CD8 | CD25 | CTLA-4 | FASL | gp100 | HLA-A2 | IFN-γ | IL-2 | MART-1 | NY-ESO-1 | TRAIL | tumour-necrosis factor-α
The following terms in this article are linked online to:
FURTHER INFORMATION
Steven A. Rosenberg’s lab: http://ccr.cancer.gov/Staff/Staff.asp?StaffID=615
References
- 1.Shastri N, Schwab S, Serwold T. Producing nature’s gene-chips: the generation of peptides for display by MHC class I molecules. Annu Rev Immunol. 2002;20:463–493. doi: 10.1146/annurev.immunol.20.100301.064819. [DOI] [PubMed] [Google Scholar]
- 2.Renkvist N, Castelli C, Robbins PF, Parmiani G. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother. 2001;50:3–15. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins PF, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185–1192. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jager E, et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. 1998;187:265–270. doi: 10.1084/jem.187.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawakami Y, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawakami Y, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–3968. [PubMed] [Google Scholar]
- 7.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 8.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg SA, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nature Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber J, et al. Granulocyte-macrophage-colony-stimulating factor added to a multipeptide vaccine for resected Stage II melanoma. Cancer. 2003;97:186–200. doi: 10.1002/cncr.11045. [DOI] [PubMed] [Google Scholar]
- 11.Wick M, et al. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med. 1997;186:229–238. doi: 10.1084/jem.186.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prevost-Blondel A, et al. Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. J Immunol. 1998;161:2187–2194. [PubMed] [Google Scholar]
- 13.Ochsenbein AF, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 14.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 15.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nature Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 17.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nature Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. This report describes adoptive cell transfer with TILs following non-myeloablative chemotherapy for treatment of patients with melanoma, and represents the first successful use of this combination therapy for the treatment of solid malignancy in patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rooney CM, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein–Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555. This paper describes the generation and clinical use of lymphocyte cultures for ACT therapy of patients with metastatic cancer. [PubMed] [Google Scholar]
- 20.Overwijk WW, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of ‘self’-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. Demonstration in an animal model that immune manipulation can result in efficacious tumour treatments mediated through unmutated melanocyte-differentiation antigens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai K, Chang AE, Shu SY. Phenotype analyses and cellular mechanisms of the pre-effector T-lymphocyte response to a progressive syngeneic murine sarcoma. Cancer Res. 1990;50:4371–4376. [PubMed] [Google Scholar]
- 22.Surman DR, Dudley ME, Overwijk WW, Restifo NP. Cutting edge: CD4+ T cell control of CD8+ T cell reactivity to a model tumor antigen. J Immunol. 2000;164:562–565. doi: 10.4049/jimmunol.164.2.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang AE, et al. Clinical observations on adoptive immunotherapy with vaccine-primed T- lymphocytes secondarily sensitized to tumor in vitro. Cancer Res. 1993;53:1043–1050. [PubMed] [Google Scholar]
- 24.Peng L, Shu S, Krauss JC. Treatment of subcutaneous tumor with adoptively transferred T cells. Cell Immunol. 1997;178:24–32. doi: 10.1006/cimm.1997.1124. [DOI] [PubMed] [Google Scholar]
- 25.Zeh HJ, 3rd, Perry–Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–994. [PubMed] [Google Scholar]
- 26.Aruga A, Shu S, Chang AE. Tumor-specific granulocyte/macrophage colony-stimulating factor and interferon gamma secretion is associated with in vivo therapeutic efficacy of activated tumor-draining lymph node cells. Cancer Immunol Immunother. 1995;41:317–324. doi: 10.1007/BF01517220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barth RJ, Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991;173:647–658. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cameron RB, Spiess PJ, Rosenberg SA. Synergistic antitumor activity of tumor-infiltrating lymphocytes, interleukin 2, and local tumor irradiation. Studies on the mechanism of action. J Exp Med. 1990;171:249–263. doi: 10.1084/jem.171.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheever MA, Greenberg PD, Fefer A, Gillis S. Augmentation of the anti-tumor therapeutic efficacy of long-term cultured T lymphocytes by in vivo administration of purified interleukin 2. J Exp Med. 1982;155:968–980. doi: 10.1084/jem.155.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou T, Bertera S, Chang AE, Shu S. Adoptive immunotherapy of microscopic and advanced visceral metastases with in vitro sensitized lymphoid cells from mice bearing progressive tumors. J Immunol. 1988;141:1775–1781. [PubMed] [Google Scholar]
- 31.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. This seminal paper describes the use of TILs, together with cyclophosphamide conditioning, to treat advanced tumours in mice. [DOI] [PubMed] [Google Scholar]
- 32.Bristol JA, Schlom J, Abrams SI. Persistence, immune specificity, and functional ability of murine mutant ras epitope-specific CD4(+) and CD8(+) T lymphocytes following in vivo adoptive transfer. Cell Immunol. 1999;194:78–89. doi: 10.1006/cimm.1999.1489. [DOI] [PubMed] [Google Scholar]
- 33.Overwijk WW, et al. Tumor regression and autoimmunity following reversal of a functionally tolerant state of self-reactive CD8+ cells. J Exp Med. doi: 10.1084/jem.20030590. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Awwad M, North RJ. Cyclophosphamide-induced immunologically mediated regression of a cyclophosphamide-resistant murine tumor: a consequence of eliminating precursor L3T4+ suppressor T-cells. Cancer Res. 1989;49:1649–1654. [PubMed] [Google Scholar]
- 35.Berenson JR, Einstein AB, Jr, Fefer A. Syngeneic adoptive immunotherapy and chemoimmunotherapy of a Friend leukemia: requirement for T cells. J Immunol. 1975;115:234–238. [PubMed] [Google Scholar]
- 36.Cheever MA, Greenberg PD, Fefer A. Specificity of adoptive chemoimmunotherapy of established syngeneic tumors. J Immunol. 1980;125:711–714. [PubMed] [Google Scholar]
- 37.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comoli P, et al. Infusion of autologous Epstein–Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood. 2002;99:2592–2598. doi: 10.1182/blood.v99.7.2592. [DOI] [PubMed] [Google Scholar]
- 39.Haque T, et al. Treatment of Epstein–Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet. 2002;360:436–442. doi: 10.1016/S0140-6736(02)09672-1. [DOI] [PubMed] [Google Scholar]
- 40.O’Reilly RJ, et al. Biology and adoptive cell therapy of Epstein–Barr virus-associated lymphoproliferative disorders in recipients of marrow allografts. Immunol Rev. 1997;157:195–216. doi: 10.1111/j.1600-065x.1997.tb00983.x. [DOI] [PubMed] [Google Scholar]
- 41.Lucas KG, Small TN, Heller G, Dupont B, O’Reilly RJ. The development of cellular immunity to Epstein–Barr virus after allogeneic bone marrow transplantation. Blood. 1996;87:2594–2603. [PubMed] [Google Scholar]
- 42.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982;155:1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science. 1984;225:1487–1489. doi: 10.1126/science.6332379. [DOI] [PubMed] [Google Scholar]
- 44.Rosenberg SA, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–1492. doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- 45.Lum LG, LeFever AV, Treisman JS, Garlie NK, Hanson JP., Jr Immune modulation in cancer patients after adoptive transfer of anti-CD3/anti-CD28-costimulated T cells-phase I clinical trial . J Immunother. 2001;24:408–419. doi: 10.1097/00002371-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 46.Curti BD, et al. Phase I trial of anti-CD3-stimulated CD4+ T cells, infusional interleukin-2, and cyclophosphamide in patients with advanced cancer. J Clin Oncol. 1998;16:2752–2760. doi: 10.1200/JCO.1998.16.8.2752. [DOI] [PubMed] [Google Scholar]
- 47.Kim JA, et al. Cellular immunotherapy for patients with metastatic colorectal carcinoma using lymph node lymphocytes localized in vivo by radiolabeled monoclonal antibody. Cancer. 1999;86:22–30. [PubMed] [Google Scholar]
- 48.Rosenberg SA, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. 1993;85:622–632. doi: 10.1093/jnci/85.8.622. [DOI] [PubMed] [Google Scholar]
- 49.Chang AE, et al. Phase II trial of autologous tumor vaccination, anti-CD3-activated vaccine-primed lymphocytes, and interleukin-2 in stage IV renal cell cancer. J Clin Oncol. 2003;21:884–890. doi: 10.1200/JCO.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 50.To WC, et al. Systemic adoptive T-cell immunotherapy in recurrent and metastatic carcinoma of the head and neck: a phase 1 study. Arch Otolaryngol Head Neck Surg. 2000;126:1225–1231. doi: 10.1001/archotol.126.10.1225. [DOI] [PubMed] [Google Scholar]
- 51.Plautz GE, et al. T-cell adoptive immunotherapy of metastatic renal cell carcinoma. Urology. 1999;54:617–623. doi: 10.1016/s0090-4295(99)00303-9. [DOI] [PubMed] [Google Scholar]
- 52.Meijer SL, et al. Adoptive cellular therapy with tumor vaccine draining lymph node lymphocytes after vaccination with HLA-B7/beta2-microglobulin gene-modified autologous tumor cells. J Immunother. 2002;25:359–372. doi: 10.1097/00002371-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 53.Meidenbauer N, et al. Survival and tumor localization of adoptively transferred melan-a-specific T cells in melanoma patients. J Immunol. 2003;170:2161–2169. doi: 10.4049/jimmunol.170.4.2161. [DOI] [PubMed] [Google Scholar]
- 54.Maus MV, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nature Biotechnol. 2002;20:143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 55.Thomas AK, Maus MV, Shalaby WS, June CH, Riley JL. A cell-based artificial antigen-presenting cell coated with anti-CD3 and CD28 antibodies enables rapid expansion and long-term growth of CD4 T lymphocytes. Clin Immunol. 2002;105:259–272. doi: 10.1006/clim.2002.5277. [DOI] [PubMed] [Google Scholar]
- 56.Oelke M, et al. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nature Med. 2003;9:619–624. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 57.Sili U, et al. Large-scale expansion of dendritic cell-primed polyclonal human cytotoxic T-lymphocyte lines using lymphoblastoid cell lines for adoptive immunotherapy. J Immunother. 2003;26:241–256. doi: 10.1097/00002371-200305000-00008. [DOI] [PubMed] [Google Scholar]
- 58.Walter EA, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. This influential paper describes the use of CMV-specific cloned lymphocytes for the treatment of bone-marrow-transplant recipients, and their efficacy in prevention of CMV-induced disease. [DOI] [PubMed] [Google Scholar]
- 59.Dudley ME, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 60.Dudley ME, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. The failure of non-myeloablative chemotherapy followed by administration of highly avid T-cell clones for treatment of patients with melanoma is documented. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. An excellent example of the failure of highly avid T-cell clones to induce objective clinical responses in patients with metastatic solid tumours. T-cell function, transport and persistence were monitored. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan R, et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood. 1999;93:1506–1510. [PubMed] [Google Scholar]
- 63.Brodie SJ, et al. in vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nature Med. 1999;5:34–41. doi: 10.1038/4716. [DOI] [PubMed] [Google Scholar]
- 64.Marzo AL, et al. Tumor-specific CD4+ T cells have a major ‘post-licensing’ role in CTL mediated anti-tumor immunity. J Immunol. 2000;165:6047–6055. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 65.Donawho CK, Pride MW, Kripke ML. Persistence of immunogenic pulmonary metastases in the presence of protective anti-melanoma immunity. Cancer Res. 2001;61:215–221. [PubMed] [Google Scholar]
- 66.Topalian SL, et al. Immunotherapy of patients with advanced cancer using tumor- infiltrating lymphocytes and recombinant interleukin-2: a pilot study. J Clin Oncol. 1988;6:839–853. doi: 10.1200/JCO.1988.6.5.839. [DOI] [PubMed] [Google Scholar]
- 67.Kradin RL, et al. Tumour-infiltrating lymphocytes and interleukin-2 in treatment of advanced cancer. Lancet. 1989;1:577–580. doi: 10.1016/s0140-6736(89)91609-7. [DOI] [PubMed] [Google Scholar]
- 68.Dillman RO, et al. Continuous interleukin-2 and tumor-infiltrating lymphocytes as treatment of advanced melanoma. A national biotherapy study group trial. Cancer. 1991;68:1–8. doi: 10.1002/1097-0142(19910701)68:1<1::aid-cncr2820680102>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 69.Arienti F, et al. Adoptive immunotherapy of advanced melanoma patients with interleukin-2 (IL-2) and tumor-infiltrating lymphocytes selected in vitro with low doses of IL-2. Cancer Immunol Immunother. 1993;36:315–322. doi: 10.1007/BF01741170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Figlin RA, et al. Treatment of metastatic renal cell carcinoma with nephrectomy, interleukin-2 and cytokine-primed or CD8(+) selected tumor infiltrating lymphocytes from primary tumor. J Urol. 1997;158:740–745. doi: 10.1097/00005392-199709000-00012. [DOI] [PubMed] [Google Scholar]
- 71.Quattrocchi KB, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neurooncol. 1999;45:141–157. doi: 10.1023/a:1006293606710. [DOI] [PubMed] [Google Scholar]
- 72.Valmori D, et al. Circulating tumor-reactive CD8(+) T cells in melanoma patients contain a CD45RA(+)CCR7(−) effector subset exerting ex vivo tumor-specific cytolytic activity. Cancer Res. 2002;62:1743–1750. [PubMed] [Google Scholar]
- 73.Finke JH, et al. Characterization of tumor-infiltrating lymphocyte subsets from human renal cell carcinoma: specific reactivity defined by cytotoxicity, interferon-gamma secretion, and proliferation. J Immunother Emphasis Tumor Immunol. 1994;15:91–104. doi: 10.1097/00002371-199402000-00002. [DOI] [PubMed] [Google Scholar]
- 74.Hom SS, Rosenberg SA, Topalian SL. Specific immune recognition of autologous tumor by lymphocytes infiltrating colon carcinomas: analysis by cytokine secretion. Cancer Immunol Immunother. 1993;36:1–8. doi: 10.1007/BF01789124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoshino T, et al. HLA class-I-restricted and tumor-specific CTL in tumor-infiltrating lymphocytes of patients with gastric cancer. Int J Cancer. 1997;70:631–638. doi: 10.1002/(sici)1097-0215(19970317)70:6<631::aid-ijc1>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 76.Nakamura H, Ishiguro K, Mori T. Different immune functions of peripheral blood, regional lymph node, and tumor infiltrating lymphocytes in lung cancer patients. Cancer. 1988;62:2489–2497. doi: 10.1002/1097-0142(19881215)62:12<2489::aid-cncr2820621207>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 77.Schwartzentruber DJ, Solomon D, Rosenberg SA, Topalian SL. Characterization of lymphocytes infiltrating human breast cancer: specific immune reactivity detected by measuring cytokine secretion. J Immunother. 1992;12:1–12. doi: 10.1097/00002371-199207000-00001. [DOI] [PubMed] [Google Scholar]
- 78.Itoh K, Tilden AB, Balch CM. Interleukin 2 activation of cytotoxic T-lymphocytes infiltrating into human metastatic melanomas. Cancer Res. 1986;46:3011–3017. [PubMed] [Google Scholar]
- 79.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142:3714–3725. [PubMed] [Google Scholar]
- 80.Yannelli JR, et al. Growth of tumor-infiltrating lymphocytes from human solid cancers: summary of a 5-year experience. Int J Cancer. 1996;65:413–421. doi: 10.1002/(SICI)1097-0215(19960208)65:4<413::AID-IJC3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 81.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosenberg SA, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–1166. doi: 10.1093/jnci/86.15.1159. A comprehensive summary of the clinical experience with TIL for the treatment of patients with melanoma in the absence of non-myeloablative conditioning. [DOI] [PubMed] [Google Scholar]
- 83.Rosenberg SA, et al. Gene transfer into humans: immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 84.Maine GN, Mule JJ. Making room for T cells. J Clin Invest. 2002;110:157–159. doi: 10.1172/JCI16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fry TJ, Mackall CL. Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001;22:564–571. doi: 10.1016/s1471-4906(01)02028-2. [DOI] [PubMed] [Google Scholar]
- 86.Phan GQ, Attia P, Steinberg SM, White DE, Rosenberg SA. Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J Clin Oncol. 2001;19:3477–3482. doi: 10.1200/JCO.2001.19.15.3477. [DOI] [PubMed] [Google Scholar]
- 87.Yee C, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma. Direct evidence of T cell-mediated vitiligo. J Exp Med. 2000;192:1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Overwijk WW, et al. Vaccination with a recombinant vaccinia virus encoding a ‘self’ antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phan GQ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clay TM, et al. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513. [PubMed] [Google Scholar]
- 92.Hwu P, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55:3369–3373. [PubMed] [Google Scholar]
- 93.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nature Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 94.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–4266. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 95.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol. 2001;167:6356–6365. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.