

Abstract
The dramatic tumor regression observed following adoptive T cell transfer in some patients has led to attempts to identify novel Ags to understand the nature of these responses. Nearly complete regression of multiple metastatic melanoma lesions was observed in patient 1913 following adoptive transfer of autologous tumor-infiltrating lymphocytes. The autologous 1913 melanoma cell line expressed a mutated HLA-A11 class I gene product that was recognized by the bulk tumor-infiltrating lymphocytes as well as a dominant T cell clone derived from this line. A second dominant T cell clone, T1D1, did not recognize the mutated HLA-A11 product, but recognized an allogeneic melanoma cell line that shared expression of HLA-A11 with the parental tumor cell line. Screening of an autologous melanoma cDNA library with clone T1D1 T cells in a cell line expressing the mutated HLA-A11 gene product resulted in the isolation of a p14ARF transcript containing a 2-bp deletion in exon 2. The T cell epitope recognized by T1D1, which was encoded within the frameshifted region of the deleted p14ARF transcript, was also generated from frameshifted p14ARF or p16INK4a transcripts that were isolated from two additional melanoma cell lines. The results of monitoring studies indicated that T cell clones reactive with the mutated HLA-A11 gene product and the mutated p14ARF product were highly represented in the peripheral blood of patient 1913 1 wk following adoptive transfer, indicating that they may have played a role in the nearly complete tumor regression that was observed following this treatment.
A variety of mechanisms have been found to give rise to tumor-associated Ags. The melanocyte differentiation Ags are expressed on melanomas and normal cells with a limited tissue distribution, whereas cancer testis Ags are limited in their expression to selected cancers as well as to the cells in the testis that lack expression of HLA class I and II Ags (1). Genes such as PRAME and FGF-5 are overexpressed in a variety of tumor types (2, 3), and the relatively low levels of expression in normal tissues may not be sufficient to trigger T cell responses. A variety of genetic alterations that include point mutations, nucleotide deletions, as well as chromosomal translocations have been shown to be involved with generating tumor-specific T cell epitopes (4–8).
Although, in the majority of cases, individual mutations appear to be limited to tumors derived from a single patient, particular codons within certain genes have been found to be mutated in tumors isolated from multiple patients, presumably because they encode a protein with an altered function that confers an advantage on cells that express this product. A mutated β-catenin epitope that is naturally recognized by tumor-reactive T cells was expressed in tumor isolated from multiple patients (9), and candidate peptides that encompass a common point mutation present in the ras oncogene have been used to generate tumor-reactive T cells (10). Additional examples of genes that are frequently altered in tumor cells as a result of short nucleotide deletions or insertions include the CDKN2A locus that encodes two tumor suppressor gene products, p14ARF and p16INK4a (11). These two proteins do not contain any sequence similarities, because they are encoded by distinct exon 1 sequences and share a single exon 2 sequence that is, however, translated in two alternative open reading frames (ORFs).2 These two gene products regulate the cell cycle through independent means, because the p16INK4a product binds to the cyclin-dependent kinase 4 or 6 and cyclin D1 complex that negatively regulates progression of mammalian cells through the cell cycle (12), whereas the p14ARF product interacts with MDM2, resulting in stabilization of p53 and G1 arrest (13). A number of studies have demonstrated that germline mutations of p14ARF and/or p16INK4a are associated with melanoma susceptibility, and in addition, both germline and somatic mutations of these two genes appear to predispose individuals to the development of other tumor types (14–17). Within this locus, relatively small deletions or insertions have been observed to occur at frequencies as high as 59% in certain tumor types as well as their premalignant progenitors (14).
In this study, a tumor-infiltrating lymphocyte (TIL) that appeared to result in the nearly complete regression of multiple metastatic lesions following adoptive transfer was shown to recognize a mutated class I HLA gene product, as well as a peptide encoded by frameshifted p14ARF and p16INK4a transcripts. The analysis of peripheral blood samples suggests that T cells reactive with these two mutated Ags may have played an important role in the nearly complete tumor regression that was observed following adoptive TIL transfer.
Materials and Methods
Cell lines
The 1913 melanoma (mel) cell line was established from a metastatic lesion resected in 2001 from a 37-year-old female patient, and a TIL culture was established from the same tumor by culturing dissociated cells in 6000 IU/ml rIL-2. Tumor-reactive T cell clones were derived from this cell line by limiting dilution cloning as previously described (18). Briefly, bulk TIL cultures were plated at 0.6 cells per well in 96-well U-bottom plates in 200 μl of complete medium (CM; RPMI 1640 containing 10% human serum), 30 ng/ml OKT-3 (Ortho Biotech, Raritan, NJ), 300 IU/ml IL-2, and 5 × 104 allogeneic irradiated (34 Gy) PBMC per well. After 7 days of culture, IL-2 was added to all wells to a final concentration of 300 IU/ml. After 14 days of culture, growth-positive microwells were screened for specific recognition of autologous melanoma cells using IFN-γ cytokine release assay. For this assay, one-half of the cells from each microwell were split between replicate plates, one of which contained 2.5 × 104 autologous tumor cells, and the other of which contained 2.5 × 104 HLA-A2+ allogeneic tumor cells. The T cell clones were incubated overnight with the tumor cells, and IFN-γ release into the culture supernatants was determined by ELISA (Pierce/Endogen, Rockford, IL). Ag-reactive microcultures were expanded for further functional screening using a modified rapid expansion protocol with allogeneic irradiated PBMC, IL-2, and OKT-3 in CM as previously described (19). Additional cytokine release assays were conducted as described above using 104 T cells and 2.5 × 104 stimulator cells.
Generation of HLA-A11-expressing cell lines
Initially, an HLA-A11 gene product was isolated from 1913 mel and PBL obtained from patient 1913 by carrying out RT-PCR with the Platinum Taq DNA Polymerase High Fidelity kit (Invitrogen, Carlsbad, CA) using the forward primer EX1-A6 (5′-CAGACGCCGAGGATGGCC) and the reverse primer A-EX8-1 (5′-CACACAAGGCAGCTGTCTCACA) (20) that were designed to amplify HLA-A locus gene products. The HLA-A11 gene product amplified from 1913 lymphocytes corresponded to the normal A*11011 allele, but the product amplified from 1913 mel contained a C-to-T mutation at codon 11 of the mature HLA-A11 gene product that resulted in a substitution of a phenylalanine residue for a serine residue. The normal and mutated HLA-A11 gene products were then cloned into the retroviral vector pCLPCX4 (−). This vector was generated from the pCLNCX vector (21) by digestion with HindIII, followed by insertion of a double-stranded oligonucleotide that destroyed the original HindIII site but that encoded the XhoI, NotI, HindIII, NsiI, BamHI, EcoRI, and BglII restriction sites. The neomycin phosphotransferase gene was then removed by digestion with the SbfI and PmeI restriction endonucleases, followed by treatment with mung bean nuclease and calf intestinal phosphatase (Invitrogen) to generate a blunt-end dephosphorylated fragment. The puromycin gene was removed from pMSCVPure (BD Biosciences, Mountain View, CA) by digestion with HindII and ClaI and a blunt-end fragment generated using Klenow (Invitrogen) amd mung bean nuclease, followed by ligation to the digested vector. Retroviral supernatants were generated by transient transfection of the 293-GP packaging cell line, as previously described (22). Retrovirally transduced cells were selected by incubation with medium containing 1 μg/ml puromycin. A transcript encoding the full-length HLA-A11 gene product that contained the single mutation at codon 11 described above was isolated from the 1913 mel library that had been generated in the expression vector pCMV-Sport6 (Invitrogen) and was used to perform the majority of the transient transfection experiments.
Construction and screening of cDNA library
A cDNA expression library was constructed according to the manufacturer's procedures using 5 μg of poly(A)+ RNA that was isolated from 1913 mel by using the PolyATtract mRNA Isolation Systems kit (Promega, Madison, WI). The cDNA library was divided in pools of ∼100 bacterial colonies per well, and 0.3 μg of DNA was transfected into 45,000 COS cells that were stably transfected with the mutated HLA-A11 gene construct in 96-well plates by using Lipofectamine 2000 (Invitrogen), and 18 h after transfection, 2 × 104 T1D1 T cells were added to each well. Supernatants from each well were then harvested 24 h later and tested for IFN-γ by ELISA. Individual cDNAs isolated from positive wells were tested as above, sequenced using the Big-Dye Terminator V 1.1 kit (Applied Biosystems, Foster City, CA) and analyzed using an ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems).
Peptide synthesis, purification testing
Peptides were synthesized using a solid-phase method based on standard F-moc chemistry on a multiple peptide synthesizer (Gilson, Worthington, OH). The purity of the peptides, as analyzed by mass spectrometry (Tufts Core Facility, Boston, MA) was estimated to be >90%. Lyophilized peptides were resuspended in DMSO and incubated at varying concentrations with target cells, which were then used to stimulate T1D1 T cells in an overnight coculture assay containing 2 × 104 cells, and IFN-γ secretion was measured by ELISA.
RT-PCR and sequence analysis
To identify the p14ARF T cell epitope, two cDNA fragments comprising the region 5′ and 3′ of the mutation site identified in the transcript was cloned from the 1913 mel cell line. Amplification of the upstream region was conducted using a forward primer that encompassed the normal p14ARF translational start site, p14-ARF-NF (5′-CACCCGAGAACATGGTGCGC-3′), and a reverse primer, p14-ARF-NR (5′-GAGGTTGAGTCCGCGACA-3′). Amplification of the region located downstream of the mutation site was conducted using the forward primer p14-ARF-MF (5′-CACCATGAGTTGGAGCTGC-3′), which encompassed residues 217–231 of the mutated p14ARF transcript isolated from 1913 mel and contained a consensus start site to allow efficient translation, along with the reverse primer p14-ARF-MR (5′-TGGCCCAGCTCCTCAGCC-3′), which was complementary to bases 396–413 of the mutated p14 transcript. Amplification of the full-length cDNA encoding p14ARF was conducted using the forward p14-ARF-FF (5′-CACCCGGAGTAGGGCAGG-3′) and reverse p14-ARF-FR (5′-TGGCCCAGCTCCTCAGCC-3′) primers. A truncated cDNA was amplified comprising the region encoding aa 1–123 of the normal p16INK4a using the forward primer p16-INK4a-E3F (5′-GCTGCGGAGAGGGGGAGAGCAGGCA-3′) along with the reverse primer p16-INK4a-E3R (5′-TGGCCCAGCTCCTCAGCC-3′). The reverse-transcriptase reaction was conducted for 1 h at 50°C and 5 min at 85°C, followed by a PCR consisting of a 4-min incubation at 94°C, followed by 35 cycles comprised of incubations of 1 min at 94°C, 1 min at 58°C, and 1 min and 30 s at 72°C, and a final incubation at 72°C for 10 min. The RT-PCR products were then cloned into expression vectors from pcDNA3.1 Directional TOPO or pcDNA3.1/V5-His vectors (Invitrogen). Sequence reactions were conducted using the Big-Dye Terminator V 1.1 kit (Applied Biosystems) using a standard T7 forward primer or a BGH reverse primer and were analyzed using the ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems).
5′-RACE analysis of BV gene expression
Amplification of TCR BV region sequences was conducted using the SMART RACE cDNA Amplification kit (BD Biosciences). Briefly, first-strand cDNA was synthesized from 0.1 to 1 μg of total RNA using an oligo(dT) primer, and amplification was conducted using a primer complementary to the TCR β-chain C region 5′-CTCTTGACCATGGCCATC-3′ and an anchor primer according to the manufacturer's instructions. Following amplification, 5′-RACE products were cloned into the pCDNA3.1 TA cloning vector (Invitrogen).
Results
Recognition of a mutated HLA class I gene product by TIL 1913
Patient 1913 had failed conventional therapy but subsequently demonstrated nearly complete regression of multiple visceral and s.c. metastatic lesions following adoptive transfer of the autologous TIL 1913. Preliminary assays indicated that TIL 1913 failed to recognize the dominant HLA-A2-restricted melanoma Ags MART-1 and gp100 (23), but the gene encoding the second HLA-A allele expressed by 1913 mel cells, HLA-A11, contained a C-to-T alteration that resulted in a substitution of a phenylalanine for a serine residue at aa 11 of the mature HLA-A11 gene product. This represented a mutated gene product and was designated HLA-A11mut, because T cells isolated from patient 1913 expressed a protein that corresponded to the normal HLA-A11 sequence, which was designated HLA-A11N. Additional studies demonstrated that the bulk TIL 1913 recognized COS-7 cells transfected with the HLA-A11Mut but not HLA-A11N (data not shown), and a T cell clone isolated from TIL 1913, T2G10, recognized COS cells transfected with a construct encoding the HLA-A11Mut product but not constructs encoding either the normal HLA-A11N, HLA-A2, or HLA-A3 gene products (Fig. 1).
FIGURE 1.
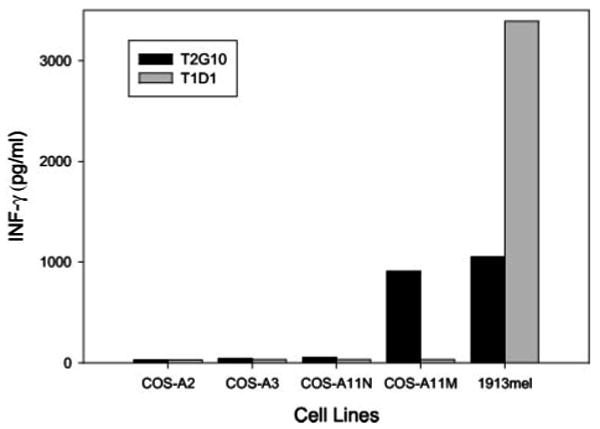
Recognition of HLA-A11Mut by clone T2G10. COS-7 cells were transfected for 24 h with HLA-A11Mut, HLA-A11N, HLA-A3, or HLA-A2. A coculture was then conducted with either clone T2G10 or T1D1, and the release of IFN-γ was measured 18 h later. The autologous 1913 mel cell line was also included in the coculture panel as a positive control.
T cells from TIL 1913 recognized mutated products derived from the CDKN2A locus
A second CD8+ T cell clone derived from this TIL by limiting dilution, T1D1, failed to recognize target cells expressing HLA-A11Mut (Fig. 1). However, this T cell recognized an allogeneic melanoma, 1011 mel, that shared expression of HLA-A11 but not any additional HLA class I alleles with 1913 mel (Table I). The T1D1 T cells failed to recognize EBV-transformed B cells that expressed the normal HLA-A11 gene product, as well as several other melanoma and renal carcinoma cell lines that expressed HLA-A11. A total of 384 pools that were generated from an autologous melanoma cDNA library was then screened by transfection of COS-A11Mut target cells, followed by the addition of clone T1D1 and measurement of IFN-γ release. The sequence of a single cDNA that was isolated from one of the six positive pools that were identified in the initial screen corresponded to a mutated p14ARF transcript that contained a 2-bp deletion in exon 2 as well as a C-to-T alteration that flanked this deletion (Fig. 2A). The same mutation was observed in a sample of fresh uncultured 1913 tumor cells, but the transcript that was isolated from autologous T cells corresponded to the normal p14 sequence, indicating that this represented a somatic mutation that arose during tumor development. In addition, no normal p14ARF transcripts were identified in 1913 mel cells, demonstrating that loss of heterozygosity had occurred at this locus. The CDKN2A locus encodes two gene products, p14ARF and p16INK4a, that result from the splicing of two distinct exon 1 sequences to a single exon 2 that is translated in two alternative ORFs (11). The 2-bp deletion in the CDKN2A exon 2 sequence that was cloned from the 1913 mel would therefore result in the generation of two chimeric gene products (Fig. 2A). The product that was initially cloned from the 1913 mel cDNA library, which represented a fusion of aa 1–73 of the normal p14ARF product with 85 aa that were translated from the third exon 2 ORF (ORF3), was designated p14ARF-ORF3. A second product that would also presumably be expressed in this cell line represented a fusion of aa 1–58 of the normal p16INK4a product with aa 74–132 of the normal p14ARF protein and was designated p16INK-p14ARF. Based on previous studies (24, 25), the p14ARF and p16INK4a gene products expressed in the 1913 mel cell line should not be capable of functioning normally, indicating that this mutation may have had a role in the development of this tumor.
Table I.
Clone T1D1 response
| Cell Types | Cell Lines | HLA-A11 | IFN-γ (pg/ml) |
|---|---|---|---|
| None | 0a | ||
| Melanomas | |||
| 697 | +b | 0 | |
| 836 | +b | 0 | |
| 1011 | +b | 581 | |
| 1913 | +b | 1564 | |
| 1770 | +b | 0 | |
| 1228 | +b | 0 | |
| 1278 | +b | 0 | |
| 1833 | +b | 0 | |
| 1851 | +b | 0 | |
| 1927 | +b | 0 | |
| 2048 | +b | 0 | |
| 624 | 0 | ||
| 888 | 0 | ||
| 526 | 0 | ||
| 1909 | 0 | ||
| 1909 | +c | 0 | |
| 1087 | +c | 0 | |
| B lymphocytes | |||
| EBVBs | 697 | + | 0 |
| 836 | + | 0 | |
| 1743 | + | 0 | |
| B-CD40L | 1913 | + | 0 |
| Renal carcinoma cells (RCC) | |||
| 2206R | +c | 0 | |
| UOK130 | +c | 0 | |
| 2192R | +c | 0 | |
| 1743 | +c | 0 | |
| 2193R | +c | 0 |
T1D1 T cells (104) were incubated with the indicated target cells (2.5 × 104) for 18 h followed by the measurement of IFN-γ release.
Cell lines derived from HLA-A11+ patients tested by genome typing.
Cell lines were transduced with retroviral vector-expressed HLA-A11 Mut, which was isolated from 1913 mel.
FIGURE 2.
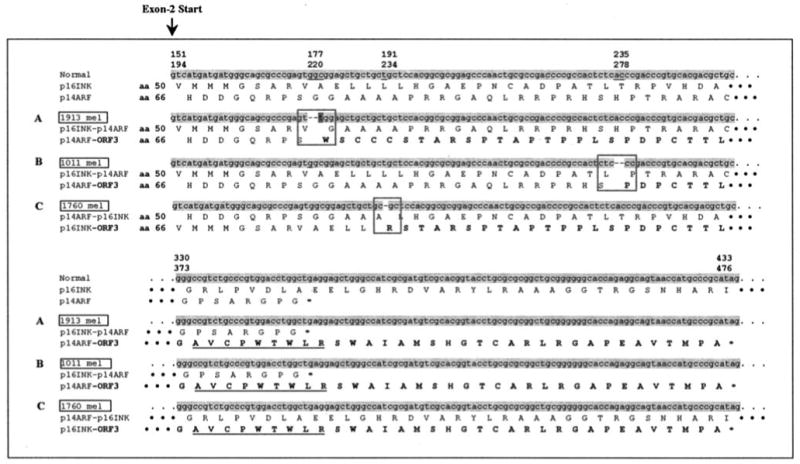
Sequences of frameshifted p14-ARF and p16-INK4a gene products expressed by melanoma cells. The partial nucleotide and amino acid sequences of the p14-ARF and p16-INK4a transcripts that were cloned from the three melanoma cell lines 1913 mel (A), 1011 mel (B), and 1760 mel (C) are presented in this figure. Deleted nucleotides are indicated by dashes, and the point mutation is highlighted by the dark gray. Amino acid sequences encoded within ORF3 are noted in bold. The first two amino acid sequences represent those that are encoded by the normal p16INK4a and p14ARF transcript, and the lower set of amino acid sequences represent those that are encoded by the frameshifted transcripts. The amino acids that flank the mutation sites are boxed, and the sequence of the T cell epitope is underlined.
Identification of the p14ARF-ORF3 T cell epitope
Studies were then conducted to identify the T cell epitope in the p14ARF-ORF3 gene product that was recognized by T1D1 T cell. In an attempt to narrow down the region that encoded the epitope, two constructs that corresponded to the regions of the p14ARF-ORF3 transcript that were located either upstream or downstream of the mutation site were generated by RT-PCR and transfected into COS cells expressing HLA-A11Mut. The results of a cytokine stimulation assay indicated that target cells transfected with a construct encoding the mutated region of the p14ARF-ORF3 transcript stimulated 550 pg/ml IFN-γ from T1D1 T cells, whereas cells transfected with a construct encoding the normal region of the p14ARF-ORF3 transcript stimulated 33 pg/ml IFN-γ from T1D1 T cells. Based on this result, peptides derived from the frameshifted region of this transcript that corresponded to the previously described HLA-A11 binding motif were then synthesized and tested for their ability to sensitize HLA-A11-positive targets. A single peptide, AVCPWTWLR, that corresponded to aa 125–133 of the p14ARF-ORF3 protein (p14ARF-ORF3125∼133) and that was encoded within the frameshifted region of this product, was recognized by 1D1 T cells (Fig. 3A). A minimum concentration of between 1 and 10 pM p14ARF-ORF3125–133 peptide was required to sensitize COS-7 target cells expressing the HLA-A11Mut gene product for recognition by 1D1 T cells, whereas between 1 and 10 nM was required to sensitize COS-7 cells expressing the normal HLA-A11N gene product (Fig. 3B). Similarly, a minimum concentration of between 1 and 10 nM p14ARF-ORF3125–133 peptide was required to sensitize an HLA-A11-positive allogeneic EBV-transformed B cell line for recognition by T1D1 T cells. These results indicate either that the peptide has higher binding affinity to the HLA-A11Mut, or that the structure of the peptide-HLA-A11Mut complex is preferentially recognized by the TCR. Target cells that were pulsed with the six additional peptides containing either deletions or extensions of the N or C terminus of the p14-ORF3125–133 peptide stimulated significantly less cytokine from T1D1 T cell than the p14-ORF3125–133 peptide, indicating that the peptide AVCPWTWLR represents the optimal epitope recognized by T1D1 T cells (Fig. 3C).
FIGURE 3.
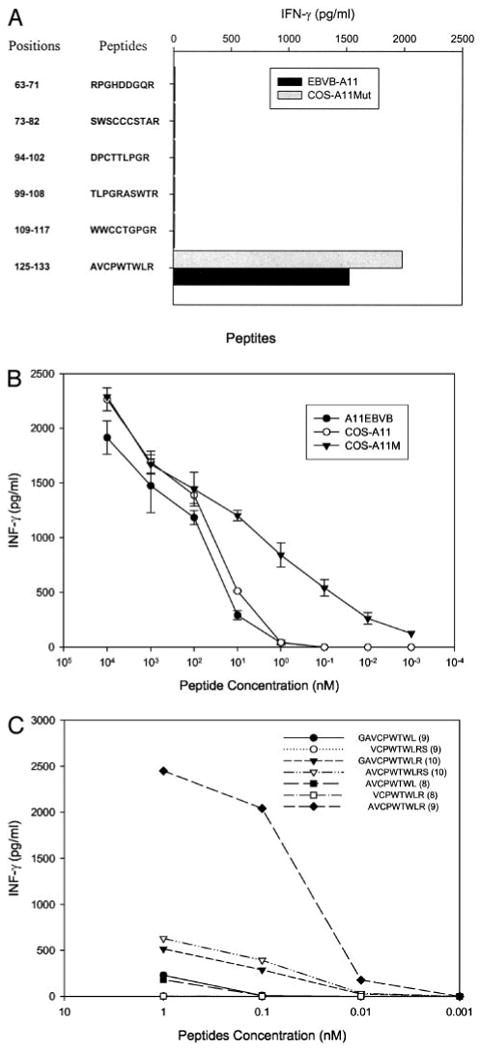
Identification and titration of the p16INK4a/p14ARF peptide recognized by clone T1D1. A, Peptide screening. Six peptides were synthesized and incubated for 2 h with A11-EBVB and COS-A11Mut cells at a concentration of 100 nM. Target cells were then cocultured with clone T1D1, and IFN-γ release was measured 18 h later. B, Peptide titration. Either HLA-A11-expressing EBVB, COS-A11N, or COS-A11Mut cells were loaded with the indicated concentrations of the AVCPWTWLR peptide for 2 h; clone T1D1 cells were added; and IFN-γ release was measured 18 h later. C, Testing of peptide variants. Variants of the peptide AVCPWTWLR either lacking or containing an additional N- or C-terminal residue were incubated with COS-A11Mut cells at indicated concentrations for 2 h. Clone T1D1 T cells were then added, and IFN-γ release was measured 18 h later.
Response to normal and mutated p14ARF and p16INK4a gene products
The observation that the T cell epitope was encoded by an alternative ORF generated as a result of a 2-nt deletion in the p14ARF gene raised the issue of whether or not a similar mechanism was responsible for the recognition of the allogeneic 1011 mel cell line. Amplification of the p14ARF transcript that was expressed in the 1011 mel cell line (Fig. 2B) revealed that these cells expressed a single transcript containing a 2-bp deletion that was located 58 bp downstream from the region of the deletion identified in the 1913 mel (Fig. 2A). The transcript isolated from the 1011 mel cells should also encode the p14ARF-ORF3125–133 peptide, because this deletion was also located upstream of the region that encoded the T cell epitope (Fig. 2B). The screening of 18 additional melanoma cell lines resulted in the identification of a transcript derived from the HLA-A11-negative 1760 mel cell line that contained a deletion of a single base pair in exon 2. Although the T cell epitope would not be encoded by a p14ARF transcript containing a single base pair deletion, this deletion would generate a p16INK4a-ORF3 transcript that encoded the peptide p16INK4a-ORF3111∼119, which is identical with the p14ARF-ORF3125∼133 peptide AVCPWTWLR (Fig. 2C).
Assays were then set up to examine the ability of cells that expressed the normal and frameshifted p14ARF and p16INK4a products to stimulate T cells reactive with the p14ARF-ORF3125∼133/p16INK4a-ORF3111–119 peptide, as well as to further evaluate T cell recognition of target cells expressing either the HLA-A11Mut or A11N products. As a result of difficulties encountered with the propagation of T1D1 T cells, additional T cell clones were screened for their ability to recognize the p14ARF-ORF3125–133 or p16INK4a-ORF111∼119 peptide. One additional clone isolated from TIL 1913, CL131, that expressed a TCR BV family member that was distinct from that expressed by T1D1, 5S1, was also found to recognize target cells pulsed with the peptide AVCPWTWLR (data not shown). Plasmids encoding the p14ARF-ORF3 and p16INK4a-ORF3 products were transiently transfected into COS-A11Mut, COS-A11N, or COS-A2 cells, and tested for their ability to stimulate CL131 T cells. The results indicated that COS-A11Mut cells that were transfected with either the p14ARF-ORF3 or p16INK4a-ORF3 construct stimulated strong and relatively comparable levels of cytokine release from CL131 T cells, whereas responses to cells transfected with a control gp100 construct were undetectable (Fig. 4A). The levels of cytokine release stimulated by COS-A11N cells that were transfected with either of these constructs were consistently lower than those stimulated by the COS-A11Mut transfectants, whereas transfectants of COS-A2 target cells failed to stimulate the CL131 cells, similar to results seen with the T1D1 clone (Fig. 4, A and B, and data not shown). The ability of HLA-A11-expressing COS cells transfected with the normal p14ARF or p16INK4a constructs to stimulate cytokine release from CL131 T cells was then compared with that of the p14ARF-ORF3 construct. The results indicated that cells expressing either the normal p14ARF or p16INK4a products stimulated levels of IFN-γ release from CL131 T cells that were 10–15% of those stimulated by cells transfected with the p14ARF-ORF3 construct (Fig. 4B). As before, COS-A11Mut cells transfected with the p14ARF-ORF3 construct stimulated significantly higher levels of cytokine release from CL131 T cells than COS-A11N cells. Taken together, these results indicate that the expression of frameshifted p14ARF or p16INK4a products that encode the p14ARF-ORF3125–133 peptide in combination with the mutated HLA-A11 gene product is required for optimal recognition by T1D1 T cells.
FIGURE 4.
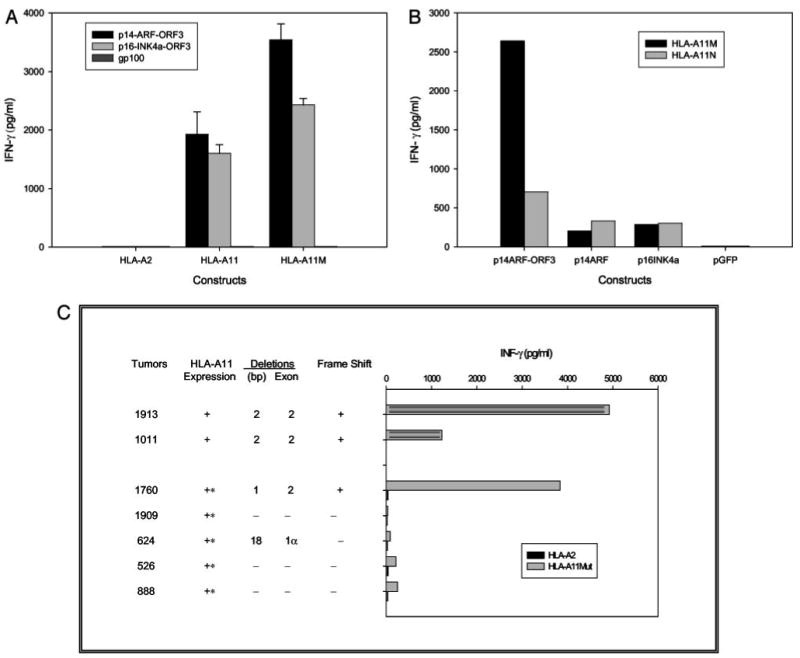
T1D1/CL131 recognition of melanomas and transfected cells with frameshifted and normal p16INK4a and p14ARF gene products. A, CL131 recognition of cells transfected with constructs encoding p14ARF-ORF3 or p16INK4a-ORF3. COS-A2, COS-A11N, and COS-A11Mut cells were transiently transfected with constructs encoding p14ARF-ORF3, p16INK4a-ORF3, or gp100; 24 h later, clone CL131 T cells were added; and IFN-γ release was measured 18 h later. B, Relative recognition of target cells transfected with constructs encoding p14ARF-ORF3, normal p14ARF, or normal p16INK4a by T1D1. The COS-A11N and COS-A11Mut lines were transfected with the indicated constructs; 24 h later, clone T1D1 T cells were added; and IFN-γ release was measured 18 h later. C, Recognition of melanoma cell lines that express frameshifted or nonframeshifted p14ARF or p16INK4a transcripts by T1D1 cells. The 1913, 1011, and 1760 melanoma cell lines, which express frameshifted transcripts, as well as four additional melanoma cell lines, 1909, 624, 526, and 888, which did not express frameshifted transcripts, were incubated with T1D1 T cells, and IFN-γ release was measured 18 h later. The 1760, 1909, 624, 526, and 888 mel cell lines, which do not naturally express HLA-A11, were transduced with recombinant retroviral constructs that encode either HLA-A2 or the HLA-A11mut gene isolated from 1913 mel, as indicated.
Additional cell lines that expressed either normal or mutated products of the CDKN2A locus were then examined for their ability to stimulate T1D1 T cells following transduction with a retrovirus encoding HLA-A11, because only ∼10% of melanoma patients expressed HLA-A11. To examine this issue, several additional melanoma cell lines that failed to express HLA-A11, were transduced with retroviral constructs encoding either the HLA-A11Mut product or a control plasmid encoding HLA-A2 and tested for their ability to stimulate T1D1 T cells. The results indicated that T1D1 cells were strongly stimulated by 1913 and 1011 mel cells, as well as 1760 mel cells that were transduced with the mutated HLA-11 gene product (Fig. 4C). The ability of 1760 mel cells that were transfected with the mutated HLA-A11 construct to stimulate T1D1 T cells presumably results from expression in these cells of a p16INK4a-ORF3 transcript that encodes the T cell epitope, as demonstrated in Fig. 2C. The 1909, 526, 624, and 888 mel cells did not express frameshifted p14ARF or p16INK4a transcripts, and the low levels of stimulation observed following transduction of these cells with HLA-A11Mut presumably resulted from the recognition of the relatively low levels of translated products that were derived from alternative ORFs (Fig. 4C).
In vivo persistence of tumor-reactive T cell clones from 1913 TIL
The adoptive transfer of TIL 1913, which was conducted following a nonmyeloablative chemotherapy regimen, resulted in the nearly complete regression of a multiple s.c. as well as visceral tumor deposits. As summarized in a recent report, this regimen resulted in enhanced engraftment of adoptively transferred T cells and in addition appeared to result in enhanced clinical responses (23). The potential role of tumor-reactive T cells in the tumor regression that was observed following adoptive transfer of TIL 1913 was then examined by evaluating the in vivo persistence of individual clones present in this bulk population of T cells. To evaluate the prevalence of individual T cell clones in TIL and PBL samples, the TCR BV sequences expressed by TIL and PBL samples were initially amplified using a 5′-RACE technique that provides an unbiased sample of the TCR repertoire. Between 79 and 135 products were sequenced from individual samples to obtain an estimate of the relative distribution of cells that express these transcripts, which represent essentially clonal T cell markers as a result of the extensive diversity generated by recombination of multiple TCR V, D, and J regions. The alignment of sequences of clones that were present at a level of 5% or more in TIL 1913 with sequences isolated from PBL samples demonstrated that a dominant clone within 1913 TIL expressed a BV1S1 sequence that corresponded to that of clone T1D1, whereas a second dominant clone expressed a BV6S2 sequence that corresponded to that of clone T2G10 (Table II). Two additional TCR sequences derived from T cells of unknown specificity were also detected in TIL as well as PBL samples obtained following adoptive transfer. The T2G10 clone appeared to comprise ∼20% of the infused TIL, 25% of the peripheral T cells 9 days following adoptive transfer, and ∼14% of the peripheral T cells 154 days following transfer. The T1D1 clone represented ∼16% of the infused T cells, 13% of the T cells 5 days following transfer, and 3% of the T cells 9 days following transfer, but appeared to represent <1% of the T cells 154 days after transfer. These results suggest that T cells that recognize the mutated HLA-A11 gene product and the frameshifted p14ARF tumor suppressor gene product may have contributed to the clinical response that was observed following adoptive transfer to patient 1913.
Table II.
TCR-BV gene transcripts of dominant clones in patient treatment TIL and PBLs
| Percentage (%) of Sequences in TIL and PBLa | |||||||
|---|---|---|---|---|---|---|---|
| TCR-BV Sequenceb | T Cell Clones | Ag Specificity | TIL | PBLc (day 5) | PBL (day 9) | PBL (day 55) | PBL (day 154) |
| 9S1 | — | — | 19 | 6 | 7 | 3 | <1 |
| 6S2 | T2G10 | HLA-A11Mut | 20 | 17 | 25 | 16 | 14 |
| 1S1 | T1D1 | p14ARF/p16INK4a-ORF3 | 16 | 13 | 3 | <1 | <1 |
| 4S1 | — | — | 5 | 9 | 9 | 4 | 5 |
A total of between 79 and 135 TCR-BV gene transcripts were analyzed for each of these TIL or PBLs, and the percentage of each of the indicated sequences obtained from TIL or PBL is indicated.
The four sequences that were detected at a level of 5% or greater in the TIL 1913 sample that was used for treatment are indicated separately on each row of the table. For two of the sequences that were derived from the 6S2 and 1S1 germline BV sequences, tumor-reactive clones were identified. Clones corresponding to the additional two sequences presented in the table were not isolated from TIL 1913, as indicated by the dashes.
PBL samples were obtained at the indicated number of days following adoptive transfer.
Discussion
Extensive studies are now being conducted on adoptively transferred tumor-reactive T cells in an attempt to identify characteristics of those cells that are associated with tumor regression. The results of several studies suggest that HLA class I-restricted T cells that recognize nonmutated shared Ags such as the melanocyte differentiation Ags MART-1 and gp100 play a role in tumor regression in some patients (23, 26). It is more difficult to assess the role of T cells that are reactive with mutated Ags in tumor regression, because individual mutations are generally restricted to only one or a relatively small percentage of tumors. Nevertheless, TIL that are associated with tumor regression have been found to contain T cells that recognize mutated Ags (27), suggesting that they may in some cases play a role in this process.
This study presents the analysis of a TIL 1913 that was associated with a nearly complete regression of multiple metastatic lesions following adoptive immunotherapy. The dominant tumor-reactive T cell clones present in this TIL recognized two mutated gene products. One of these Ags, a mutated HLA-A11 class I gene product, was recognized by a T cell clone that comprises ∼20% of the injected T cells, and represents the most highly prevalent clone in the peripheral blood 9 days as well as 5 mo following adoptive transfer. Mutated HLA class I products resemble alloantigens, and the potent T cell responses that are generated by these products may generally prevent the outgrowth of tumor cells that express these product; however, in one report, T cells isolated from a renal cancer patient were shown to recognize a mutated HLA-A2 gene product expressed by the autologous tumor (6). It is not clear at this time whether T2G10 T cells recognize the mutated HLA-A11Mut transcript alone or recognize a specific peptide or set of peptides that are bound to this HLA class I allele. Preliminary results indicate that a tetramer composed of the mutated p14ARF-ORF3 peptide did not bind to T2G10 T cells but did bind to CL131 T cells, indicating that T2G10 T cells may recognize an unknown peptide or set of peptides in the context of the HLA-A11Mut class I molecule (data not shown).
A second tumor-reactive T cell clone that comprised ∼16% of the infused TIL also persisted at relatively high levels for the first week following adoptive transfer but was not detected in the peripheral blood of this patient 5 mo following transfer. This clone may also have played a role in initiation of the tumor regression, because the relatively slow regression of visceral tumor masses observed in patient 1913 over a period of > 1 year following adoptive transfer may not reflect the period of time needed to resorb the residual tumor mass. Tumor regression may have resulted either from the direct lysis of tumor targets by the CD8+ T cell clones or the release of inflammatory cytokines in the tumor environment that leads to tumor destruction. The significance of long-term T cell persistence in peripheral blood is also not entirely clear, because tumor-reactive T cells may migrate to sites of tumor, leading to the depletion of cells from the peripheral circulation. Screening of a cDNA library with this clone resulted in the isolation of a mutated transcript that contained a deletion of 2 bp in exon 2 of the p14ARF tumor suppressor gene as well as a single point mutation flanking the site of the deletion. The p14ARF gene is derived from the CDKN2A locus, which also encodes a second tumor suppressor gene, p16INK4a. The products of this locus are translated from two distinct exon 1 sequences and share a single exon 2 sequence that is translated from a different ORF in the two products (11). The deletion observed in 1913 mel cells resulted in the generation of a frameshifted product, termed p14ARF-ORF3, that encoded the T cell epitope AVCPWTWLR. A second melanoma cell line that expressed a p14ARF transcript containing a distinct 2-bp deletion, 1011 mel, was also found to express the p14ARF-ORF3 T cell epitope. A third melanoma, 1760 mel, expressed a CDKN2A transcript containing a single base pair deletion in exon 2 that resulted in the generation of a frameshifted p16INK4a product, p16INK4a-ORF3, which also encoded this T cell epitope. The T cell epitope is encoded within a region that encompasses the last 22 nt of the normal p14ARF ORF, and thus nearly all of the mutations within this locus that result in a frameshift would generate this epitope, either from transcripts initiated from the p14ARF promoter or from the p16INK4a promoter.
The expression of the two mutated gene products in the 1913 mel cell line appeared to facilitate T cell recognition, because a cell line expressing the mutated HLA-A11 gene product was capable of presenting the p14ARF-ORF3 peptide at concentrations that were 100- to 1000-fold lower than a cell line that expressed the normal HLA-A11 gene product. Nevertheless, expression of a mutated HLA-A11 was not required for recognition of the endogenously processed epitope, because the 1011 mel cell line that naturally expressed the normal HLA-A11 product along with a frameshifted p14ARF transcript was also recognized by T cells reactive with this epitope.
Prior studies have demonstrated that a variety of T cell epitopes are encoded by alternative ORFs. Transcripts of the TGFβ RII and BAX genes containing frameshift mutations were found to encode epitopes recognized by tumor-reactive T cells (8). In addition, T cells have been shown to recognize nonmutated TRP-1 (28) and NY-ESO-1 (29) gene products representing protein transcripts initiating from alternative AUG start codons located downstream of the natural start site. Transfectants expressing the normal p14ARF or p16INK4a products only weakly stimulated T cells that recognized the p14ARF-ORF3125–133 or p16INK4a-ORF3111–119 peptide, however, indicating that, although this epitope can be readily processed from frameshifted transcripts, ORFs that use alternative start codons present in the normal p14ARF and p16INK4a gene products are not translated efficiently.
Disruptions of the ARF-MDM2-p53 and INK4a-CDK4/6-Rb cell cycle control pathways appear to be critically involved in the progression of a variety of tumor histologies. Tumor-associated mutations in exon 2 of CDKN2A locus were found to be present in ∼65% of acute T lymphoblastic leukemia, 56% of bladder squamous cell carcinoma, and 20% of melanomas that were analyzed (14–17, 30). In primary tumors, the majority of mutations that have been identified occur in the shared exon 2 region and appear to alter the function of both the p14ARF and p16INK4a gene products. These products are frequently inactivated by small homozygous deletions of <200 bp, but not by point mutation (24, 25). In studies conducted on a variety of tumor samples, 72 of the 268 (27%) alterations in exon 2 of the CDKN2A locus that were identified contained deletions or insertions that resulted in frameshifts (14, 15, 31). Our investigation indicated that 3 of 18 melanoma cell lines that were analyzed expressed a CDKN2A transcript containing deletions in exon 2 (17%) that resulted in the generation of a frameshifted transcript and demonstrated loss of heterozygosity at this locus. One additional tumor in this panel expressed a single p16INK4a transcript that contained a deletion of 18 bp from exon 1α that, on the basis of previous studies (32), would be expected to encode a nonfunctional protein. The deletion of sequences between aa 83 and 101 of p14ARF has been shown to result in the failure of these products to localize properly to the nucleolus, and also reduced the ability of the mutated gene products to stabilize p53 (31). The 1913 mel, 1760 mel, and 1011 mel cell lines contain deletions in the expressed p14ARF gene products that alter the reading frame following aa 73, 78, and 92, respectively, and should therefore be functionally inactive in these cell lines. This region is also critical for maintenance of the normal function of p16INK4a. Artificial deletion of 24 bp of p16INK4a that encode aa 62–69 was found to result in the generation of a product that lost binding activity to CDK4 or CDK6 (33). The deletions present in the p16INK4a transcripts isolated from 1913 mel, 1011 mel, and 1760 mel cell lines would result in alteration of the ORF following aa 58, 63, and 78 of the respective protein transcripts, thereby presumably also resulting in the functional inactivation of these products.
In conclusion, these results demonstrate that a TIL associated with tumor regression contains dominant clones of tumor-reactive T cells that recognize a mutated HLA-A11 class I gene product, as well as a frameshifted product of the p14ARF tumor suppressor gene, and support the hypothesis that T cells that recognize mutated gene products may mediate potent in vivo antitumor responses. These results also provide an impetus to identify additional immunogenic T cell epitopes that may be encoded within the frameshifted regions of CDKN2A transcripts that, due to their occurrence at relatively high frequency in certain tumor types, would represent shared mutated Ags.
Footnotes
Abbreviations used in this paper: ORF, open reading frame; TIL, tumor-infiltrating lymphocyte.
References
- 1.Robbins PF, Wang RF, Rosenberg SA. Tumor antigens recognized by cytotoxic lymphocytes. In: Sitkovsky MV, Henkart PA, editors. Cytotoxic Cells: Basic Mechanisms and Medical Applications. Lippincott Williams & Wilkins; Philadelphia: 2000. p. 363. [Google Scholar]
- 2.Ikeda H, Lethe B, Lehmann F, van Baren N, Baurain JF, de Smet C, Chambost H, Vitale M, Moretta A, Boon T, Coulie PG. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199. doi: 10.1016/s1074-7613(00)80426-4. [DOI] [PubMed] [Google Scholar]
- 3.Hanada K, Perry-Lalley DM, Ohnmacht GA, Bettinotti MP, Yang JC. Identification of fibroblast growth factor-5 as an overexpressed antigen in multiple human adenocarcinomas. Cancer Res. 2001;61:5511. [PubMed] [Google Scholar]
- 4.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 5.Mandruzzato S, Brasseur F, Andry G, Boon T, van der Bruggen P. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med. 1997;186:785. doi: 10.1084/jem.186.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandle D, Brasseur F, Weynants P, Boon T, Van den Eynde B. A mutated HLA-A2 molecule recognized by autologous cytotoxic T lymphocytes on a human renal cell carcinoma. J Exp Med. 1996;183:2501. doi: 10.1084/jem.183.6.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang RF, Wang X, Rosenberg SA. Identification of a novel major histocompatibility complex class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4+ T cells. J Exp Med. 1999;189:1659. doi: 10.1084/jem.189.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeterdal I, Bjorheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, Nesland JM, Eriksen JA, Moller M, Lindblom A, Gaudernack G. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA. 2001;98:13255. doi: 10.1073/pnas.231326898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 10.Linard B, Bezieau S, Benlalam H, Labarriere N, Guilloux Y, Diez E, Jotereau F. A ras-mutated peptide targeted by CTL infiltrating a human melanoma lesion. J Immunol. 2002;168:4802. doi: 10.4049/jimmunol.168.9.4802. [DOI] [PubMed] [Google Scholar]
- 11.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 12.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 13.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA. 1998;95:8292. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 15.Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (p16) gene and human cancer. Mol Med. 1997;3:5. [PMC free article] [PubMed] [Google Scholar]
- 16.Papp T, Schipper H, Pemsel H, Bastrop R, Muller KM, Wiethege T, Weiss DG, Dopp E, Schiffmann D, Rahman Q. Mutational analysis of N-ras, p53, p16INK4a, p14ARF, and CDK4 genes in primary human malignant mesotheliomas. Int J Oncol. 2001;18:425. doi: 10.3892/ijo.18.2.425. [DOI] [PubMed] [Google Scholar]
- 17.Sun S, Pollock PM, Liu L, Karimi S, Jothy S, Milner BJ, Renwick A, Lassam NJ, Hayward NK, Hogg D, et al. CDKN2A mutation in a non-FAMMM kindred with cancers at multiple sites results in a functionally abnormal protein. Int J Cancer. 1997;73:531. doi: 10.1002/(sici)1097-0215(19971114)73:4<531::aid-ijc13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 18.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, Schwartzentruber DJ, Hwu P, Marincola FM, Sherry R, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 20.Bettinotti MP, Hadzikadic L, Ruppe E, Dhillon G, Stroncek DS, Marincola FM. New HLA-A, -B, and -C locus-specific primers for PCR amplification from cDNA: application in clinical immunology. J Immunol Methods. 2003;279:143. doi: 10.1016/s0022-1759(03)00233-3. [DOI] [PubMed] [Google Scholar]
- 21.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70:5701. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapointe R, Royal RE, Reeves ME, Altomare I, Robbins PF, Hwu P. Retrovirally transduced human dendritic cells can generate T cells recognizing multiple MHC class I and class II epitopes from the melanoma antigen glycoprotein 100. J Immunol. 2001;167:4758. doi: 10.4049/jimmunol.167.8.4758. [DOI] [PubMed] [Google Scholar]
- 23.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11:210. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 25.Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee WC, Altomare DA, Nobori T, Olopade OI, Buckler AJ, Testa JR. p16 alterations and deletion mapping of 9p21–p22 in malignant mesothelioma. Cancer Res. 1994;54:5547. [PubMed] [Google Scholar]
- 26.Kawakami Y, Rosenberg SA. Immunobiology of human melanoma antigens MART-1 and gp100 and their use for immuno-gene therapy. Int Rev Immunol. 1997;14:173. doi: 10.3109/08830189709116851. [DOI] [PubMed] [Google Scholar]
- 27.Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA. A mutated β-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang RF, Parkhurst MR, Kawakami Y, Robbins PF, Rosenberg SA. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J Exp Med. 1996;183:1131. doi: 10.1084/jem.183.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang RF, Johnston SL, Zeng G, Topalian SL, Schwartzentruber DJ, Rosenberg SA. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol. 1998;161:3598. [PubMed] [Google Scholar]
- 30.Obana K, Yang HW, Piao HY, Taki T, Hashizume K, Hanada R, Yamamoto K, Tanaka Y, Toyoda Y, Takita J, et al. Aberrations of p16INK4A, p14ARF and p15INK4B genes in pediatric solid tumors. Int J Oncol. 2003;23:1151. [PubMed] [Google Scholar]
- 31.Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- 32.Lilischkis R, Sarcevic B, Kennedy C, Warlters A, Sutherland RL. Cancer-associated mis-sense and deletion mutations impair p16INK4 CDK inhibitory activity. Int J Cancer. 1996;66:249. doi: 10.1002/(SICI)1097-0215(19960410)66:2<249::AID-IJC19>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 33.Hashemi J, Platz A, Ueno T, Stierner U, Ringborg U, Hansson J. CDKN2A germ-line mutations in individuals with multiple cutaneous melanomas. Cancer Res. 2000;60:6864. [PubMed] [Google Scholar]