

Abstract
Helper T cells are triggered by molecular complexes of antigenic peptides and class II proteins of the major histocompatibility complex . The formation of stable complexes between class II major histocompatibility complex proteins and antigenic peptides is often accompanied by the formation of a short-lived complex. In this report, we describe T cell recognition of two distinct complexes, one short-lived and the other long-lived, formed during the binding of an altered myelin basic protein peptide to I-Ak. One myelin basic protein-specific T cell clone is triggered by only the short-lived complex, and another is triggered by only the stable complex. Thus, a single peptide bound to a particular class II molecule can activate different T cells depending on the conditions of the binding reaction.
The binding of a peptide to class II major histocompatibility complex (MHC) requires extension of a peptide from an unstructured configuration to a conformation with a polyproline twist. This process is driven by the formation of a conserved set of hydrogen bonds between the peptide backbone and the MHC protein, as well as by interactions between specific peptide side chains and polymorphic protein residues (1–3). Compared with many receptor-ligand pairs, this binding reaction is slow, with a rate constant on the order of 10–100 M−1⋅s−1 (4–7).
In many cases, the binding of a peptide to MHC results in isomeric products, which differ in terms of complex stability and resistance to SDS/PAGE denaturation (8–13). Although structures of several stable peptide–MHC complexes have been solved by x-ray crystallography (1–3), little is known about the structures of the short-lived isomers. In most, but not all, short-lived isomers, the peptide appears to bind within the classical peptide-binding groove of MHC (8, 13).
Some short-lived isomers may be obligatory reaction intermediates in the formation of the stable terminal complex (11). In these cases, the short-lived complexes may lack some of the hydrogen bonds between the MHC protein and the peptide backbone that are conserved in stable terminal complexes, with one end of the peptide flopping around in solution and the other end rigidly bound in the binding groove. Another possibility is that some short-lived isomers may differ from stable isomers in the configuration of certain amino acid side chains in the peptide-binding groove or that a conformational change in the protein may be required to generate the stable terminal complex (9). In this report, we used three complementary approaches—peptide–MHC binding kinetics, T cell activation assays, and molecular modeling—to investigate the structure and biological activity of a particular short-lived isomer of a peptide–MHC complex.
MATERIALS AND METHODS
Peptides and MHC Protein.
All peptides were synthesized using standard fluorenylmethoxycarbonyl chemistry, purified by reverse-phase high performance liquid chromatography, and characterized by mass spectroscopy. For peptide–MHC binding experiments, MBP Ac1–14 and mutant MBP peptides were labeled on their carboxy termini with a cysteine residue tagged with 5,6-carboxyfluorescein. HEL 46–61 (NTDGSTDYGILQINSR) was labeled with 5,6-carboxyfluorescein at its amino terminus. I-Ak was obtained from BW5147.G.1.4 cells transfected with I-Ak cDNA (14) and purified as described (15, 16).
Quantitation of Peptide–MHC Binding.
Peptide–MHC complex formation was measured as described (15). In brief, 300 μM of fluorescently labeled peptide was incubated with 200 nM of I-Ak in PBS/1 mM dodecyl maltoside (pH 7.0) at 37°C. Excess peptide was removed from the sample at 4°C with Sephadex G50-SF spin columns. Complex was separated from the remaining unbound peptide by high performance size exclusion chromatography and quantitated using a Shimadzu RF-551 spectrofluorometric detector and a standard UV detector connected in series. Peptide–MHC complex dissociation was determined by first isolating complex from a spin column and then incubating it in the absence of added peptide in PBS/1 mM dodecyl maltoside (pH 7.0) at 37°C.
T Cell Clones and Proliferation Assays.
The myelin basic protein (MBP)-specific F2 and E3 T cell clones (full names B10A.F2 and B10A.E3) were obtained from lymph node cells of B10.A mice immunized s.c. at the base of the tail with 200 μg of rat MBP Ac1–11 in a 50:50 emulsion of complete Freund’s adjuvant as described (17). T cell clones were maintained by stimulation every 2 weeks with 40–80 μM of wild-type MBP peptide and a 10-fold excess of irradiated (3000 rad) B10.A splenocytes. All assays were performed between day 12 and day 14 after antigenic stimulation. For proliferation assays, T cells (5 × 104) and irradiated (3000 rad) B10.A spleen cells (5 × 105) were incubated with serial dilutions of peptides in a 96-well plate. [3H]thymidine (1 μCi) was added at 48 h, and cell DNA was harvested at 64 h.
Microphysiometry.
Acid release was measured as described (18). T cells (4–8 × 106) were mixed with 2–4 × 105 I-Ak-transfected L cells (19), pelleted, and resuspended in 80 μl of medium, which was then mixed with 22 μl of low temperature melting agarose (Molecular Devices). The agarose–cell mixture (7 μL) was immediately spotted onto the membrane of a Cytosensor cell capsule (Molecular Devices) and cooled to 4°C in a refrigerator. After 5 min, the cell capsule was assembled and loaded in the microphysiometer sensing chamber maintained at 37°C. The chamber was perfused (50 μL/min) with low buffer RPMI 1640 medium (Molecular Devices) containing 1 mM sodium phosphate, 1 mg/ml endotoxin-free bovine serum albumin (Calbiochem), and no bicarbonate (pH 7.4). Acid release rates were determined with 20-s potentiometric pH measurements after a 58-s pump cycle and a 10-s delay (90-s total cycle time). Basal acid release rates ranged from 60 to 120 μV⋅ s−1.
Molecular Modeling.
Molecular modeling was performed using the look interface (Molecular Applications Group, Palo Alto, CA) as described (15, 20).
RESULTS AND DISCUSSION
Peptide–MHC Binding Kinetics.
MBP is a primary T cell target in multiple sclerosis and experimental allergic encephalomyelitis, an animal model for multiple sclerosis. In B10.A mice with experimental allergic encephalomyelitis, the T cell response is mainly toward the acetylated N-terminal peptide of MBP presented by I-Ak (the wild-type MBP peptide; Table 1). This peptide binds very weakly to I-Ak (7, 14). According to a recent proposal, this weak binding is due both to an unfavorable interaction between residue 4 of the peptide (a lysine) and the positively charged p6 pocket of the MHC protein and to incomplete occupancy of the MHC binding groove (C.L., M.N.L., K.T., C.B., P. P. Jones, and H.M.M., unpublished results). Mutation of residue 4 from lysine in the wild-type peptide to alanine removes the unfavorable contact, resulting in a peptide (the MBP 4A peptide) with higher MHC affinity and much greater potency in triggering T cells (14). However, the MBP 4A peptide still fills only a portion of the MHC binding groove and lacks favorable anchoring interactions. Therefore, it also dissociates rapidly from I-Ak compared with most antigenic peptides.
Table 1.
Peptides studied and their predicted alignment on I-Ak
| I-Ak pocket | p1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild-type MBP peptide | Ac | A | S | Q | K | R | P | S | Q | R | H | G | S | K | Y | |
| MBP 4A peptide | – | – | S | – | A | – | – | – | – | – | – | – | – | – | – | |
| MBP 2K 4A peptide | – | – | K | – | A | – | – | – | – | – | – | – | – | – | – |
The altered peptide studied in detail in this report, the MBP 2K 4A peptide (Table 1), has both the 4A mutation and an additional mutation at position 2 from serine to lysine. The binding and dissociation of fluorescently labeled MBP 4A and MBP 2K 4A to isolated, detergent solubilized I-Ak is shown in Fig. 1. The wild-type MBP peptide is not included because its binding is too weak to detect by these methods (7, 14). Also included is a positive control peptide that binds strongly to I-Ak, residues 46–61 of hen egg lysozyme (HEL).
Figure 1.

The MBP 2K 4A peptide binds to I-Ak to form two isomeric complexes. (A) Binding of fluorescently labeled HEL (46–61) (triangles), MBP 4A (squares), and MBP 2K 4A (circles) to detergent-solubilized I-Ak. (B) Dissociation of the complexes isolated after the following incubation times: HEL, 14 h; MBP 4A, 2.5 h; and MBP 2K 4A, 14 h. Data were fit to a single exponential decay with t1/2 equal to 280 h for HEL, 32 h for MBP 2K 4A, and 0.26 h for MBP 4A. (C) Rapid, biphasic dissociation of the MBP 2K 4A complex formed after a short incubation period (0.7 h) compared with the dissociation of the MBP 4A complex formed after a 2.5-h incubation. MBP 4A dissociation was fit to a single exponential decay, and MBP 2K 4A dissociation was fit to a double exponential decay. (D) The ratio of long-lived to short-lived MBP 2K 4A complex increases with increasing incubation time. Labeled MBP 2K 4A peptide was incubated with detergent-solubilized I-Ak for the time indicated on the x axis, complex was isolated and quantitated, and the ratio of long-lived to short-lived complex was determined by the fraction of complex that remained after a 30-min dissociation reaction. Data were fit by linear regression with the slope = 0.9 h−1.
The serine-to-lysine substitution at residue 2 of MBP has only a small effect on the total amount of peptide–MHC complex formed (Fig. 1A). However, although MBP 4A complex accumulates relatively rapidly, MBP 2K 4A complex accumulates very slowly. Figs. 1 B and C show the dissociation of the HEL, MBP 4A, and MBP 2K 4A complexes with I-Ak. As shown in Fig. 1B, after a long (14-h) incubation with I-Ak, most of the MBP 2K 4A complex formed is >100 times more stable than the MBP 4A complex (t1/2 = 32 h vs. 0.26 h). In contrast, after a short (40-min) incubation with I-Ak, most of the MBP 2K 4A complex is less stable than the MBP 4A complex (Fig. 1C). The dissociation curves of HEL and MBP 4A complexes do not depend on the duration of the association reaction (M.L. and C.B., unpublished results). The decay of the MBP 2K 4A complex formed after the short incubation is biphasic. Thus, at least two isomeric complexes are formed. The rapid complex decay during the first 6 min indicates that one complex has a dissociation half-time of <3 min. The stable amount of complex over the next 40 min shows that the remaining complex has a half-life of greater than several hours.
These data are consistent with either of the following reaction mechanisms: (i) Sequential reactions. The MBP 2K 4A peptide (p) reacts with empty I-Ak (M) to form first a short-lived intermediate complex (Mp)i which infrequently converts to a long-lived terminal complex (Mp)t.
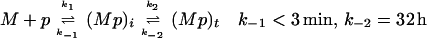 |
And (ii) Competing reactions. The short-lived complex (Mp)1 and the long-lived complex (Mp)2 are on independent reaction pathways.
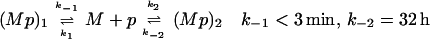 |
Both of the above reaction mechanisms imply that the ratio of the concentration of long-lived complex to the concentration of short-lived complex increases approximately linearly with time during the first few hours of the reaction (Fig. 1D). In the case of sequential reactions (scheme 1), the slope of this increase provides an estimate of the rate constant k2 of 1 h−1. In the case of competing reactions (scheme 2), formation of the long-lived complex occurs at <10% of the rate of formation of the short-lived complex (i.e., k2 < 0.1 k1; for details, see Appendix I).
T Cell Recognition.
To investigate the biological activity of the two MBP 2K 4A complexes, we first determined the proliferation response of MBP-specific T cells to the MBP 2K 4A peptide and then used an assay of early T cell activation to determine the kinetics of these responses. The relative potency of the wild-type MBP, MBP 4A, and MBP 2K 4A peptides in causing the proliferation of two different MBP-specific T cell clones (F2 and E3) derived from B10.A mice with experimental allergic encephalomyelitis is shown in Fig. 2 A and B. The two clones differ in Vβ gene usage (F2 is Vβ8, and E3 is Vβ13; K.T., unpublished results) and respond quite differently to altered MBP peptides with mutations at MBP residues 2 and 3 (Fig. 2 C and D). However, both clones give a full proliferation response to MBP 2K 4A peptide at a peptide concentration of ≈10 μM.
Figure 2.

Proliferation response of MBP-specific T cell clones to altered MBP peptides (A–D) The indicated T cell clone was incubated with the indicated altered MBP peptide and B10.A splenocytes. Proliferation was measured by 3H-thymidine incorporation. All peptides are named by their difference from the wild-type (WT) Ac1–14 MBP peptide. Similar data have been obtained in at least three independent experiments with each peptide and T cell clone and using fluorescently labeled MBP 2K 4A peptide.
The kinetics of these T cell responses were determined using a silicon-based biosensor, the Cytosensor microphysiometer. The microphysiometer detects early biochemical events in T cell activation based on increases in the rate of acid release of a mixture of T cells and antigen-presenting cells (APCs) (18). These responses are antigen-specific; they can be blocked by either antagonist peptide or anti-MHC antibody (17, 21). One unique feature of the microphysiometer is that T cell responses can be quantitated both as peptide is being added and while peptide is being washed away. Thus the microphysiometer can provide information about the binding and dissociation reactions between peptides and MHC and between peptide–MHC complexes and T cell receptors (17). Previous work has shown that the response of the F2 clone to the wild-type MBP peptide declines rapidly as the peptide is washed away. In contrast, the response to the MBP 4A peptide declines slowly, consistent with its longer dissociation half-time of 15 min (17). Similar responses also occur in the E3 clone (Appendix II).
The acid release response of a mixture of F2 or E3 T cells and I-Ak-transfected APCs to the MBP 2K 4A peptide is shown in Fig. 3 A and B. Addition of 10 μM of MBP 2K 4A results in a gradual increase in acid release in both clones. In the E3 clone, this response is faster and larger than the response to wild-type MBP peptide, consistent with the MBP 2K 4A peptide being more potent than wild-type in causing proliferation. However, in the F2 clone, the response is markedly slower than the response to wild-type. The slow nature of this response suggests that it might be due to slow accumulation of the long-lived complex, with no F2 response to the short-lived complex. In addition, the prolonged induction phase preceding the first detectable increase in acid release by the F2 clone is consistent with an obligatory reaction intermediate preceding formation of the long-lived MBP 2K 4A complex.
Figure 3.

Kinetics of T cell responses to addition (A and B) and washout (C and D) of the MBP 2K 4A peptide. (A and B) The acid release of a mixture of the indicated T cell clone and L cells transfected with I-Ak was measured as either 10 μM wild-type MBP peptide or 10 μM MBP 2K 4A peptide was added. (C and D) A mixture of the indicated T cell clone and L cells transfected with I-Ak was activated by addition of either wild-type MBP peptide (circles) or 2K 4A MBP peptide (squares) for 6 min (t = −6 min to t = 0 min). Subsequently, the unbound peptide was washed away, and the acid release response was recorded. Peptide concentrations: C, WT = 50 μM, 2K 4A = 200 μM; D, WT = 200 μM, 2K 4A = 50 μM; 100% acid release represents the acid release rate before peptide addition. Similar results to each panel have been obtained in at least two independent experiments.
A reliable indicator of the stability of a stimulatory peptide–MHC complex is how rapidly the microphysiometer response decreases when peptide is washed away (Appendix II). The effect of washing away the MBP 2K 4A peptide on F2 and E3 acid release is shown in Fig. 3 C and D. In both cases, a T cell–APC mixture was stimulated by addition of a large concentration of wild-type MBP peptide or MBP 2K 4A peptide for a short time, and the T cell response was recorded as the peptides were washed away. As shown in Fig. 3C, the response of the F2 clone to the MBP 2K 4A peptide does not decline at all after peptide washout. A stable F2 clone response to the MBP 2K 4A peptide was found at all MBP 2K 4A concentrations tested, ranging from 10 μM to 200 μM (data not shown). Thus, the F2 clone recognizes only the long-lived complex of the MBP 2K 4A peptide with I-Ak. In contrast, the response of the E3 clone to both the MBP 2K 4A peptide and the wild-type peptide declines rapidly after peptide washout (Fig. 3D) independent of peptide concentration added (concentrations from 10 to 100 μM were tested; data not shown). Thus, the E3 clone recognizes only the short-lived complex of the MBP 2K 4A peptide with I-Ak.
To confirm that the F2 clone recognizes the stable MBP 2K 4A complex and that the E3 clone recognizes only the short-lived MBP 2K 4A complex, we tested the F2 and E3 proliferation responses to APCs prepulsed with MBP 2K 4A peptide. The prepulse protocol is to incubate the APCs with peptide for 100 min at 37°C, wash with media, incubate for 20 min at 37°C in fresh media to allow dissociation of short-lived peptide–MHC complexes, wash again, and add the APCs to T cells (22). As shown in Fig. 4, APCs prepulsed with the MBP 2K 4A peptide trigger the proliferation of the F2 T cell clone but cause no proliferation of the E3 clone, confirming that the E3 clone recognizes the short-lived complex while the F2 clone recognizes the stable complex.
Figure 4.

The F2 but not the E3 T cell clone proliferates in response to APCs prepulsed with MBP 2K 4A peptide. The indicated T cell clone was incubated with B10.A splenocytes never exposed to peptide (Left), in the presence of 25 μM MBP 2K 4A (Center), or prepulsed with 200 μM MBP 2K 4A for 100 min and then washed to remove unbound peptide (Right). Plotted is mean ± SE of assays performed in triplicate. Similar data have been obtained in two independent experiments. In control experiments, APCs prepulsed with 10 μM of MBP 4A peptide caused a full proliferation response in both the E3 and the F2 clones whereas APCs prepulsed with 200 μM of wild-type MBP peptide caused no proliferation of either clone (data not shown).
The large concentration of the MBP 2K 4A peptide required to cause E3 T cell clone proliferation (Fig. 2B) is consistent with the E3 clone recognizing only the short-lived MPB 2K 4A complex. In addition, the large concentration of the MBP 2K 4A peptide required to cause F2 T cell clone acid release after a brief exposure to peptide (Fig. 3) is consistent with the infrequent formation of the long-lived MBP 2K 4A complex (Fig. 1A). However, the much larger concentration of MBP 2K 4A than MBP 4A peptide required to cause F2 T cell clone proliferation is not consistent with the similar amounts of MBP 4A complex and long-lived MBP 2K 4A complex formed after long incubation times (Fig. 1A). A possible explanation for this result is that F2 T cell receptor recognizes the long-lived MBP 2K 4A complex as a partial agonist (21).
Structural Implications.
The ability of the short-lived MBP 2K 4A complex to trigger the E3 T cell clone, which also responds to wild-type MBP and MBP 4A, implies that the antigenic surface of the short-lived complex resembles that of the wild-type MBP complex (C.L., M.N.L., K.T., C.B., P. P. Jones, and H.M.M., unpublished results). Similarly, the ability of the long-lived MBP 2K 4A complex to trigger the F2 T cell clone implies that the antigenic surface of the long-lived complex is also similar to the wild-type MBP complex. Thus, in both isomeric complexes, the MBP 2K 4A peptide likely lies in the MHC binding groove in the extended polyproline configuration that has been observed in all class II MHC–peptide x-ray crystal structures (1–3). To align the MBP 2K 4A peptide on I-Ak, we have applied a molecular modeling algorithm that fixes the peptide and protein backbones but allows full amino acid side chain mobility (15, 20). This approach takes advantage of the fact that, although MHC proteins are highly polymorphic, in x-ray crystal structures the amino acid backbones of different class II MHC proteins align very closely (1–3, 23, 24). Using this method, we found that the antigenic surface of the MBP 2K 4A peptide bound to I-Ak resembles that of wild-type MBP bound to I-Ak only when MBP 2K 4A, like wild-type MBP, places residue 2 in the p4 protein pocket. Therefore, the isomeric complexes we observe, in contrast to those proposed previously (34, 35) and observed by Quaratino et al. (36) using a much longer peptide, differ only in terms of conformation, not in terms of alignment of peptide in the MHC binding groove.
These findings are of interest in terms of protein-folding reaction kinetics because they provide an example of a protein–peptide complex that folds into two distinct, biologically active conformations. Recently, several proteins have been identified that fold into a biologically active intermediate structure that slowly converts into more stable structure via a gross conformational change (25, 26). The MBP 2K 4A complexes differ from these proteins in that the different MBP 2K 4A complexes likely differ in the position of only a small number of protein and peptide amino acid side chains.
Potential in Vivo Role of Short-Lived Isomers.
Whole protein immunization activates only a subset of the T cells that are capable of responding to peptide fragments of the protein (27). One reason for this is that some antigenic peptide fragments are never presented by MHC after intracellular protein processing. Another reason is that sometimes a particular peptide–MHC complex may trigger a T cell clone if the complex is formed by free peptide binding to MHC but not if the complex is formed by protein processing (28, 29). Our discovery that T cells can specifically recognize a short-lived kinetic isomer in the binding of peptide to MHC suggests that presentation of short-lived isomers may help explain differences between peptide and protein immunization. Most short-lived complexes may not be presented after protein immunization because the complex may either dissociate or convert to a more stable structure before the peptide–MHC complex reaches the APC cell surface. The lifetime of short-lived isomers may be particularly brief in the endosomal compartment because of low pH and the peptide-exchange catalyst DM (30–32). Because short-lived isomers may not be presented after conventional intracellular processing, T cells specific for these conformations may escape negative selection (29). Thus, T cell responses to short-lived isomers, which could potentially be presented by peptide binding to cell surface MHC or by a class II recycling pathway (33), may potentially contribute to the breakdown of self-tolerance.
Acknowledgments
We thank Pat Jones and her laboratory for the E3 and F2 T cell clones and advice and Tom Anderson, Lutz Schmitt, Emil Unanue, and Mark Davis for valuable discussions. This work was supported by the Medical Scientist Training Program (J.D.R.), the Franklin Veatch Fellowship (M.L.), the American Cancer Society (C.L.), the Cancer Research Institute (C.B.), and grants from the National Institutes of Health to H.M.M. Peptides were characterized at the Mass Spectroscopy Facilities at the University of California, San Francisco.
ABBREVIATIONS
- APC
antigen presenting cell
- HEL
hen egg lysozyme
- MBP
myelin basic protein
- MHC
major histocompatibility complex
Appendix
I. Kinetic Rate Constants.
The reaction schemes for peptide binding to MHC presented in the text ignore that the MHC protein used in our experiments is initially filled with endogenous peptides, which complicates quantitation of the rate of MHC–peptide complex formation (7), preventing exact determination of k1 or k2 for either scheme. However, in the case of scheme 1, the particular circumstances of the reaction of MBP 2K 4A with I-Ak allow us to bound k2 by the inequality S < k2 < 2S where S is the rate of increase in the ratio of long-lived to short-lived complex during the first 4 h of the association reaction, i.e., the slope of Fig. 1D. In the case of scheme 2, S < k2 (k−1/k1) < 2S, which implies k2 < 0.1 k1 as S = 1 h−1 and k−1 > 20 h−1. Qualitatively speaking, these inequalities are valid because of the following: The short-lived complex dissociates so rapidly that it always remains at pseudo-steady-state with the empty MHC, the long-lived complex forms too slowly to deplete the pool of empty MHC (see Fig. 1A), and the long-lived complex dissociates very slowly (Fig. 1B). The specific quantitative assumptions are the following (symbols defined as in the text; M is empty MHC): M ≫ (Mp)total (see Fig. 1A); t1/2-short-lived ≪ t ≪ t1/2-long-lived; M′> 0; M"< 0.
II. Interpretation of Microphysiometer Data.
A limitation of using the microphysiometer to determine the stability of a stimulatory peptide–MHC complex is that the rate of change in the T cell acid release response is limited by a cellular step unrelated to peptide–MHC reactions with a half-time of ≈2 min (17). Therefore, the microphysiometer response when peptide is washed away may be identical for a stimulatory complex with a half-life of a few minutes, vs. one with a half-life of only seconds (e.g., E3 clone response to wild-type MBP vs. MBP 2K 4A; Fig. 3D). Given this limitation, the decrease in the microphysiometer response when peptide is washed away is taken to be a reliable indicator of the stability of a stimulatory peptide–MHC complex. This assertion is supported by experiments comparing F2 T cell clone acid release responses to wild-type MBP vs. MBP 4A (17) as discussed in the text. To provide further evidence for this hypothesis, both the F2 and the E3 clones were stimulated with the MBP 4A and MBP 4E peptides. The latter binds to I-Ak more stably than MBP 4A (t1/2 > 10 h; M.L., unpublished results). In both clones, the acid release response to the MBP 4E peptide does not decline detectably after peptide washout whereas the response to the MBP 4A peptide declines appoximately linearly, with a 50% decline occurring over ≈1 h (Fig. 5). Because T cell acid release response is proportional to the logarithm of peptide concentration, this slow rate of decline in the acid release response to the MBP 4A peptide is consistent with its 15-min dissociation half-time [although T cell receptor binding may somewhat inhibit peptide dissocation from MHC, as discussed in detail by Beeson et al. (17)]. Thus, in both clones, the rate of decrease in the acid release response when peptide is washed away is indeed a reliable indicator of the stability of the stimulatory peptide MHC complex.
Figure 5.

Evidence that the kinetics of the T cell response to peptide washout reflect the dissociation kinetics of the stimulatory peptide–MHC complex. (A and B) The acid release of a mixture of the indicated T cell clone and L cells transfected with I-Ak was measured as either 1 μM of MBP 4A peptide or 1 of μM of MBP 4E peptide was added and subsequently washed away. The duration of peptide addition is indicated by the shaded box.
References
- 1.Stern L J, Brown J H, Jardetzky T S, Gorga J C, Urban R G, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh P, Amaya M, Mellins E, Wiley D C. Nature (London) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 3.Fremont D H, Hendrickson W A, Marrack P, Kappler J. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 4.Buus S, Sette A, Colon S M, Jenis D M, Grey H M. Cell. 1986;47:1071–1077. doi: 10.1016/0092-8674(86)90822-6. [DOI] [PubMed] [Google Scholar]
- 5.Roche P A, Cresswell P. J Immunol. 1990;144:1849–1856. [PubMed] [Google Scholar]
- 6.Stern L J, Wiley D C. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- 7.Mason K, McConnell H M. Proc Natl Acad Sci USA. 1994;91:12463–12466. doi: 10.1073/pnas.91.26.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadegh-Nasseri S, McConnell H M. Nature (London) 1989;337:274–276. doi: 10.1038/337274a0. [DOI] [PubMed] [Google Scholar]
- 9.Sadegh-Nasseri S, Stern L J, Wiley D C, Germain R N. Nature (London) 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- 10.Witt S N, McConnell H M. Biochemistry. 1994;33:1861–1868. doi: 10.1021/bi00173a032. [DOI] [PubMed] [Google Scholar]
- 11.Beeson C, McConnell H M. Proc Natl Acad Sci USA. 1994;91:8842–8845. doi: 10.1073/pnas.91.19.8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beeson C, Anderson T G, Lee C, McConnell H M. J Am Chem Soc. 1995;117:10429–10433. [Google Scholar]
- 13.Tompkins S M, Moore J C, Jensen P E. J Exp Med. 1996;183:857–866. doi: 10.1084/jem.183.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason K, Denney D W, McConnell H M. J Immunol. 1995;154:5216–5227. [PubMed] [Google Scholar]
- 15.Liang M N, Lee C, Xia Y, McConnell H M. Biochemistry. 1996;35:14734–14742. doi: 10.1021/bi961725b. [DOI] [PubMed] [Google Scholar]
- 16.Watts T, Brian A A, Kappler J W, Marrack P, McConnell H M. Proc Natl Acad Sci USA. 1984;81:7564–7568. doi: 10.1073/pnas.81.23.7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beeson C, Rabinowitz J, Tate K, Gutgemann I, Chien Y H, Jones P P, Davis M M, McConnell H M. J Exp Med. 1996;184:777–782. doi: 10.1084/jem.184.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McConnell H M, Wada H G, Arimilli S, Fok K S, Nag B. Proc Natl Acad Sci USA. 1995;92:2750–2754. doi: 10.1073/pnas.92.7.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tate K M, Lee C, Edelman S, Carswell-Crumpton C, Liblau R, Jones P P. Int Immunol. 1995;7:747–761. doi: 10.1093/intimm/7.5.747. [DOI] [PubMed] [Google Scholar]
- 20.Lee C, McConnell H M. Proc Natl Acad Sci USA. 1995;92:8269–8273. doi: 10.1073/pnas.92.18.8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinowitz J D, Beeson C, Wulfing C, Tate K, Allen P M, Davis M M, McConnell H M. Immunity. 1996;5:125–135. doi: 10.1016/s1074-7613(00)80489-6. [DOI] [PubMed] [Google Scholar]
- 22.Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue E R. J Immunol. 1996;156:450–458. [PubMed] [Google Scholar]
- 23.Brown J H, Jardetzky T S, Gorga J C, Stern L J, Urban R G, Strominger J L, Wiley D C. Nature (London) 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 24.Jardetzky T S, Brown J H, Gorga J C, Stern L J, Urban R G, Chi Y I, Stauffacher C, Strominger J L, Wiley D C. Nature (London) 1994;368:711–718. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 25.Baker D, Agard D A. Biochemistry. 1994;33:7505–7509. doi: 10.1021/bi00190a002. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Mottonen J, Goldsmith E J. Biochemistry. 1996;35:16443–16448. doi: 10.1021/bi961214p. [DOI] [PubMed] [Google Scholar]
- 27.Sercarz E E, Lehmann P V, Ametani A, Benichou G, Miller A, Moudgil K. Annu Rev Immunol. 1993;11:729–766. doi: 10.1146/annurev.iy.11.040193.003501. [DOI] [PubMed] [Google Scholar]
- 28.Viner N J, Nelson C A, Unanue E R. Proc Natl Acad Sci USA. 1995;92:2214–2218. doi: 10.1073/pnas.92.6.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viner N J, Nelson C A, Deck B, Unanue E R. J Immunol. 1996;156:2365–2368. [PubMed] [Google Scholar]
- 30.Weber D A, Evavold B D, Jensen P E. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 31.Kropshofer H, Vogt A B, Moldenhauer G, Hammer J, Blum J S, Hammerling G J. EMBO J. 1996;15:6144–6154. [PMC free article] [PubMed] [Google Scholar]
- 32.van Ham S M, Gruneberg U, Malcherek G, Broker I, Melms A, Trowsdale J. J Exp Med. 1996;184:2019–2024. doi: 10.1084/jem.184.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinet V, Vergelli M, Martin R, Bakke O, Long E O. Nature (London) 1995;375:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 34.Bhayani H, Paterson Y. J Exp Med. 1989;170:1609–1625. doi: 10.1084/jem.170.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurata A, Berzofsky J A. J Immunol. 1990;144:4526–4535. [PubMed] [Google Scholar]
- 36.Quaratino S, Thorpe C J, Travers P J, Londei M. Proc Natl Acad Sci USA. 1995;92:10398–10402. doi: 10.1073/pnas.92.22.10398. [DOI] [PMC free article] [PubMed] [Google Scholar]