

Abstract
Recent epidemiological studies indicate beneficial effects of moderate ethanol consumption in ischemic heart disease. Most studies, however, focus on the effect of long-term consumption of ethanol. In this study, we determined whether brief exposure to ethanol immediately before ischemia also produces cardioprotection. In addition, because protein kinase C (PKC) has been shown to mediate protection of the heart from ischemia, we determined the role of specific PKC isozymes in ethanol-induced protection. We demonstrated that (i) brief exposure of isolated adult rat cardiac myocytes to 10–50 mM ethanol protected against damage induced by prolonged ischemia; (ii) an isozyme-selective ɛPKC inhibitor developed in our laboratory inhibited the cardioprotective effect of acute ethanol exposure; (iii) protection of isolated intact adult rat heart also occurred after incubation with 10 mM ethanol 20 min before global ischemia; and (iv) ethanol-induced cardioprotection depended on PKC activation because it was blocked by chelerythrine and GF109203X, two PKC inhibitors. Consumption of 1–2 alcoholic beverages in humans leads to blood alcohol levels of ≈10 mM. Therefore, our work demonstrates that exposure to physiologically attainable ethanol levels minutes before ischemia provides cardioprotection that is mediated by direct activation of ɛPKC in the cardiac myocytes. The potential clinical implications of our findings are discussed.
Keywords: translocation inhibitor, peptide, preconditioning
Epidemiological and animal studies demonstrate that moderate ethanol consumption correlates with decreased morbidity and mortality from ischemic heart disease (1–3). The cardioprotective effect of ethanol has been attributed to modulation of blood lipoproteins and reduced platelet activation and thrombosis (4). However, other studies suggest a direct protective effect of ethanol on the heart muscle (1, 5–8).
What are the molecular mechanisms by which ethanol could exert direct cardioprotective effects? Although ethanol causes changes in the biophysical and biochemical properties of membranes, ethanol also interacts directly with membrane proteins to alter their activity. In a variety of cell models, ethanol exposure modulates both cAMP-dependent protein kinase-mediated signal transduction (9, 10) and protein kinase C (PKC)-mediated signal transduction (11–15). Changes in cAMP-mediated signaling were found in cultured neuronal cells (16). However, we found no effect of acute or prolonged exposure to 100–300 mM ethanol on basal, norepinephrine-, or adenosine-stimulated cAMP levels in cultured cardiac myocytes (17). We therefore focused on the effect of ethanol on PKC-mediated signal transduction in the heart.
It was previously reported that (8) protection from global ischemia in hearts isolated from adult guinea pigs fed with ethanol for 12 weeks yielding blood alcohol levels of 10 ± 2 mg/dl (≈20 mM). Protection was abolished if the PKC inhibitor chelerythrine (10 μM) was included in the perfusate 10 min before the ischemic period (8). Selective translocation of ɛPKC as evidenced by Western blot analysis and immunofluorescence staining in these experiments suggested that ɛPKC may mediate ethanol-induced cardioprotection. However, the role of ɛPKC in cardiac protection could not be tested directly in these studies because the available isozyme-selective PKC inhibitors could not be used in intact heart. In addition, in vivo studies could not exclude a role for nonmyocyte cells in ethanol-induced protection. Finally, the previous studies did not determine whether a brief exposure to ethanol, immediately before ischemia, provided cardioprotection. To address these questions, in the present study we used an isolated adult rat myocyte model of cardiac ischemia and determined the effect of isozyme-selective PKC inhibitors developed in our laboratory on ethanol-mediated protection.
Isozyme-selective peptide inhibitors of PKC have been used successfully in a variety of cell systems to determine the function of particular isozymes (18) and work by competing for binding of activated isozymes to their anchoring proteins, termed RACKs or receptors for activated C-kinase (19, 20). Relevant to this study, we have recently demonstrated a role for ɛPKC in cardioprotection of neonatal cultured cardiac myocytes (21). Earlier studies in vivo and in isolated cells demonstrated that a short period of ischemia before the prolonged ischemia causes a significant decrease in damage to heart cells (22–24). This protection, termed preconditioning, is likely to occur in humans (25–29). Therefore, means to activate this form of protection without the use of a brief ischemic insult, a potentially harmful procedure per se, is highly desired. We showed that, in neonatal cardiomyocytes, protection after ischemic preconditioning is abolished by inhibition of ɛPKC with the translocation inhibitor peptide ɛV1-2 (21), suggesting a role for ɛPKC in cardioprotection.
Here we determined whether acute exposure to ethanol mimics preconditioning and produces cardioprotection and what the minimal ethanol concentration is that produces this protection. Using the isozyme-specific inhibitors that we developed, we also determined the role of specific PKC isozymes in this ethanol-induced cardioprotective effect. Our results demonstrate direct protective effects after a 10- to 20-min exposure of as little as 10 mM ethanol on intact heart and on adult cardiomyocytes and indicate that activation of ɛPKC is essential for ethanol-induced cardioprotection from ischemic injury. The effect of acute exposure to physiologically attainable concentrations of ethanol on the heart opens the possibility for therapeutic use of ethanol before impending ischemia.
Materials and Methods
Peptide Preparation and Delivery.
ɛV1-2 peptide [amino acids 14–21 of ɛPKC (30)], and βC2-4 peptide [amino acids 218–226 of βPKC; (31)] were synthesized at the Stanford Protein and Nucleic Acid Facility (Stanford, CA), and a Cys residue was added to their amino termini. The peptides were purified (>95%) and cross-linked via an N-terminal Cys-Cys bond to the Drosophila Antennapedia homeodomain-derived carrier peptide (C-RQIKIWFQNRRMKWKK) (32, 33). The peptides (0.1–1 μM; applied concentration) were introduced into cells as carrier-peptide conjugates (32, 33) with a carrier-carrier dimer as control. Previous studies indicated that the intracellular concentration of the peptides did not exceed 10% of the applied concentration and that the majority of cells contained the introduced peptides (not shown).
Cardiac Myocyte Isolation.
Hearts from adult male Wistar rats (250–300 g) were isolated and perfused on a Langendorff apparatus as described (8). Myocyte isolation was carried out as established by Downey and collaborators for rabbit heart (23, 34, 35). Perfusion was performed at constant pressure of 85 mmHg (1 mmHg = 133 Pa) at 37°C by using Krebs-Henseleit buffer containing 118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, and 10 mM glucose (pH 7.4) for 5 min. The perfusate was continuously bubbled with 95% O2/5% CO2. After the initial 5-min perfusion, the perfusate was changed to Ca+2-free Krebs-Henseleit buffer for 10 min and then Krebs-Henseleit buffer containing 1 mg/ml collagenase (Worthington) for 15 min. Ventricular myocytes were isolated by maceration and centrifugation for 4 min at 100 × g. The myocyte pellet then was resuspended in incubation buffer containing 118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 1.2 mM KH2PO4, 1.2 mM MgSO4, 30 mM Hepes, 60 mM taurine, 20 mM creatine, 0.68 mM glutamine, 1% Basal Medium Eagle amino acids, 1% Eagle’s MEM non-essential amino acids, 1% Basal Medium Eagle vitamins, and 2% BSA (pH 7.4). Typically, cell viability was >60% after isolation as determined by exclusion of trypan blue in hypotonic solution. There was a <10% increase in cell death after incubation under normoxic conditions (Fig. 1).
Figure 1.

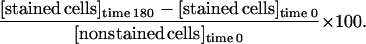 |
Simulated Ischemia of Isolated Cardiac Myocytes.
Immediately after isolation, myocytes were treated with ethanol and were co-incubated with the PKC inhibitors chelerythrine, GF109203X (both from Alexis Biochemicals, San Diego, CA), or isozyme-selective PKC inhibitory peptides (18) for 10–15 min. Cells then were pelleted by low speed centrifugation for 1 min and were washed twice with incubation buffer. (Under these conditions, most of the nonmyocyte cells remain in the supernatant, producing a pellet that contains >95% myocytes.) For the simulated ischemia, cells were transferred to microcentrifuge tubes, were washed twice with degassed glucose-free incubation buffer, and were pelleted as before. Each cell pellet occupied a volume of ≈150 μl. After the last centrifugation, 90% of the supernatant was removed, and microballoons (Sig Manufacturing, Montezuma, IA) were layered over the remaining buffer to create an air-tight environment. In the experiments described in Fig. 4, cell pellets were overlayed with degassed hypoxic and nitrogen-saturated buffer up to the rim of the tube, and the tubes were sealed with an airtight top. Tubes then were incubated at 37°C as above for 180 min. Similar results were obtained by using either procedure. Each experiment was repeated at least three times on separate days using different animals.
Figure 4.

Ten millimolar ethanol is sufficient to produce inhibition of ischemic damage. Adult rat cardiac myocytes were isolated and subjected to control normoxic conditions (Normoxia), simulated ischemia (Ischemia), and simulated ischemia after preincubation or preincubation followed by co-incubation with 10–50 mM ethanol as described in Materials and Methods. Cell damage then was determined as in Fig. 1, and data are mean ± SEM of three independent experiments (*, P < 0.05; **, P < 0.005).
Assessment of Cell Viability.
Ischemia-induced cell damage was assessed by an osmotic fragility test that has been previously used to measure the effectiveness of preconditioning intervention (34, 35). Myocytes were incubated in a hypotonic (85 milliosmolar) trypan blue solution (36). During simulated ischemia, the cells become progressively more fragile, as evidenced by an increasing percentage of myocytes stained with trypan blue. Normoxic control cells were centrifuged as above, except the cells were resuspended in 3 ml of incubation buffer and were incubated at 37°C, exposed to atmospheric oxygen (normoxia control). Cells that did not exclude the trypan blue dye were considered nonviable because of membrane fragility. Three- to four-hundred cells were counted in each experiment. Blind scoring (done in the majority of the study) did not alter the results.
Western Blot Analyses.
Isolated myocytes were initially divided into three groups and were treated with vehicle (0.05% DMSO), 0–50 mM ethanol, or 100 nM 4β-phorbol 12-myristate 13-acetate (PMA) (Alexis Biochemicals) for 15 min at 37 °C. Myocytes were homogenized, and PKC distribution in the 100,000 × g supernatant (cytosolic) fraction and 1% Triton X-100 extract of the pellet were determined as described (21) and were quantitated by densitometric analysis of digitized autoradiograms using nih image 1.58 (http://rsb.info.nih.gov./nih-image/-Default.html).
Ischemia-Reperfusion of Isolated Adult Rat Heart and Creatine Kinase Assay.
Hearts from adult male Wistar rats (250–300 g) were isolated and perfused on a Langendorff apparatus (37). Perfusion was performed at constant pressure of 85 mmHg at 37°C by using Krebs-Henseleit buffer containing 118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 1.2 mM KH2PO4, 1.2 mM MgSO4,1.8 mM CaCl2, and 5 mM glucose (pH 7.4). The perfusate was oxygenated by continuous bubbling of 95% O2/5% CO2. After an initial 10-min equilibration period, hearts were perfused for 20 min with treatments of 10 mM ethanol and/or 10 μM chelerythrine (Alexis Biochemicals), followed by 45 min of no-flow ischemia. Immediately after ischemia, the hearts were subjected to 30 min of reperfusion. Samples of coronary venous out flow were collected every 2.5 min during the reperfusion period (total of 12 fractions). Creatine kinase release was assayed by using a colorimetric determination kit (Sigma) as a measure of cardiac injury.
Results
Protection Against Simulated Ischemia After Acute Exposure to Ethanol in Isolated Adult Cardiac Myocytes.
We first determined whether ethanol-induced protection from ischemic injury is attributable to direct action of ethanol on cardiac myocytes and whether a brief exposure to ethanol produces this protection. The sensitivity of isolated adult rat myocytes to ischemic injury was measured by incubating pellet of cardiomyocytes with a small volume of degassed medium lacking glucose. Cell damage then was determined by measuring osmotic fragility. This model, termed “simulated ischemia,” is well established and is thought to mimic ischemic injury of whole heart (34). Under normoxic conditions, only 9 ± 7% of the cells demonstrated osmotic fragility (mean ± SEM; n = 7; P not significant; Fig. 1A) over the 3 hours of incubation, indicating that isolated myocytes remained healthy. However, during the same period, when cells were incubated under simulated ischemic conditions, 74 ± 8% demonstrated osmotic fragility (Fig. 1A; n = 7; P < 0.05). This increased fragility induced by simulated ischemia was inhibited by a 10-min exposure of cells to 50 mM ethanol before the ischemia from ≈75% in the absence of ethanol to 50 ± 5% in its presence (Fig. 1A; n = 7; P < 0.05 vs. untreated ischemic cells). Note that, although ethanol was not included during prolonged ischemia, there was an ≈35% reduction in cell damage. Importantly, ethanol had no effect on the osmotic fragility of cells incubated under normoxic conditions (not shown). Ischemia-induced damage also can be assessed by rounding of the cardiac myocytes (Fig. 1B Upper), an indicator of irreversible damage (38), and by staining of nuclei with propidium iodide (Fig. 1B Lower), an indicator of loss of membrane integrity (39). Because ischemic damage (as assessed by all of these assays) was significantly reduced after a 10-min incubation with ethanol of isolated myocytes, we conclude that ethanol exerts a direct effect on cardiac myocytes that renders them more resistant to ischemic injury.
Role of PKC in the Ethanol-Mediated Protection Against Simulated Ischemia.
Because PKC has been implicated in cardioprotection against global ischemia (8, 21–24, 40), and because PKC is activated by ethanol (8, 11, 13, 41, 42), we determined whether the protection observed in isolated myocytes after a brief exposure to ethanol is also mediated by PKC. Using chelerythrine or GF109203X, we first showed that these PKC inhibitors have effect on osmotic fragility under normoxic conditions (Fig. 1A). However, the presence of 1 or 10 μM chelerythrine during the 10-min co-incubation with ethanol before the simulated ischemia abolished ethanol-induced protection (Fig. 1; n = 3; P < 0.05). In addition, GF109203X (10 μM) also abolished protection induced by pretreatment with 50 mM ethanol (Fig. 1; n = 3; P < 0.05). These data indicate that the beneficial effect of ethanol in protecting cardiac myocytes from ischemic injury is mediated specifically through PKC.
Western Blot Analysis.
There are multiple PKC isozymes in cardiac myocytes (43–47), but ɛPKC has been previously implicated as a mediator of cardioprotection because its translocation correlates with preconditioning-induced protection or protection induced after 8–12 weeks of exposure to ethanol (8, 47). Using Western blot analysis, we therefore first determined whether translocation of ɛPKC occurs also after a short exposure to ethanol in isolated adult rat myocytes. PKC activation by hormones or phorbol esters is associated with translocation of individual PKC isozymes from the soluble fraction to the particulate fraction (48, 49). Fig. 2A shows that a 15-min incubation of myocytes with either 50 mM ethanol or 100 nM 4β-phorbol 12-myristate 13-acetate (PMA) caused translocation of ɛPKC from the soluble to the particulate fraction. In addition, translocation of ɛPKC by ethanol was concentration-dependent. The amount of ɛPKC in the particulate fraction increased from 36 ± 7% of the total detectable ɛPKC in the absence of ethanol to 56 ± 10% at 1 mM ethanol, 72 ± 11% at 10 mM ethanol, and 70 ± 8% at 50 mM ethanol (n = 4, P < 0.05); a representative experiment is shown in Fig. 2B. Maximum translocation of ɛPKC isozyme by ethanol was reached at 10 mM, a physiologically relevant concentration of ethanol.
Figure 2.

Ethanol induces translocation of ɛPKC. Shown is a Western blot analysis of duplicate sets of subcellular fractions from isolated adult rat cardiac myocytes. (A) Myocytes were treated with vehicle (Con), 50 mM ethanol (EtOH), or 100 nM 4β-phorbol 12-myristate 13-acetate (PMA) for 15 min. Soluble and particulate fractions from equal numbers of myocytes (75–100 mg protein per lane) were subjected to SDS/PAGE, were transferred to nitrocellulose, and were probed with an ɛPKC isozyme-selective antibody. Results are representative of two independent experiments. (B) Myocytes were treated with 1 mM, 10 mM, or 50 mM ethanol for 15 min. Soluble and particulate fractions from equal numbers of control (Con) and treated myocytes (75–100 μg protein per lane) were subjected to SDS/PAGE, were transferred to nitrocellulose, and were probed with δPKC or ɛPKC selective antibody. Results are representative of three to four independent experiments.
Isozyme-Selective Inhibition Studies.
We next determined whether activation of ɛPKC is required for the protective effect of acute exposure to ethanol in the isolated myocyte model by using an ɛPKC isozyme-selective inhibitory peptide, ɛV1-2 (30). In previous studies, we showed that this eight-amino acid peptide, derived from the V1 region of ɛPKC, inhibits activation-induced translocation and function of ɛPKC in cardiac myocytes (30). Relevant to this study, ɛV1-2 inhibited preconditioning- and PMA-induced cardioprotection from hypoxia-induced cell death in cultured neonatal cardiac myocytes (21). As discussed earlier, because, in this study, our aim is to determine whether ethanol-induced cardioprotection in adult animals is also mediated by activation of ɛPKC, we used here adult myocytes and delivered the peptides into the cells by conjugating them to the Drosophila Antennapedia-derived cell-permeable carrier peptide (32, 33). We found that ɛV1-2 (1 μM), chelerythrine (10 μM), or GF109203X (10 μM) almost completely abolished ethanol induced protection in adult cardiomyocytes (Fig. 3; n = 3; P < 0.05). In contrast, incubation with a control peptide (carrier peptide dimer), or the selective inhibitor peptide of the classical PKC isozymes, βC2-4 (31), had no effect on ethanol-induced protection (Fig. 3). Ethanol also caused translocation of δPKC (Fig. 2). Therefore, the role of δPKC in ethanol-induced protection of adult cardiac myocytes from hypoxia-induced death cannot be excluded. However, because ethanol-induced protection was almost completely abolished by the ɛPKC-selective inhibitor, ɛV1-2, these data indicate that cardioprotection by acute ethanol treatment is mediated largely, if not exclusively, by activation of ɛPKC in the cardiac myocytes.
Figure 3.

Inhibition of ethanol protection by an ɛPKC isozyme-selective antagonist peptide, ɛV1-2. Ethanol-induced protection of cardiac myocytes from damage induced by simulated ischemia was determined in the presence of chelerythrine (10 μM), GF109203X (10 μM), the ɛPKC selective inhibitor ɛV1-2, or the cPKC-selective inhibitor βC2-4, each conjugated to the cell-permeable Drosophila Antennapedia carrier peptide and applied at 1 μM. The carrier peptide alone (1 μM) also was used as a negative control. After co-incubation with 50 mM ethanol for 10 min, cells were washed and subjected to simulated ischemia for 180 min. Data, presented as percent inhibition of maximal protection by 50 mM ethanol, are mean ± SEM of three experiments (*, P < 0.05).
Protection Against Ischemic Damage Also Occurs at 10 mM Ethanol.
Because exposure of cardiac myocytes to 10 mM ethanol is sufficient to cause ɛPKC translocation in these cells (Fig. 2), we determined whether 10 mM ethanol also can provide cardioprotection in the ischemic model. In these experiments, osmotic fragility was increased by 50 ± 4% after incubation for 180 min under hypoxic conditions (Fig. 4; n = 3); this increased fragility was inhibited by 42% (to 29 ± 5% cell damage) after a transient 10-min preexposure of cells to 50 mM ethanol (Fig. 4; n = 3; P < 0.05 vs. untreated ischemic cells). Protection of cardiac myocytes by a transient exposure to 10 mM ethanol before the ischemic condition was not significant (not shown). However, when ethanol was included also during the prolonged ischemia, a 56% inhibition in cell damage was observed even with 10 mM ethanol (Fig. 4); only 22 ± 9% of the cells pretreated with 10 mM ethanol showed damage as compared with 50 ± 4% of untreated ischemic cells (n = 3; P < 0.05 vs. untreated ischemic cells). Importantly, ɛV1-2 (1 μM) added together with the ethanol abolished the above protection of 10 mM ethanol by 90 ± 4%, and 0.5 μM ɛV1-2 caused a 73 ± 11% inhibition of this ethanol protection (n = 2). Therefore, cardioprotection induced by 10 mM ethanol depends on ɛPKC activation.
Perfusion with 10 mM Ethanol Protects Adult Rat Heart from No-Flow (Global) Ischemia-Reperfusion Damage.
Finally, to determine whether such a brief exposure to 10 mM ethanol also can provide protection of the intact heart, we used a no-flow (global) ischemia model and exposed the hearts to ethanol just before the ischemic insult. Hearts perfused on a Langendorff apparatus were subjected to a 20-min perfusion in the absence or presence of 10 mM ethanol (Fig. 5A). After 45 min of ischemia, we reperfused the hearts, fractions of perfusate were collected, and the levels of creatine kinase (CK) released from the damaged myocardium were determined. We found a 4-fold increase in CK release after ischemia (Isc) as compared with hearts maintained in normoxic conditions (Nor; unfilled symbols) that was sustained for 30 min of reperfusion (Fig. 5A). This increase in CK release was greatly attenuated in hearts that were preperfused with 10 mM ethanol (Isc/EtOH) for 20 min before the ischemic insult (Fig. 5A); an average of ≈70% reduction in CK release after ischemia-reperfusion was noted in hearts from rats exposed briefly to ethanol as compared with control ischemic hearts (Fig. 5B; P < 0.05). Importantly, chelerythrine (Che; 10 μM) co-perfused with 10 mM ethanol greatly inhibited the protection of intact heart from ischemic-reperfusion damage [Fig. 5 A (Isc/EtOH/Che) and B], yet neither ethanol alone or chelerythrine alone cause CK release under normoxic conditions (Fig. 5A; unfilled symbols). Therefore, even a brief exposure of intact hearts to 10 mM ethanol produces protection from ischemic insult that depends on PKC activity.
Figure 5.

Ex vivo exposure of intact heart to 10 mM ethanol is sufficient to produce inhibition of ischemic reperfusion damage. (A) Hearts were perfused without or with 10 mM ethanol (EtOH) for 20 min before the ischemic damage. Where indicated, the PKC inhibitor, chelerythrine (Che) (10 μM) also was perfused. Ischemia-reperfusion injury then was determined by colorimetric reaction of creatine kinase (CK) released from the myocardium as described in Materials and Methods. Numbers presented are OD reading at 520 nm of the 2.5-min fraction collected during the 30-min reperfusion period after a 45-min no-flow ischemia (Isc) or continuous perfusion of oxygenated buffer for the same period of time (Nor). Data are each from a single heart. (B) Ischemia-reperfusion injury is presented as units of CK released during the entire 30-min reperfusion period. Perfusion protocol and treatments of ethanol and chelerythrine was identical as in A. Results are mean ± SEM obtained from each of the three animals (*, P < 0.05).
Discussion
In this study, we show that acute exposure of isolated adult rat cardiac myocytes (Figs. 1, 3, and 4) or intact heart (Fig. 5) to ethanol induces cardioprotective effects by acting directly on cardiac myocytes. Using isolated myocytes, we found a >50% protection of these cells from ischemic injury after a 10-min exposure to as little as 10 mM ethanol before and during the ischemic event (Fig. 4). This model of cardiac ischemia has proven to be a very useful approach to identification of potential cellular mediators of preconditioning because it permits direct comparison of control and acutely treated myocytes using cells obtained from the same animal without the complications of other organ effects. Indeed, the same concentration of ethanol caused a 70% reduction in CK release in a model of global ischemia of the intact heart (Fig. 5). The increased protection in the intact organ (Fig. 5; ≈70% protection) as compared with the isolated myocytes (Fig. 4; ≈50% protection) may be attributable to ethanol-induced autocrine activity [e.g., of adenosine (37)] in the intact organ that enhances PKC activation and cardiac protection.
We also demonstrated that the protection from ischemia obtained by acute exposure to ethanol is attributable to activation of PKC in the cardiac myocytes themselves and that ɛPKC is the isozyme required for this protective effect. This is in accordance with studies using other models of cardioprotection and suggesting a role for ɛPKC in cardioprotection induced by an exposure to a brief ischemic event or direct activation of PKC by PMA (see, for example, refs. 22–24, 40, 50, and 51). Three lines of evidence support a critical role for ɛPKC activation and translocation in this ethanol-mediated cardioprotection of freshly isolated adult rat cardiomyocytes. First, the presence of the general PKC inhibitors chelerythrine or GF109203X during ethanol pretreatment of myocytes abolished ethanol-induced protection during subsequent ischemia (Fig. 1). Second, treatment of cardiac myocytes with 10–50 mM ethanol approximately doubled the amount of ɛPKC that translocates from the cell soluble to the cell particulate fraction compared with controls (Fig. 2). Finally, the presence of the ɛPKC-selective inhibitor peptide, ɛV1-2, during ethanol preconditioning of isolated rat cardiac myocytes selectively abolished ethanol-induced protection during subsequent ischemia (Fig. 3 and see text). In contrast, an inhibitor of the classical PKC isozymes, βC2-4, had no effect (Fig. 3). Therefore, protection from ischemia obtained by acute exposure to 10 mM ethanol is not only correlated with activation of specific PKC isozymes (Fig. 2) but would not occur if activation of ɛPKC is inhibited (Fig. 3).
The ability of ethanol to activate PKC in general and ɛPKC in particular has been observed in several other cell systems. Ethanol-induced increases in PKC activity and amount of δ and ɛPKC isozymes were found in the pheochromocytoma cell line, PC12, cultured in 200 mM ethanol for 6 days (52, 53). Importantly, an ɛPKC-selective inhibitor prevented ethanol-induced neurite extension in this cell model (54). In addition, increases in the amount of α, δ, and ɛPKC were found in NG108-15 neural cells after prolonged ethanol exposure (42). Moreover, Gordon et al. have shown that this chronic exposure to ethanol causes translocation of δ and ɛPKC in these cells (42). Therefore, other responses induced by ethanol are likely to be mediated, at least in part, by activation of ɛPKC. Future studies, using nonpeptide analogs of ɛPKC specific inhibitors and activators, should determine whether ɛPKC mediates protection of the intact heart from global ischemia. However, our findings using the isolated myocyte model of simulated ischemia are highly supportive of this possibility.
The current recommendation for moderate daily consumption [one drink per day for women and two drinks per day for men (Dietary guidelines, Human Nutrition Information Service, www.americanheart.org)] roughly translates to 10 mM blood alcohol levels 1 hour after consumption. Our study illustrates that cardioprotection can be induced by exposure to 10 mM ethanol immediately before and during the ischemic insult. We also found that protection of 25–50 mM ethanol occurred after prolonged ethanol feeding in vivo guinea pig model (8). A notable difference between the two studies is the exposure time and dosage of ethanol. In the in vivo model, animals were exposed to ethanol via their drinking water for 8–12 weeks, resulting in a maximal blood alcohol level of ≈20 mM that fluctuated during the day. Here, isolated cardiac myocytes exposed to a single dose of as little as 10 mM demonstrated a significant protection from damage induced by 3 hours of ischemia. In addition, the dependency of ethanol-induced protection on ɛPKC activation was demonstrated here by using isozyme-selective inhibitors. In contrast, in the previous study, there was only a correlation between ɛPKC activation and protection; causality using isozyme-selective inhibitors could not be demonstrated because the peptide inhibitors cannot be used in vivo. Finally, a protection of intact heart by ex vivo exposure of rat hearts to 10 mM ethanol for 20 min also was demonstrated here.
Each year, in the United States alone, 600,000 adults and 12,000 children undergo open heart operations using cardiopulmonary bypass, during which the heart is subjected to periods of controlled ischemia ranging from several minutes to well over 1 hour. Despite advances in cardiac protection, myocardial dysfunction during the immediate postoperative period remains a leading cause of morbidity and mortality in these patients. Because the exact timing of the ischemic insult is known ahead of time in these patients, the potential exists to significantly reduce myocardial damage by inducing a protective response in the hours or minutes before surgery. Although a brief period of ischemia before the prolonged ischemia provides protection, this method is not without danger. Future studies examining the protection induced by ethanol exposure in whole animals should determine whether there is a benefit of exposing humans to low levels of ethanol (e.g., 10 mM) immediately before scheduled ischemia, such as that occurring during angioplasty and in cardiac surgery.
Finally, our findings support the epidemiological studies demonstrating the benefits of moderate ethanol consumption for ischemic heart disease (1–3, 55). However, the data provided here suggest that rather than merely lowering the rate of atherosclerosis by regular exposure to ethanol over many years (4), at least some of the ethanol-induced protection can be produced acutely, within a few minutes. Moreover, the protection is obtained by physiologically attainable blood ethanol levels observed after one to two alcoholic drinks. Finally, we showed that this protection is mediated by a direct effect of the ethanol on the myocardium. Identification of the role of ɛPKC in the cardioprotective effect of ethanol may result in development of a new therapeutic agent, an ɛPKC agonist, that mimics the ethanol effect and provides protection against ischemic injury without the detrimental medical and social effects of drinking.
Acknowledgments
We thank Drs. James Downey and Guang-Shun Liu for help with the myocyte model, Tamar Liron and Leon Chen for help with the study, and Dr. Downey for advice on the manuscript. This work was supported by funds from National Institutes of Health (Grant AA11147 to D.M.R.).
Abbreviations
- PKC
protein kinase C
- PMA
4β-phorbol 12-myristate 13-acetate
- CK
creatine kinase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Parratt J R. Trends Pharmacol Sci. 1994;15:19–25. doi: 10.1016/0165-6147(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 2.De Labry L O, Glynn R J, Levenson M R, Hermos J A, LoCastro J S, Vokonas P S. J Stud Alcohol. 1992;53:25–32. doi: 10.15288/jsa.1992.53.25. [DOI] [PubMed] [Google Scholar]
- 3.Ahlawat S K, Siwach S B. Int J Cardiol. 1994;44:157–162. doi: 10.1016/0167-5273(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 4.Langer R D, Criqui M H, Reed D M. Circulation. 1992;85:910–915. doi: 10.1161/01.cir.85.3.910. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi H, Ashraf M, Rahamathulla P M, Minami M. Pathol Res Pract. 1987;182:810–816. doi: 10.1016/S0344-0338(87)80047-X. [DOI] [PubMed] [Google Scholar]
- 6.Sheehy T W. Postgrad Med J. 1992;91:271–277. doi: 10.1080/00325481.1992.11701294. [DOI] [PubMed] [Google Scholar]
- 7.Lynch C., III . In: Mechanisms of Anesthetic Action in Skeletal, Cardiac, and Smooth Muscle. Blanck T J J, Wheeler D M, editors. New York: Plenum; 1991. [Google Scholar]
- 8.Miyamae M, Rodriguez M M, Camacho S A, Diamond I, Mochly-Rosen D, Figueredo V M. Proc Natl Acad Sci USA. 1998;95:8262–8267. doi: 10.1073/pnas.95.14.8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamond I, Wrubel B, Estrin W, Gordon A. Proc Natl Acad Sci USA. 1987;84:1413–1426. doi: 10.1073/pnas.84.5.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman P L, Valverius P, Kwast M, Tabakoff B. Alcohol Alcoholism. 1987. , Suppl. 1, 749–754. [PubMed] [Google Scholar]
- 11.Messing R O, Peterson P J, Henrich C J. J Biol Chem. 1991;266:23428–23432. [PubMed] [Google Scholar]
- 12.Lovinger D M, Zhou Q. Neuropharmacology. 1994;33:1567–1572. doi: 10.1016/0028-3908(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 13.Hoek J B, Rubin R, Thomas A P. Biochem J. 1988;251:865–871. doi: 10.1042/bj2510865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoek J B, Thomas A P, Rooney T A, Higashi K, Rubin E. FASEB J. 1992;6:2386–2396. [PubMed] [Google Scholar]
- 15.Dildy-Mayfield J E, Harris R A. J Neurochem. 1995;15:3162–3171. doi: 10.1523/JNEUROSCI.15-04-03162.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochly-Rosen D, Chang F-H, Cheever L, Kim M, Diamond I, Gordon A S. Nature (London) 1988;333:848–850. doi: 10.1038/333848a0. [DOI] [PubMed] [Google Scholar]
- 17.Figueredo V M, Mochly-Rosen D. In: Alcohol and Signal Transduction in the Myocardium. Wassef M, Zakhari S, editors. Washington, DC: CRS; 1996. pp. 263–277. [Google Scholar]
- 18.Souroujon M C, Mochly-Rosen D. Nat Biotechnol. 1998;16:919–924. doi: 10.1038/nbt1098-919. [DOI] [PubMed] [Google Scholar]
- 19.Mochly-Rosen D. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 20.Mochly-Rosen D, Gordon A S. FASEB J. 1998;12:35–42. [PubMed] [Google Scholar]
- 21.Gray M O, Karliner J S, Mochly-Rosen D. J Biol Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell M B, Parker C G, Meng X, Brew E C, Ao U, Brown J M, Harken A H, Banerjee A. Circulation. 1993;88:I633. [Google Scholar]
- 23.Ytrehus K, Liu Y, Downey J M. Am J Physiol. 1994;266:H1145–H1152. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]
- 24.Speechly-Dick M E, Mocanu M M, Yellon D M. Circ Res. 1994;75:586–590. doi: 10.1161/01.res.75.3.586. [DOI] [PubMed] [Google Scholar]
- 25.Walker D M, Walker J M, Pugsley W B, Pattison C W, Yellon D M. J Mol Cell Cardiol. 1995;27:1349–1357. doi: 10.1016/s0022-2828(05)82397-1. [DOI] [PubMed] [Google Scholar]
- 26.Speechly-Dick M E, Grover G J, Yellon D M. Circ Res. 1995;77:1030–1035. doi: 10.1161/01.res.77.5.1030. [DOI] [PubMed] [Google Scholar]
- 27.Schlaifer J D, Kerensky R A. Clin Cardiol. 1997;20:602–606. doi: 10.1002/clc.4960200705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kloner R A, Yellon D. J Am Coll Cardiol. 1994;24:1133–1142. doi: 10.1016/0735-1097(94)90880-x. [DOI] [PubMed] [Google Scholar]
- 29.Kloner R A, Shook T, Pryzklenk K, Davis V G, Junio L, Matthews R V, Burstein S, Gibson M, Poole W K, Cannon C P, et al. Circulation. 1995;91:37–45. doi: 10.1161/01.cir.91.1.37. [DOI] [PubMed] [Google Scholar]
- 30.Johnson J A, Gray M O, Chen C-H, Mochly-Rosen D. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 31.Ron D, Luo J, Mochly-Rosen D. J Biol Chem. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 32.Derossi D, Joliot A H, Chassaing G, Prochiantz A. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 33.Théodore L, Derossi D, Chassaing G, Llirbat B, Kubes M, Jordan P, Chneiweiss H, Godement P, Prochiantz A. J Neurosci. 1995;15:7158–7167. doi: 10.1523/JNEUROSCI.15-11-07158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armstrong S, Downey J M, Ganote C E. Cardiovasc Res. 1994;28:72–77. doi: 10.1093/cvr/28.1.72. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Ytrehus K, Downey J M. J Mol Cell Cardiol. 1994;26:661–668. doi: 10.1006/jmcc.1994.1078. [DOI] [PubMed] [Google Scholar]
- 36.Weinbrenner C, Liu G S, Cohen M V, Downey J M. J Mol Cell Cardiol. 1997;29:2383–2391. doi: 10.1006/jmcc.1997.0473. [DOI] [PubMed] [Google Scholar]
- 37.Miyamae M, Diamond I, Weiner M W, Camacho S A, Figueredo V M. Proc Natl Acad Sci USA. 1997;94:3235–3239. doi: 10.1073/pnas.94.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stern M D, Chein A M, Capogrossi M C, Pelto D J, Lakatta E G. Circ Res. 1985;56:899–903. doi: 10.1161/01.res.56.6.899. [DOI] [PubMed] [Google Scholar]
- 39.Mackay K, Mochly-Rosen D. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- 40.Tosaki A, Maulik N, Engelman D T, Engleman R M, Das D K. J Cardiovasc Pharmacol. 1996;28(5):723–731. doi: 10.1097/00005344-199611000-00016. [DOI] [PubMed] [Google Scholar]
- 41.Harris R A, McQuilkin S J, Paylor R, Abeliovich A, Tonegawa S, Wehner J M. Proc Natl Acad Sci USA. 1995;92:3658–3662. doi: 10.1073/pnas.92.9.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordon A S, Yao L, Wu Z-L, Coe I R, Diamond I. Mol Pharmacol. 1997;52:554–559. doi: 10.1124/mol.52.4.554. [DOI] [PubMed] [Google Scholar]
- 43.Puceat M, Hilal-Dandan R, Strulovici B, Brunton L L, Brown J H. J Biol Chem. 1994;269:16938–16944. [PubMed] [Google Scholar]
- 44.Disatnik M-H, Buraggi G, Mochly-Rosen D. Exp Cell Res. 1994;210:287–297. doi: 10.1006/excr.1994.1041. [DOI] [PubMed] [Google Scholar]
- 45.Steinberg S F, Goldberg M, Rybin V O. J Mol Cell Cardiol. 1995;27:141–153. doi: 10.1016/s0022-2828(08)80014-4. [DOI] [PubMed] [Google Scholar]
- 46.Rybin V O, Buttrick P M, Steinberg S F. Am J Physiol. 1997;272:H1636–H1642. doi: 10.1152/ajpheart.1997.272.4.H1636. [DOI] [PubMed] [Google Scholar]
- 47.Ping P, Zhang J, Qiu Y, Tang X-L, Manchikalapudi S, Cao X, Bolli R. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 48.Sando J J, Maurer M C, Bolen E J, Grisham C M. Cell Signaling. 1992;4:595–609. doi: 10.1016/0898-6568(92)90041-6. [DOI] [PubMed] [Google Scholar]
- 49.Newton A C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell M B, Meng X, Ao L, Brown J M, Harken A H, Banerjee A. Circ Res. 1995;76:73–81. doi: 10.1161/01.res.76.1.73. [DOI] [PubMed] [Google Scholar]
- 51.Kitakaze M, Takashima S, Morioka T, Sato H, Hori M. Circulation. 1993;8(I):213. doi: 10.1161/01.res.73.3.524. [DOI] [PubMed] [Google Scholar]
- 52.Messing R O. Alcohol Alcoholism. 1993. Suppl. 2, 289–293. [PubMed] [Google Scholar]
- 53.Roivainen R, Hundle B, Messing R O. Proc Natl Acad Sci USA. 1995;92:1891–1895. doi: 10.1073/pnas.92.6.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hundle B, McMahon T, Dadgar J, Chen C-H, Mochly-Rosen D, Messing R O. J Biol Chem. 1997;272:15028–15035. doi: 10.1074/jbc.272.23.15028. [DOI] [PubMed] [Google Scholar]
- 55.Miller G J, Beckles G L, Maude G H, Carson D C. Int J Epidemiol. 1990;19:923–930. doi: 10.1093/ije/19.4.923. [DOI] [PubMed] [Google Scholar]