

Abstract
Cysteine-scanning mutagenesis (SCAM) and computer-based modeling were used to investigate key structural features of the S6 transmembrane segment of the calcium-activated K+ channel of intermediate conductance IKCa. Our SCAM results show that the interaction of [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET) with cysteines engineered at positions 275, 278, and 282 leads to current inhibition. This effect was state dependent as MTSET appeared less effective at inhibiting IKCa in the closed (zero Ca2+ conditions) than open state configuration. Our results also indicate that the last four residues in S6, from A283 to A286, are entirely exposed to water in open IKCa channels, whereas MTSET can still reach the 283C and 286C residues with IKCa maintained in a closed state configuration. Notably, the internal application of MTSET or sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES) caused a strong Ca2+-dependent stimulation of the A283C, V285C, and A286C currents. However, in contrast to the wild-type IKCa, the MTSET-stimulated A283C and A286C currents appeared to be TEA insensitive, indicating that the MTSET binding at positions 283 and 286 impaired the access of TEA to the channel pore. Three-dimensional structural data were next generated through homology modeling using the KcsA structure as template. In accordance with the SCAM results, the three-dimensional models predict that the V275, T278, and V282 residues should be lining the channel pore. However, the pore dimensions derived for the A283–A286 region cannot account for the MTSET effect on the closed A283C and A286 mutants. Our results suggest that the S6 domain extending from V275 to V282 possesses features corresponding to the inner cavity region of KcsA, and that the COOH terminus end of S6, from A283 to A286, is more flexible than predicted on the basis of the closed KcsA crystallographic structure alone. According to this model, closure by the gate should occur at a point located between the T278 and V282 residues.
Keywords: cysteine, calcium-activated potassium channel, channel pore, HeLa cells, thiol reagents
INTRODUCTION
Calcium-activated potassium channels of intermediate conductance (IKCa) play a prominent role in a large variety of cellular events such as the endothelium-derived hyperpolarizing factor (EDHF)*-based regulation of vascular tone (Yamamoto et al., 1999), the sustained efflux of Cl− ions in secretory epithelia (Devor et al., 1996; Singh et al., 2001) and the proliferation of T-lymphocytes (Grissmer et al., 1993; Logsdon et al., 1997; Jensen et al., 1999; Ghanshani et al., 2000) and other cell types (Langille et al., 1986; Rane, 2000). IKCa channels belong to one of the three main classes of calcium-activated potassium channels (K[Ca2+]) identified to date on the basis of their permeation properties and pharmacology (Vergara et al., 1998). These include the charybdotoxin (ChTX)- and iberiotoxin-sensitive MaxiK channels of large conductance (150–220 pS), the 20–50 pS IKCa channels inhibited by ChTX (Cai et al., 1998) and clotrimazole (Rittenhouse et al., 1997), and the apamine-sensitive and -insensitive SK channels of small conductance (<10 pS) (Kohler et al., 1996). MaxiK, SK, and IKCa channels differ also in terms of their permeation and gating properties. For instance, in contrast to the MaxiK, both SK and IKCa channels are voltage insensitive and demonstrate a nonohmic current/voltage relationship with a conductance two to three times higher for inward than outward currents. Furthermore, the Ca2+ sensitivity of the SK and IKCa arises from the Ca2+ binding protein, calmodulin, constitutively bound to the channels (Xia et al., 1998; Khanna et al., 1999), and not as documented for the MaxiK channel from the direct binding of Ca2+ to an integral part of the channel structure (Schreiber and Salkoff, 1997).
IKCa channels are mostly expressed in nonexcitable cells (Sauvé et al., 1988; Brugnara et al., 1993; Grissmer et al., 1993; Marchenko and Sage, 1996; Sauvé et al., 1986; Logsdon et al., 1997; Schmid-Antomarchi et al., 1997) although reports have documented the presence of IKCa in vascular smooth muscles where they may contribute to muscle plasticity (Neylon et al., 1999). These channels have been recently cloned from different tissue preparations (Ishii et al., 1997; Joiner et al., 1997; Logsdon et al., 1997; Vandorpe et al., 1998; Warth et al., 1999) leading to a gene product comprising 425–427 amino acids with a predicted topology of six transmembrane segments S1–S6, and a pore motif between S5 and S6. Amino acid sequence alignments have revealed furthermore that the human IKCa channels identified so far are 42–45% identical and 50–55% conserved as compared with SK channels, indicating that IKCa are more closely related to SK than MaxiK. Little is known, however, on the three-dimensional structural organization of IKCa. Homology modeling and toxin binding studies have already provided evidence for topological similarities between the external vestibule region of IKCa and other K+ channels (Rauer et al., 2000). In addition, the crystal structure of the rat SK2 calmodulin binding domain (residues 395–430) has been reported recently, enabling for the first time molecular modeling of the IKCa–calmodulin complex which underlies channel gating (Schumacher et al., 2001). However, despite the importance of theses results, structural data concerning the IKCa channel pore and gating regions are still lacking.
The first direct structural data for pore formation in K+ channels came with the solved structure of the KcsA channel, a bacterial potassium channel with two transmembrane domains (Doyle et al., 1998). In the crystal structure of the pore region, the P loop of KcsA forms the ion selectivity filter and the α-helical M2 lines the long inner vestibule between the selectivity filter and the cytoplasm. Of equal interest is the observation that on the intracellular side, the M2 helices are joined together by the residues T107 and A111 to form a pore with a diameter smaller than 4.5 Å (Doyle et al., 1998; Bernèche and Roux, 2000). Although the passage of a dehydrated cation through this narrow entrance is sterically possible, electrostatic calculations show that such a process is energetically prohibitive, indicating that the intracellular entrance of the crystallographic structure of the KcsA channel is effectively closed (Roux et al., 2000). By analogy, these results suggest a key role of the S6 segments to the pore formation in IKCa and a contribution of the residues in the COOH-terminal segment of S6 to the channel gate.
The present study aims to investigate the structural features of the S6 segment from an IKCa channel cloned from HeLa cells. Fig. 1 shows an alignment of the 42 amino acids spanning the S6 segment in IKCa, rSK2, KcsA, and voltage-gated Shaker channels. Although there is little amino acid identity between IKCa, Shaker, and KcsA in the S6 COOH-terminal region, it is possible to align the upper part by using the conserved GYGD motif to constrain the entire alignment. This analysis indicates that there is a correspondence between the valine and threonine at positions 275 and 278 in IKCa and the cavity-lining residues I100 and F103 in KcsA. A similar equivalence also prevails between the T278 and V282 residues in IKCa and the I470 and V474 in Shaker, two amino acids reported to contribute to pore lining (Liu et al., 1997). These observations strongly suggest that the region spanning from V275 to A286 in IKCa is most likely to play a crucial role in the IKCa channel pore formation and gating mechanism. The binding of one calmodulin molecule to two adjacent monomers, as described by Schumacher et al. (2001) in their work on the closely related rSK2 channel, argues for structural changes in the IKCa S6 segment different from those prevailing during channel opening for voltage-dependent channels. A study was thus undertaken in which we examined the effects of the water-soluble methanethiosulfonate (MTS) derivatives sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES) and [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET) on the single channel conductance and gating properties of cysteine IKCa mutants for the region comprised between V275 and A286. These results and the structural three-dimensional data generated by homology modeling using templates as structures for the closed and open KcsA channel point toward distinct domains for the S6 segment; a first region extending from V275 to V282 with features corresponding to the inner cavity region of KcsA, and a second region in the COOH terminus end of S6, from A283 to A286 that would be more flexible than predicted on the basis of the closed KcsA crystallographic structure alone.
Figure 1.

Amino acid sequence alignment of IKCa, rSK2, KcsA, and Shaker showing sequence similarities within the S6 transmembrane segment. The sequence alignment of the Pore + S6 region of IKCa, rSK2, KcsA, and Shaker was based on the conserved GYGD pore motif. Sequence similarities of 100% and 75% are shaded in black and gray, respectively. The S6 segment in IKCa is presented as extending from V266 to the A286.
MATERIALS AND METHODS
Cloning, Sequencing, and Site-directed Mutagenesis of the IKCa Channel
IKCa channel cDNAs were obtained from HeLa cells by PCR using two oligonucleotides designed from the reported hKCa4 sequence (EMBL/GenBank/DDBJ accession no. AF022150). These two oligonucleotides (from 1–23 and 1,270–1,284) were chosen to yield a 1,281-bp fragment containing the entire coding sequence of the human IKCa channel. The PCR fragments were cloned into the pMT21 mammalian expression vector (Klein et al., 1999) and sequenced by the dideoxynucleotide chain termination method using the T7 Sequenase kit (USB) with synthetic oligonucleotides as primers. Sequencing the PCR product obtained from HeLa cells revealed a 100% identity with the sequences reported for the hKCa4, hIK1 (EMBL/GenBank/DDBJ accession no. AF022150), and hSK4 (EMBL/GenBank/DDBJ accession no. AF000972) channels. Site-directed mutagenesis of IKCa channels were performed using the QuikChange site-directed mutagenesis kit (Stratagene). To obtain the 10 X/C consecutive point mutations, amino acid changes were introduced by using 25 mer mutated oligonucleotides (V275/C, T278/C, A279/C, L280/C, L281/C, V282/C, A283/C, V284/C, V285/C, A286/C) and wild-type IKCa as template. All mutations were confirmed by sequencing the entire codon region in both directions on both strands.
Oocytes
Mature oocytes (stage V or VI) were obtained from Xenopus laevis frogs anesthetized with 3-aminobenzoic acid ethyl ester. The follicular layer was removed by incubating the oocytes in a Ca2+-free Barth's solution containing collagenase (1.6 mg/ml; Sigma-Aldrich) for 45 min. The composition of the Barth's solution was (in mM): 88 NaCl, 3 KCl, 0.82 MgSO4, 0.41 CaCl2, 0.33 Ca(NO3)2, and 5 HEPES (pH 7.6). Defolliculated oocytes were stored at 18°C in Barth's solution supplemented with 5% horse serum, 2.5 mM Na pyruvate, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Oocytes were studied 3–5 d after coinjection of 0.92–9.2 ng of the cDNA coding for IKCa in pMT21 and 1.38 ng of cDNA coding for a green fluorescent protein that was used as a marker for nuclear injection (Klein et al., 1999).
Prior to patch-clamping, defolliculated oocytes were kept in a hypertonic solution containing (in mM) 250 KCl, 1 MgSO4, 1 EGTA, 50 sucrose, and 10 HEPES buffered at pH 7.4 with KOH. The vitelline membrane was then peeled off using forceps, and the oocyte was transferred to a superfusion chamber for patch-clamp measurements.
Patch-Clamp Recording
Single channel recordings were performed in the inside-out patch-clamp configuration using an Axopatch 200A amplifier (Axon Instruments, Inc.). Patch pipettes were pulled from borosillicate capillaries using a Narishige pipette puller (Model PP-83) and used uncoated. The resistance of the patch electrodes ranged from 4 to 10 MΩ. Unless specified otherwise, the membrane potential is expressed as −Vp, where Vp is the pipette applied potential. Data acquisition was performed using a Digidata 1320A acquisition system (Axon Instruments, Inc.) at a sampling rate of 2.0 kHz with filtering at 500 Hz. When required, the open channel probability, PO, was estimated from current amplitude histograms on the basis of a binomial distribution as described elsewhere (Morier and Sauvé, 1994). The stationarity of the control recordings was tested according to the criteria defined in a previous work (Denicourt et al., 1996). Experiments were performed at room temperature (24°C).
Data Analysis
The water accessibility of the targeted cysteine residues was estimated from the time constant of the MTS-dependent inhibition (τi) or activation (τa) of the IKCa-induced K+ currents, whereas structural changes resulting from MTS binding to the protein were inferred from the change in current amplitude and open probability measured at the single channel level.
The state dependency of the MTSET action was tested in site protection experiments in which the modulatory effect of MTSET on the open channel (3 μM internal Ca2+) was measured after an initial MTSET application in nominally Ca2+-free conditions. Our approach consisted essentially in measuring the ratio <I>(test)/<I>(ctr), where <I>(ctr) and <I>(test) correspond, respectively, to the mean current values before and after MTSET application and washout in 3 μM Ca2+, with (see Fig. 9 A) or without (see Fig. 3) a preapplication of MTSET in zero Ca2+ conditions. Experiments in which a MTSET pretreatment in Ca2+-free conditions failed to modify the <I>(test)/<I>(ctr) ratio as compared with control (no pretreatment) were interpreted as indicating a targeted cysteine less accessible to MTSET when channels are maintained in a closed state configuration. Mean current values were computed using the QuB single channel software (Qin et al., 1996, 1997) and the rates of modification induced by MTSET estimated from the time constants of inhibition (τi) or activation (τa) obtained by curve fitting to a single exponential the time integral of the unitary currents (Origin; Microcal Software, Inc.).
Figure 9.

State-dependent action of MTSET. (A) Inside-out recordings showing the state dependency of the MTSET action on IKCa mutants. The accessibility to MTSET of the substituting cysteine was estimated in MTSET protection experiments as discussed in materials and methods. The applied potential was maintained at −60 mV throughout. (B) Bar graph representation of the ratio [<I>(test)/<I>(ctr)] measured with (closed) or without (open) MTSET preconditioning in zero internal Ca2+ conditions. The <I>(test)/<I>(ctr) ratios obtained for the second MTSET application in the perfusion protocol illustrated in A were estimated at 0.02 ± .006 (n = 3) for V275C, 0.26 ± 0.02 (n = 2) for T278C, 0.16 ± 0.06 (n = 3) for V282C, 0.54 ± 0.05 (n = 2) for V284, and 2 ± 1 (n = 3) for V285C, respectively. These values are not significantly different from the <I>(test)/<I>(ctr) ratios measured without an initial MTSET exposure in zero internal Ca2+. In contrast the <I>(test)/<I>(ctr) values estimated for A283C and A286C mutants decreased drastically from 19 ± 5 (n = 5) and 10 ± 0.54 in the absence of MTSET preconditioning in zero external Ca2+, to 1.0 ± 0.05 (n = 5) and 1.0 ±0.02 (n = 3) for the perfusion protocol illustrated in A. These observations support a model whereby A283 and A286 remained accessible to MTSET in conditions where channels were maintained in the closed state.
Figure 3.

Inside-out recordings illustrating the action of MTSET on IKCa mutants. Inside-out current records measured in symmetrical 200 mM K2SO4 + 3 μM internal Ca2+ conditions. The pipette potential was 60 mV throughout. The effect of the MTSET reagent on channel activity was estimated from the ratio <I>(test)/<I>(ctr) where <I>(ctr) is the mean current measured in 3 μM Ca2+ before drug application (labeled line 1 in WT) and <I>(test), the mean current obtained at the same Ca2+ concentration (labeled line 3 in WT) after the sequential washout of the drug with a Ca2+-containing (3 μM) (labeled line 2 in WT) and a Ca2+-free 200 mM K2SO4 solution. The drug was systematically applied for 5 min with a total washout period of 2 min. This procedure ensures that the observed effects of the MTS reagents are resulting from a covalent binding of the drug to the targeted cysteine, and not from nonspecific channel interactions with the open or closed channel. Strong inhibition (>75%) of channel activity was observed with V275C, T278C, and V282C after exposure to MTSET (5 mM) for 5 min, with a complete inhibition recorded with the V275C and V282C mutants. The V284C channel showed a maximal inhibition of 50% despite a steady-state current value reached after 45 s. Notably, MTSET caused an increase in inward currents when applied on the A283C, A286C, and to a lesser extent V285C mutants. The current increase remained Ca2+ and clotrimazole sensitive, ruling out nonspecific effects of MTSET. There were no significant variations in mean currents with A279C suggesting that this residue may not be MTSET accessible.
Nonstationary noise analysis on current transients during MTSET application was performed by computing the current variance and mean value on successive short time segments of equal length, To. This procedure generated for each record an ensemble of Np ordered pairs [(σ1, <I>1), (σ2, <I>2), (σ3, <I>3.), … , (σNp, <I>Np)], with To Np equal to the total length of the record. The ratios σ2/<I> and σ2/<I>2 were computed for each interval To and analyzed in terms of the model presented in .
Solutions
The solution referred to as 200 K2SO4 had a composition as follows (in mM): 200 K2SO4, 1.8 MgCl2, 10 HEPES buffered with KOH at pH 7.4. Sulfate salts were used to avoid contributions coming from endogenous Ca2+-dependent chloride channels and to selectively chelate contaminant divalent cations such as Ba2+ (maximum 0.5 nM in 200 mM K2SO4). Solutions with submicromolar Ca2+ concentration were prepared from 200 K2SO4 solutions with 1 mM EGTA plus CaCl2. Unless specified otherwise we used EGTA-free standard 200 K2SO4 solutions with a contaminant Ca2+ concentration estimated at 3 μM. The final free Ca2+ concentration for all solutions was confirmed by Fura 2 measurements as described elsewhere (Simoneau et al., 1996). Solutions with 30 mM TEA contained 1.0 mM CaCl2 plus 500 μM EGTA as to minimize contaminations by divalent and trivalent cations due to use of TEA chloride at a high concentration. MTSES, MTSET, and MTSACE (Anatrace, Inc.) were added directly into the recording saline a few minutes before use. Our study was limited to the MTSET, MTSES, and MTSACE reagents as they were reported to be of comparable size and membrane impermeant (Holmgren et al., 1996).
Homology Modeling
Open KcsA structure
To complement our analysis, an approximate model of the KcsA in the open state was considered. Coordinates of the open KcsA structure were computed according to the procedure described elsewhere (Roux et al., 2000). Briefly, a crude model was constructed in agreement with the current data derived from electron paramagnetic spectroscopy (EPR) implicating that the opening and closing of the channel involve movements of the inner helices, which presumably result in a change in the diameter of the narrow pore lined by hydrophobic residues (Perozo et al., 1999). The model was refined using molecular dynamics and energy minimization in the presence of artificial harmonic energy restraints designed to increase the diameter of the pore on the inner side by 3 Å relative to the closed channel X-ray structure; an additional restraint maintained the symmetry of the tetramer (Roux et al., 2000). Structural data for the closed KcsA channel were obtained from the protein data bank 1BL8 file.
IKCa modeling
Automated homology modeling was performed with Modeller V4 (Sali and Blundell, 1993) and involved the generation of 50 models of the IKCa channel pore using either the open or closed KcsA channel structure as templates. Energy minimization was carried on the model with the lowest objective function (roughly related to the energy of the model) using Charmm (Brooks et al., 1983).
Statistics
Results are presented as mean ± SEM. Statistical significance between two independent populations was inferred from Students's t test statistics and set at P < 0.05.
RESULTS
Characterization of the Recombinant IKCa Channel Cloned from HeLa Cells
The IKCa channel cloned from HeLa cells was first characterized in a series of inside-out patch-clamp experiments performed on Xenopus laevis oocytes injected with wild-type IKCa cDNA. The single channel current voltage relationship measured in symmetrical 200 mM K2SO4 is presented in Fig. 2 A. The channel showed a clear inwardly rectifying behavior with a unitary conductance for inward and outward currents of 49 ± 1 pS (n = 3) and 20 ± 3 pS (n = 6), respectively. These values confirm that the basic permeation properties of the recombinant IKCa are identical to the native IKCa channel in HeLa cells (Sauvé et al., 1986). Inside-out single channel recordings measured in symmetrical 200 mM K2SO4 conditions for internal Ca2+ concentrations ranging from 0.1 to 1 μM are presented in Fig. 2 B. In these experiments, the voltage was maintained at −Vp = −60 mV throughout. Raising the internal Ca2+ concentration from 0.1 to 1 μM significantly increased the single channel activity as seen by the greater number of channel openings per s. The resulting dose-response curve is illustrated in Fig. 2 C. The sigmoidal curve was computed according to a Hill equation with [Ca]1/2 = 1.2 ± 0.1 μM and a Hill coefficient of 4.3 ± 0.4 (n = 4). Channel activity appeared voltage insensitive as no significant changes in the channel open probability were detected for voltages ranging from −150 to 150 mV (unpublished data).
Figure 2.

Characterization of the IKCa channel cloned from HeLa cells. (A) Current/voltage properties of the IKCa channel measured in symmetrical 200 K2SO4 conditions. The unitary conductance for inward and outward currents was estimated at 49 and 20 pS, respectively. (B) Inside-out single channel recordings of the wild-type IKCa channels expressed in Xenopus laevis oocytes for internal Ca2+ concentrations ranging from 0.1 to 1.0 μM. Experiments performed in symmetrical 200 mM K2SO4, pH 7.4 conditions. The pipette potential was maintained at 60 mV throughout. Current traces were filtered at 500 Hz and the letter c refers to the zero current level. (C) Normalized mean current at a constant Vp of 60 mV from four different experiments plotted as a function of the internal Ca2+ concentration. The sigmoide curve was computed according to a Hill equation with [Ca]1/2 = 1.2 ± 0.1 μM and Hill coefficient of 4.3 ± 0.4 (n = 4).
Effects of MTSET on S6 Segment Residues
Inside-out recordings were first undertaken to determine which residues within the V275 to A286 S6 segment are accessible to MTSET (10 Å × 5.8 Å) (Kuner et al., 1996) applied internally. As seen in Fig. 3, control experiments performed with wild-type IKCa recombinant channels showed only a transient inhibition of channel activity that could be recovered after washout of the drug. With the exception of L280C and L281C, which failed to yield detectable single channel events, all the IKCa mutants tested appeared functional and displayed an inward rectifying behavior. Fig. 3 provides direct evidence that the interaction of MTSET with V275C, T278C, V282C, and V284C leads to a significant inhibition of channel activity when the channel is maintained in the open state. In contrast, the covalent binding of MTSET to A283C, V285C, and A286C caused an increase of the IKCa currents. There was no significant effect of MTSET on the A279C mutant, suggesting either that the 279C residue was not accessible to MTSET or that the MTSET–279C interaction was silent. Globally, the recordings presented in Fig. 3 support a structural model where all the residues of the S6 segment from V282 to A286 are freely accessible to water from the cytoplasmic side of the membrane when the channel is in the open state.
A characterization of interaction of MTSET–IKCa mutants is presented in Fig. 4. As seen, a near total inhibition of channel activity was recorded with the V275C and V282C mutants, with <I>(test)/<I>(ctr) ratios of 0.05 ± 0.01 (n = 5) and 0.12 ± 0.06 (n = 4), respectively. MTSET appeared, however, less potent at inhibiting the T278C and V284C channels. This was particularly clear with the V284C mutant, for which we found a steady-state mean current ratio <I>(test)/<I>(ctr) of 0.45 ± 0.10 (n = 5), as compared with 0.25 ± 0.07 (n = 7) for T278C. As mentioned earlier, the interaction of MTSET–A283C, –A286C, and to a lesser extent –V285C caused a drastic increase in IKCa channel currents. The MTSET-induced current increases remained Ca2+ sensitive (0.1–3 μM) and could be inhibited by clotrimazole (50 μM) (unpublished data), indicating that channel activation did not result from a nonspecific effect of MTSET on the channel structure. The estimated <I>(test)/<I>(ctr) ratios ranged from 1.6 ± 0.2 (n = 5) for V285C to >20 ± 4 (n = 4) for A283C, suggesting a distinct structural organization for the residues located within the A283 to A286 S6 region (Fig. 4 A). Of particular interest is also the observation that the rate of modification for the interaction of MTSET–V285 residue was more than 10 times smaller relative to rates estimated for the A283C, V284C, and A286C mutants (Fig. 4 B). This observation is compatible with V285 being less accessible to MTSET relative to the neighboring A283, V284, and A286 residues. In addition, the rates of modification for the A283C, V284C, and A286C channels were significantly higher as compared with the residues located between V275 and V282. Globally these results provide evidence for a maximum accessibility to MTSET at the level of the cysteine at position 286.
Figure 4.

Effects of MTSET on IKCa channel mutants. (A) Histogram representation of the mean current ratio <I>(test)/<I>(ctr) obtained with MTSET for the amino acids spanning the V275-A286 domain. WT refers to the wild-type form of IKCa. Experiments performed in 3 μM internal Ca2+ (open channel). A ratio of 1 indicates a total absence of MTS-dependent effects on channel activity. C276 and C277 refer to endogenous cysteine and are labeled as *. Similarly, experiments could not be performed on the L280C and L281C mutants due to the absence of detectable single channel events in these cases. (B) Column representation of the modification rate for the MTSET-dependent channel inhibition and/or activation expressed in M−1s−1. With the exception of V285C (2.0 ± 0.5 s−1M−1; n = 5), modification rates for MTSET interactions were higher for the residues within the A283–A286 S6 region with values of 17 ± 0.5 s−1M−1 (n = 2), 17 ± 0.4 s−1M−1 (n = 5), and 54 ± 0.1 s−1M−1 (n = 2) for A283C, V284C, and A286C, respectively, as compared with 7 ± 0.8 s−1M−1 (n = 4) for V275C, 2.5 ± 1.5 s−1M−1 (n = 6) for T278C, and 2.9 ± 2 s−1M−1 (n = 4) for V282C. These results show that A286 is the residue most accessible to MTSET within the S6 segment extending from V275 to A286.
The mechanisms underlying the MTSET-dependent channel inhibition and/or activation were next investigated through single channel and noise analysis. The results presented in Fig. 5 confirm that the ratio σ2 (current variance)/<I> (mean current) remained nearly constant (from −0.7 to −0.8) during MTSET application on V275C, despite an important time-dependent decrease of the channel mean current value (see ). Also, the ratio σ2/<I>2 increased with a time constant equal to τi, the mean time of macroscopic current inhibition. Thus, the results presented in Fig. 5 support a model whereby MTSET causes a total inhibition of the V275C mutant, resulting in a gradual decrease in the number of active channels. Such a mechanism was confirmed through direct single channel recordings where the binding of MTSET was seen to irreversibly block the V275C channels with the unbound channel open probability, Po, remaining unchanged. A similar noise pattern, namely a constant σ2/<I> ratio coupled to a time-dependent increase in σ2/<I>2, was also obtained with the T278C and V282C mutants (unpublished data). In the former case, our results support a combined action of MTSET involving both a decrease in open probability plus a near complete (>70%) channel block. These observations contrast with the noise data derived for the residues located within the COOH terminus end of S6 (A283–A286). More specifically, the binding of MTSET to either 283C, 284C, 285C, or 286 produced a partial and not total inhibition of the channel unitary current amplitude (Fig. 6). For instance, the unitary current amplitude, I, measured at −140 mV for the A286C mutant was estimated at 3.1 pA in the presence of MTSET as compared with 7.4 pA before MTSET application. Also, the observation of a MTSET-dependent increase in mean current for the A283C, V285C, and A286C mutants requires that the action of the thiol reagent includes to overcome the partial inhibition in unitary conductance, either an increase in the channel open probability or an augmentation of the total number of active IKCa channels or both. For instance the fourfold decrease in σ2/<I> for the A283C mutant after internal MTSET application can in part be accounted for by the 65% inhibition in unitary current amplitude measured at the single channel level, the remaining MTSET-dependent decrease in σ2/<I> ratio being attributable to an increase in the channel open probability (Fig. 6). More importantly, the 20-fold decrease in σ2/<I>2 ratio estimated in this case correlates well with the magnitude of the observed increase in mean current from −0.7 to −16 pA, suggesting a system where PB > PO with PB and PO << 1 (see ). These results can thus be interpreted in terms either of a drastic change in the overall number of active channels, or as an increase in open probability (see Fig. 5 and ). A similar noise behavior was obtained with the A286C mutant (unpublished data), although in this particular case the single channel recordings presented in Fig. 6 support a MTSET-dependent current stimulation based mainly on an increase in open probability without an augmentation in the overall number of channels. The noise pattern for the V285C channel is also consistent with an action of MTSET mediated by an effect on the channel open probability with PB > PO and PO << 1, rather than the activation of silent V285C channels. In support of this proposal is: (a) the 4.5-fold MTSET-dependent reduction in the channel closed probability (1-Po) calculated from the decrease in σ2/<I> ratio, and (b) the 20-fold decrease in σ2/<I>2 ratio despite a maximum 180% increase in mean current value. Finally, the time-independent σ2/<I> ratio and the relatively modest increases σ2/<I>2 shown in Fig. 5 for the V284C mutant, added to the 40% inhibition unitary current measured in this case (Fig. 6) provide evidence for an effect of MTSET involving mainly both a partial inhibition of the channel unitary conductance and a twofold increase of the channel closed state probability. Globally, these results indicate two distinct regions in S6, namely: a region extending from V275 to V282 where a total inhibition of channel activity was observed, and a second region from A283 to A286 with the interactions MTSET–A283, –A286, and to a lesser extent –V285 leading to channel activation.
Figure 5.

Nonstationary noise analysis of the interacting MTSET-IKCa mutants. Relationship between the current variance σ2 and the mean current <I> during MTSET application illustrated for the V275C, A283C, V284C, and V285C mutants. <I> and σ2 were measured on successive time periods of 1 s for A283C, V284C, and V285C and 0.5 s for V275C. The V275C mutant displayed a constant σ2/<I> (left panel right scale) ratio despite an important decrease in <I> (left panel left scale), indicating an important inhibition of the channel unitary current. This proposal is also supported by the fact that the ratio σ2/<I>2 (right panel) increases in this case with a time constant equal to τi, the inhibition time constant measured for <I>. A similar noise pattern was also observed with the V284C mutant, although in this case the variations in σ2/<I> and σ2/<I>2 can be accounted for a partial inhibition of the channel unitary current plus a decrease in channel open probability. A different noise behavior is, however, observed with the A283C and V285C channels. The decrease in σ2/<I>2 observed with A283 correlates the increase in mean current with the σ2/<I> ratio remaining constant for time >7.5 s. This noise pattern would be compatible with a system where PB > PO with PO and PB << 1, thus supporting a model whereby the action of MTSET consists either to increase the channel open probability or recruit silent A283C mutants. The results obtained with V285C follow a similar pattern, although in this case the fact that the measured variation in σ2/<I>2 is more important than the mean current increase favors a system where PB > PO with PO << 1.
Figure 6.

Single channel analysis of the effect of MTSET on IKCa channel mutants. (A) Inside-out single channel recordings obtained from IKCa mutants under low Ca2+ conditions at an internal MTSET concentration of 5 mM. The applied voltage −Vp corresponded to −140 mV throughout. Large negative potentials were used in these experiments to optimize the signal to noise ratio, the current jump amplitude at −60 mV being barely detectable (<0.3 pA) after MTSET treatment except for the A286C mutant. The action of MTSET on 283C and 286C is seen to result in a substantial increase in open channel probability in accordance with the increase in mean current observed with the A283C and A286C mutants. (B) Bar graph representation of the effect of MTSET (5 mM) and MTSES (5mM) on the unitary current amplitude of IKCa mutants. I(MTSET) and I(ctr) correspond to the unitary current amplitude measured at −140 mV in the presence or absence of MTSET or MTSES (5 mM) respectively. * refers to endogenous cysteine or nonfunctional mutants as mentioned in Fig. 4. A ratio of 1 indicates an absence of MTSET- or MTSES-based effect on the channel unitary conductance. As seen, internal application of MTSET caused a near inhibition of the unitary current amplitude for the V275C, T278C, and V282C mutant channels. A partial inhibition ranging from 40% (V284C) to 70% (A283C) was observed with mutants obtained by cysteines substitution of the residues in the COOH-terminal region of S6 (A283–A286). In contrast, the application of the negatively charged MTSES reagent on A286C resulted in an increased unitary current amplitude. These observations suggest that the presence of charged groups at the cytoplasmic entrance of the pore can modulate the exit rate of K+ from the channel cavity into the cytosolic solution.
MTSET protection experiments were also conducted in which the pore structure of the MTSET-activated A283C and A286C mutant channels were investigated using the hydrophilic blocking agent TEA as probe. The control inside-out recordings presented in Fig. 7 show that the internal application of 30 mM TEA caused a near total block (>79%) of the wild-type IKCa currents. These results confirmed previous observations reported on the effect of internal TEA on the IKCa channels present in human red blood cells (Dunn, 1998). In contrast, there was no TEA-dependent block of the A283C and A286C currents following stimulation by internal MTSET. In fact, the percentage of TEA-related inhibition decreased from >79% ± 4 (n = 6) for the wild-type IKCa channel to <6.5% ± 4.6 (n = 3) for the MTSET-stimulated A283C mutant (Fig. 7 D). These observations strongly suggest that, in addition to an important effect on channel gating, the binding of MTSET to the cysteine engineered at position 283 or 286 leads to a narrowing of the pore such that TEA can no longer reach its blocking site.
Figure 7.

Protection by MTSET of TEA block for the A283C and A286C mutants. (A) Inside-out current recording illustrating the blocking action of TEA (30 mM) on the wild-type IKCa channel. (B) Inside-out recording demonstrating the lack of TEA-dependent block with the A283C mutant after application of MTSET. (C) Inside-out recording illustrating the reduced effectiveness of TEA on the A286C mutant stimulated by MTSET. (D) Histogram summarizing the effects of TEA on the wild-type IKCa channel (WT), and on the A283C and A286C mutants activated by MTSET. WT channel was blocked at 79 ± 4% (n = 6), whereas the blocking effect of TEA was reduced to 6.5 ± 4.6% (n = 3) for the A283C + MTSET mutant and to 27 ± 7% (n = 3) for the A286 mutant stimulated by MTSET.
Influence of the Charge on the MTS Channel Interactions
Experiments were next performed to evaluate if the presence of a positive charge on MTSET influences the interactions between MTSET and the cysteines engineered at positions 283–286. Inside-out recordings with the negatively charged MTSES reagent are presented in Fig. 8. As illustrated, all the mutants generated by cysteine mutagenesis for the residues spanning the A283 to A286 S6 domain were sensitive to MTSES. For instance, Fig. 8 shows a MTSES-based inhibition of the V284C channel activity resulting in a mean <I>(test)/<I>(ctr) ratio of 0.21 ± 0.04 (n = 7). Also, as observed with MTSET, internal applications of MTSES caused large increase of the A283C and A286C related currents with <I>(test)/<I>(ctr) values ranging from 3.0 ± 0.6 (n = 2) (A286C) to >7.0 ± 3 (n = 4) (A283C). Notably, the single channel recordings presented in Fig. 6 show that the application of MTSES on A286C resulted in a 150% increase in unitary current amplitude from a value of 8.0 pA in control conditions to 12 pA after MTSES treatment. These results contrast with the 60% unitary current inhibition measured with A286C when MTSET was used as thiol-modifying agent. Finally, experiments in which MTSES was applied before MTSET on A286C channels failed to demonstrate an additional stimulation by MTSET (Fig. 8). The recordings in Fig. 8 also indicate that in contrast to MTSET, the V275C mutant was not significantly affected by MTSES (<I>(test)/<I>(ctr) = 0.95 ± 0.2; n = 3). In fact, experiments in which MTSES was applied before MTSET showed no evidence for MTSES protecting V275C against the inhibitory action of MTSET (Fig. 8). These results clearly indicate that the accessibility of the 275C residue to MTS reagents of comparable size is charge specific, with channel inhibition being limited to the positively charged MTSET only. The internal application of MTSES succeeded, however, in blocking the T278C mutant with an I(test)/I(ctr) value of 0.27 ± 0.01 (n = 3) (unpublished data). Globally, these results demonstrate that the amino acids from T278 to A286 are significantly more accessible to the negatively charged MTSES reagent relative to V275C, for which a large difference in channel inhibition potency was observed between MTSET and MTSES.
Figure 8.

Effects of MTSES on IKCa mutants. Inside-out current records measured in symmetrical 200 mM K2SO4 + 3 μM internal Ca2+ conditions. The pipette potential was 60 mV throughout. In contrast to MTSET, MTSES failed to cause over a 5 min period a significant decrease of the V275C-induced currents. As observed with MTSET, however, MTSES succeeded in strongly inhibiting the V284C mutant and to stimulate the inward currents generated by A283C and A286C. Also illustrated are MTSES protection experiments performed on V275C and A286C, where MTSES (5 mM) was applied prior MTSET (5 mM). These experiments confirmed that a cysteine at position 275 is not accessible to MTSES and that the binding of MTSET to 286C prevents the interaction of MTSES–A286C.
State-dependent Effect of MTSET
Experiments were next conducted in which the action of MTSET in 3 μM internal Ca2+ conditions was tested after an initial MTSET exposure in nominally Ca2+-free conditions. Typical recordings are presented in Fig. 9 A. As seen, MTSET could inhibit the V275C mutant whether the channels were initially exposed to MTSET in zero internal Ca2+ conditions (Fig. 9) or not (Fig. 3). Similarly, there was no significant differences in the <I>(test)/<I>(ctr) ratios measured for the T278C and V282C mutants with (Fig. 9) or without (Fig. 3) MTSET preconditioning in zero internal Ca2+. These observations provide evidence for the 275C, 278C, and 282C residues becoming less accessible to MTSET in the closed as compared with the open IKCa channel structure.
State-dependent MTSET interactions were also recorded with the V284C and V285C channels. There were no significant differences in <I>(test)/<I>(ctr) ratios relative to control (no MTSET pretreatment) whether the V284C or V285C mutants were initially exposed to MTSET in zero Ca2+ or not. In contrast, the stimulatory action of MTSET on the A283C and A286C mutants clearly appeared state independent with MTSET activating both channels when applied in zero internal Ca2+ conditions. More importantly, a second MTSET application in the presence of 3 μM internal Ca2+ failed in these cases to initiate an additional current increase (unpublished data). These results suggest that the residues at positions 283 and 286 are accessible to MTSET independently of the channel open-closed configuration.
Homology Modeling
An analysis of the structural features of the S6 transmembrane segment in IKCa was performed by homology modeling using as templates the closed rigid KcsA channel structure obtained by X-ray crystallography (Doyle et al., 1998) and a computer derived structure for KcsA in the open state (Perozo et al., 1999; Roux et al., 2000). Surface and ribbon representations of the open IKCa S5-P-S6 regions are illustrated in Fig. 10, B and C, respectively, whereas a surface representation of the closed IKCa channel S5-P-S6 structure is presented in Fig. 10 A. A surface representation of a single S6 segment for the open channel is also illustrated in Fig. 10 B. The open and closed channel models first predict that with the exception of the Cys at position 267 which faces the selectivity filter, the endogenous cysteines at 269, 276, and 277 should be oriented away from the pore lumen (yellow in Fig. 10 C). Most interestingly, the models for the open (Fig. 10, B and C) and the closed (Fig. 10 A) channel suggest that V275, T278, and V282 are lining the channel pore with the V275 and T278 residues contributing to a central inner cavity similar to the one described for KcsA. The A279 residue should also be facing the pore lumen, but this prediction is not supported by our scanning mutagenesis (SCAM) experiments. Finally, the V284 and V285 residues are shown to be oriented directly opposite to the pore lumen, in contrast to the A286 side chain which is directed toward the pore central axis. A color representation of the MTSET modification rate for channel inhibition and/or activation is superimposed on the closed and open IKCa channel structures. Clearly, residues not predicted to be facing the pore lumen (A283, V284, and V285) turned out to be accessible to MTSET. This is particularly true for the valines at position 284 and 285, respectively. In addition, there is no structural correlation between the slow (blue) and intermediate (orange) rates of modification and the orientation of the residues relative to the pore central axis. For instance, the residue V284, which is predicted to be opposite to the pore lumen, led to a rate of modification the magnitude of which was comparable to that of the cavity lining residue V275. Notably, the fastest rate of modification (red) was obtained by cysteine substitution of the A286 residue, which should be located according to the open IKCa structure at the complete end of S6 facing the pore lumen. The pore cross-section area computed for the closed channel structure is presented in Fig. 10 D. The results of these calculations reveal that the channel cavity should be extending from V272 to T278, with a maximum inner width of ∼10 Å at the level of V275. A cavity of that size could easily accommodate a single MTSET molecule which averages a cross diameter of 5.8 Å (Kuner et al., 1996). Hence, for that region, our models based on the closed and open KcsA structures agree with the MTSET results. The correlation between the data and the models, however, decreases for residues in the COOH terminus end of S6 region. Indeed, the closed channel structure predicts a cross-section area for the 279–292 region too small (<2 Å) to account for the state-independent effects of MTSET observed with A283C and A286C (Fig. 9). Homology modeling finally provided information on the location of the putative gate by comparing the predicted structures for the closed and open IKCa channel (Fig. 10, A and B). Whereas the cross-section area of the pore differed by ∼25% at the level of the cavity between the open and closed channel structures, important differences in the pore inner diameter are predicted between these structures starting at T278 (Fig. 10 D). These qualitative considerations would thus support important structural changes at the level of the T278–V282 residues during gating.
Figure 10.

Homology modeling of the IKCa. Computer modeling of the IKCa channel using either the closed or open KcsA channel structure as template. Surface and ribbon representations of the open IKCa S5-P-S6 region are illustrated in B and C, respectively. B also includes a surface representation of a single S6 segment for the open channel. The molecular surface computed for the closed IKCa S5-P-S6 region is presented in A. According to the proposed models, the residues V275, T278, and V282 are lining the pore lumen resulting in a cavity 10 Å wide at the level of V275. Also, the endogenous cysteines (yellow in Fig. 9 C) at positions 269, 276, and 277 are presented as facing opposite to the pore lumen, whereas the C267 residue is predicted to be oriented toward the selectivity filter. A color representation of the MTSET modification rate for channel inhibition and/or activation is superimposed on the closed and open IKCa channel structures. Residues with slow (<5 M−1s−1), intermediate (5 M−1s−1 to 20 M−1s−1), and fast (>50 M−1s−1) rates of modification are colored in blue, orange, and red respectively. The pore lining residue A286 is seen as being the most accessible (red), whereas residues with the slowest rates of modification (blue) turned out to be located either inside (T278) or opposite to the pore lumen (V285). B also illustrates that all the residues from A283 to A286 are accessible to MTSET irrespective of their orientation relative to the pore central axis. (D) Cross-section area for residues 264–292 computed for the open and closed IKCa structures. Z refers to the channel axial distance with Z = 0 at V275. The closed channel structure predicts a central cavity extending from residues 272–278. The cross-section area for residues spanning the 282–286 region appeared under these conditions too small to accommodate an MTSET molecule of 5.8 Å diameter assuming a rigid structure for IKCa. A flexible structure extending below V282 can, however, account for the SCAM results.
DISCUSSION
This is the first report attempting to describe some of the structural features of the inner pore region of an IKCa channel. Our results clearly suggest functional differences between the residues located in the COOH-terminal region of S6 (A283–A286), and those located from V275 to V282. In support of this conclusion are the single channel observations that MTSET totally blocked the V275C, T278C, and V282C IKCa mutants while partially inhibiting channels where cysteines were substituting for the residues in the COOH terminus end of S6 (A283–A286). Furthermore, the interaction of MTSET with the cysteines at positions 283, 286, and to a lesser extent 285 led to a strong channel activation and to a narrowing of the channel pore as revealed by the absence of TEA block on the MTSET-stimulated A283C and A286C mutants. The negatively charged MTSES could also affect channel gating after binding with cysteines in the COOH-terminal region of S6, and interact with residues up to T278. Finally, the action of MTSET on the A283C and A286C mutants was found to be state independent in contrast to V275C, T278C, V282, V284, and V285, which showed a reduced MTSET accessibility for the closed IKCa structure.
Limits of the Present Study
Several assumptions are attached to the interpretation of cysteine mutagenesis. It is assumed, for instance, that the cysteine substitution does not dramatically alter the protein structure as to expose endogenous cysteines which would normally be buried. The S6 segment of IKCa contains four of the nine channel endogenous cysteines over a ten amino acid span covering the C267 to the C277 domain. We cannot currently rule out a possible contribution of those endogenous residues to the MTS-based effects reported in this work. For instance, the conservative substitution of the cysteine at position 276 by a serine led to a nonfunctional channel, indicating that the C276 residue is critical for channel proper functioning. Similarly, the C276 and C277 are located adjacent to the key V275 and T278 residues and might have contributed to the effects measured with the V275C and T278C mutants. The possible contribution of an auxiliary protein that would be MTSET sensitive cannot also be currently ruled out. In fact, the presence of an auxiliary protein has already been emphasized in a recent work on the regulation by ATP of the IKCa channel (Gerlach et al., 2001). Such an interaction is not expected, however, to modify the channel permeation properties, but may alter channel gating.
Our results show a transient noncovalent inhibition by MTSET of the wild-type IKCa channel. A noncovalent MTSET-dependent current inhibition has also been reported for the Kir6.2 channel by Cui et al. (2002). First, it should to be noticed that we never observed a partial recovery from a noncovalent block after MTSET or MTSES removal in all the experiments performed according to the protocols illustrated in Figs. 3 and 9. Furthermore, the perfusion protocol we used to measure the effect of MTS reagents includes an extensive washout period in zero and standard 3 μM internal Ca2+. This procedure insures that the estimated <I>(test)/<I>(ctr) ratios reflect an irreversible binding of the reagent to a targeted cysteine residue and not a transient non covalent interaction. A transient current block of the wild-type IKCa by MTSET is, however, to be expected if MTSET can diffuse up to the channel inner cavity. This proposal is supported by the observation that MTSET blocked the V275C mutant (Fig. 3). The absence of a noncovalent MTS contribution to the experiments illustrated in Figs. 3 and 9 can thus be seen as an indication that the covalent binding of MTS reagents to a targeted cysteine located between the cytoplasmic channel entrance and the V275 residue impairs the access of a MTS molecule to the cavity region, thus preventing a noncovalent MTS-based action. As such, the observation that the binding of MTSET to A283C and A286C impaired the blocking action of TEA (Fig. 7) provides indirect evidence that the binding of MTSET with residues in the COOH-terminal end of S6 can indeed limit the access of molecules such as MTSET to the channel pore.
The V275–V282 Region
The amino acid alignment presented in Fig. 1 locates the V275 and T278 residues of IKCa within the structurally identified hydrophobic lined cavity defined by the I100 and F103 residues in KcsA. The V275 and T278 residues in IKCa are therefore expected to contribute to the hydrophobic lumen of the IKCa channel cavity. This proposal is confirmed through homology modeling which predicts that V275, T278, and V282 should be facing the pore lumen with C276 and C277 opposite to the pore. The possibility of V275 lining the channel inner cavity has also been inferred from a molecular analysis of the interactions between IKCa and triarylmethanes such as clotrimazole (Wulff et al., 2001). The pore cross-section area for the open IKCa structure computed for the V275–V282 domain yielded values ranging from 28 to 44 Å2, with a cavity ∼10 Å wide at the level of V275. Clearly, the dimensions of the pore in the cavity should be suitable to accommodate at least a single MTSET molecule of 5.8 Å diameter and a theoretical cross-section area of 26 Å2 (Kuner et al., 1996). These computational results are thus compatible with the observed total block of V275C, T278C, and V282C measured at the single channel level in the presence of MTSET. The absence of an inhibitory action on V275C by MTSES is also expected as a structural analysis of the MTSES reagent locates the negatively charged moiety of the molecule ∼7 Å from the reactive atom, directly inside the channel inner cavity for a MTSES molecule bound to 275C. Also, the observation that MTSES succeeded at inhibiting the T278C mutant indicates that the IKCa channel can accommodate a negatively charged group up to a point corresponding to the V282 residue located 7 Å below 278C. It follows that the valine at position 282 cannot formally be a constitutive part of the channel inner cavity as the presence of a negative group inside the cavity is energetically unfavorable (Roux and MacKinnon, 1999; Roux et al., 2000). Finally, the MTSET-dependent inhibition of the V275C, T278C, and V282C mutants was found to be impaired when the channel was maintained in the closed configuration (zero Ca2+ conditions). These observations and the differences in pore dimensions from L280 to A286 between the closed and open IKCa channels support an IKCa model where the channel cavity becomes less accessible to MTSET upon channel closure.
The A283–A286 Region
The region comprising the residues A283–A286 in S6 is distinctive in many ways. First, all the residues between A283 and A286 were found to be sensitive to MTSET in the open channel configuration. The COOH-terminal region of S6 (A283–A286) is unlikely therefore to be embedded in the membrane where the accessibility to MTSET would be restricted. Second, one of the key observations of the present study concerns the increase in current triggered by MTSET and/or MTSES when applied to the A283C, A286C, and to a lesser extent V285C mutants. The A283C, A286C, and V285C channels clearly remained Ca2+ and clotrimazole (unpublished data) sensitive, indicating that the observed current stimulation did not result from some deleterious modifications of the channel structure. If, as suggested (Wulff et al., 2001), the inhibitory action of clotrimazole on IKCa involves binding to residues in the channel inner cavity, the observation that clotrimazole and not TEA (Fig. 7) block the MTSET-activated A283C and A286C currents suggests that clotrimazole has access to the cavity region through a hydrophobic pathway. In addition, this stimulatory action required the presence of internal Ca2+ and was not charge dependent, as similar effects were measured with MTSET, MTSES, and the neutral MTS reagent MTSACE (unpublished data). More importantly, our results indicate that the binding of MTSET or MTSES to A283C and A286C either activated silent IKCa channel mutants that did not contribute to the K+ current before application of the thiol modifying agent, or caused an increase of the channel open probability such that PO << PB with PO << 1. The single channel recordings presented in Fig. 6 for the A286C mutant better support, in our opinion, a model whereby PO << PB, suggesting that most of the observed current noise behavior can be explained in terms of an increase in open probability. We cannot, however, currently rule out the possibility that the A283C, and to a lesser extent the A286C IKCa mutants, represent a “rundown” form of IKCa which can be reactivated by MTSET or MTSES. Notably, the MTSET-dependent increase in current intensity recorded with A283C and A286C appeared to be state independent, thus providing evidence for a pore structure in which the A283 and A286 residues are accessible to the hydrophilic MTSET reagent when IKCa channels are in a closed state configuration. Important changes in MTSET accessibility were, however, observed with the V284C and V285C mutants, indicating that V284 and V285 become exposed to water after channel opening. These observations support a structural model whereby the S6 region from A283 to A286 undergoes an important reorganization during gating.
An additional feature of the COOH terminus end of S6 concerns the contribution of electrostatic effects to the MTSET- and MTSES-based changes in unitary current. Our results indicate that the binding of the positively charged MTS reagent MTSET to the cysteines substituting for the residues A283–A286 leads to a 40–70% inhibition of the unitary current amplitude. In contrast, we observed an increase in unitary current amplitude with the A286C and A283C (unpublished data) mutants after treatment with the negatively charged MTS reagent MTSES. These results strongly suggest that the variations in K+ currents related to the presence of either MTSET or MTSES at positions 283–286 involved electrostatic interactions such that the presence of a charged group near the cytoplasmic entrance of the pore affects the exit rate of K+ from the cavity into the cytosolic solution.
Model of the S6 Segment
Several conclusions on the structural organization of the IKCa S6 segment can be drawn on the basis of the results presented in this work. For instance, the SCAM data relative to the COOH-terminal segment of S6 (A283–A286) challenge the predicted structures for the closed IKCa channel derived from homology modeling using a rigid representation of KcsA. The observation that the residue A283 was accessible to MTSET for the closed IKCa channel argues for a pore structure in which the COOH-terminal of S6 is wider than predicted from a rigid closed KcsA structure. In fact, the proposed closed IKCa model predicts that the pore sectional area for the region extending from A279 to K292 should be <2 Å2. In addition, modeling of the closed A283C and A286C mutants indicates that the cysteines at positions 283 and 286 should be in contact with the pore lumen, suggesting that MTSET needs to diffuse inside the pore to reach the targeted cysteine residues. As such, the observation that the binding of MTSET to A283C or A286C impaired the blocking action of TEA argues for a narrowing of the channel pore resulting either from a direct occlusion by MTSET of the pore lumen or from a displacement of the S6 segment substantial enough to decrease the pore cross section area. In the former case, the results obtained with TEA provide evidence for a structure of the S6 segment where the A283 and A286 residues are facing the pore lumen in accordance with the closed and open models proposed for IKCa. Our results also indicate that MTSET caused a greater protection against TEA block with the A283C than A286C mutant. This observation can be interpreted as indicating that the channel pore in the open state is wider at the level of the A286 as compared with the A283 residue. The calculations presented in Fig. 10 D do not, however, predict important variations in cross-section area at the level of the A283 and A286 residues. The absence of correlations between the solvent exposure calculations (Fig. 10) and the SCAM results, in particular the state-independent action of MTSET with the A283C and A286C mutants, strongly suggest in our view that the similarity-based models derived from the rigid closed form of KcsA do not provide a valid description of the COOH terminus end of the S6 segment (residues 283–286).
A better correlation between the SCAM results for the A283–A286 S6 domain and the closed KcsA structure can, however, be achieved if it is hypothesized that the COOH terminus end of S6 is highly flexible. If present, this flexibility could account for the observation that MTSET reached the pore lining 283C residue in conditions where the A283C mutant was maintained in a closed state conformation, and that the pore is wider at A286 as compared with A283. Furthermore, the cross section calculations presented in Fig. 10 predict small variations in pore dimensions between the open and closed states within the S6 region extending from K264 to T278. Therefore, the results of these calculations argue for strong structural constraints for the T264–K278 S6 segment not present within the A283–A286 domain. The data derived from homology modeling and SCAM experiments would thus support a model in which the domain extending from V275 to V282 possesses features corresponding to the inner cavity region of KcsA, and where the COOH terminus end of S6, from A283 to A286, is more flexible than predicted on the basis of the closed KcsA crystallographic structure alone. Under these conditions, the channel hypothetical gate cannot be located down the A283 residue, and should be at a point restricted to a region extending from V278 to V282.
The possibility of a highly flexible region near the channel gate was also recently proposed for the Shaker channel (del Camino and Yellen, 2001). It was argued in this case that closure of the gate was taking place between the residues at positions 474 (282 IKCa) and 478 (286 IKCa), with the region below 478 showing considerable flexibility. Furthermore, results of blocker protection experiments on the Shaker V476C mutant have led to the proposal of a “bent S6” model whereby a bent in the inner S6 transmembrane segment is introduced at the level of the P473-V474-P475 residues (del Camino et al., 2000). IKCa lacks a similar P-X-P motif that could disrupt the S6 helical structure, the P-V-P sequence in Shaker corresponding to the L281-V282-A283 sequence in IKCa (Fig. 1). Our results are nevertheless in agreement with the findings reported for the Shaker channel which assign to the P473-P475 region (L281-A283) a determinant role to the channel gating process. Finally, it is clear that in our case the stimulation by MTSET of the A283C, V285C, and A286C channels will require a model whereby A283, V285, and A286 participate actively in the coupling mechanism relating the Ca2+-dependent conformational change of calmodulin to the actual opening of the pore.
Conclusion
Globally, our results would support a model of the S6 transmembrane segment of IKCa where the region extending from V275 to V282 can be correctly approximated by the crystal structure of the KcsA channel. The structural features of the COOH-terminal region of S6 appeared, however, more compatible with a flexible structural model of KcsA, with A283 and A286 participating to the activation process.
APPENDIX
Let us consider a system consisting of Nt identical channels reacting with MTSET according to the following irreversible kinetic scheme:
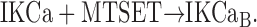 |
Let no stand for the number of unbound channels and PO, IO the associated open channel probability and unitary current amplitude. If the interaction of MTSET channel leads to a change in current amplitude (IB) and open probability PB, the current mean value <I> at any time during MTSET application will be given by:
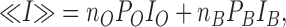 |
where nB is the number of channels bonded to MTSET given by Nt − nO. Similarly, the variance at any time will correspond to:
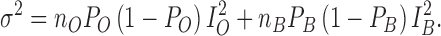 |
According to this model, the application of MTSET should lead to a time-dependent change in σ2/<I> from an initial value equal to Io (1 − PO) to a final steady-state value given by IB (1 − PB). Similarly, the σ2/<I>2 ratio is expected to vary from (1 − PO)/NtPO to (1 − PB)/NtPB in a time-dependent manner.
In conditions where the binding of MTSET causes a total inhibition of the channel unitary current amplitude, (IB = 0) the ratio σ2/<I> is predicted to be time independent with a value equal to
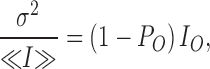 |
whereas the ratio σ2/<I>2 will be given by:
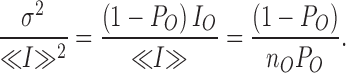 |
The ratio σ2/<I>2 will be independent of the unitary current amplitude and increase with a time constant equal to that measured for the current inhibition process.
In conditions where the binding of MTSET results in an increase of the channel open probability such that PB >> Po with Po << 1, the ratio σ2/<I> becomes approximately equal to:
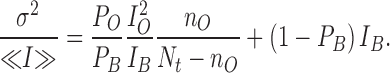 |
This equation predicts that the contribution of the first term will decrease with an apparent time constant faster than the observed relaxation time for current activation, with σ2/<I> reaching a constant value corresponding to (1 − PB) IB. Under these same conditions the ratio σ2/<I>2 will decrease from ∼1/PONt to (1 − PB)/PBNt, resulting in a (σ2/<I>2)before MTSET/(σ2/<I>2)after MTSET value greater than expected from the ratio PB /PO computed from the variations in mean current alone.
Finally, a current noise pattern where (σ2/<I>2 before MTSET/(σ2/<I>2)after MTSET ≈ PO/PB, with PO/PB computed from the variations in mean current may be indicative of a system in which both PO and PB << 1 or where the total number of functional channel varies from Ntinitial to Ntfinal with little variation in unitary current I and open probability PO. These systems cannot be discriminated on the basis of a noise analysis alone.
Acknowledgments
We thank Dr. Mats Eriksson for his valuable help with the homology modeling procedure, Ms. Julie Verner for expert oocyte preparation, and Mr. Claude Gauthier for drawings.
This work was performed with grants from the Canadian Institutes of Health Research (MOP 7769) and the Canadian Heart and Stroke Foundation to R. Sauvè and with a joint grant from the Fonds Concerté Action Recherche to R. Sauvè and L. Parent.
Note added in proof. The structure of the calcium-activated MthK K+ channel determined recently by X-ray crystallography shows that the intracellular entrance of the pore has a diameter of ∼12 Å, in good qualitative agreement with the current modeling of IKCa (Jiang et al., 2002a,b).
Footnotes
Abbreviations used in this paper: EDHF, endothelium-derived hyperpolarizing factor; EPR, electron paramagnetic spectroscopy; MTS, methanethiosulfonate, MTSES, sodium (2-sulfonatoethyl) methanethiosulfonate; MTSET, [2-(trimethylammonium)ethyl] methanethiosulfonate bromide; SCAM, scanning mutagenesis.
References
- Bernèche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K(+) channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, and M. Karplus. 1983. A program for macromolecular energy, minimization, and dynamics calculations. J. Com. Chem. 4:187–217. [Google Scholar]
- Brugnara, C., L. De Franceschi, and S.L. Alper. 1993. Ca2+-activated K+ transport in erythrocytes: comparison of binding and transport inhibition by scorpion toxins. J. Biol. Chem. 268:8760–8768. [PubMed] [Google Scholar]
- Cai, S., L. Garneau, and R. Sauvé. 1998. Single-channel characterization of the pharmacological properties of the K(Ca2+) channel of intermediate conductance in bovine aortic endothelial cells. J. Membr. Biol. 163:147–158. [DOI] [PubMed] [Google Scholar]
- Cui, Y., W. Wang, and Z. Fan. 2002. Cytoplasmic vestibule of the weak inward rectifier kir6.2 potassium channel. J. Biol. Chem. 277:10523–10530. [DOI] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage- gated K(+) channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- Denicourt, N., S. Cai, L. Garneau, M. Gagnan-Brunette, and R. Sauvé. 1996. Evidence from incorporation experiments for an anionic channel of small conductance at the apical membrane of the rabbit distal tubule. Biochim. Biophys. Acta. 1285:155–166. [DOI] [PubMed] [Google Scholar]
- Devor, D.C., A.K. Singh, R.A. Frizzell, and R.J. Bridges. 1996. Modulation of Cl− secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am. J. Physiol. 271:L775–L784. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dunn, P.M. 1998. The action of blocking agents applied to the inner face of Ca(2+)-activated K+ channels from human erythrocytes. J. Membr. Biol. 165:133–143. [DOI] [PubMed] [Google Scholar]
- Gerlach, A.C., C.A. Syme, L. Giltinan, J.P. Adelman, and D.C. Devors. 2001. ATP-dependent activation of the intermediate conductance, Ca2+-activated K+ channel, hIK1, is conferred by a C-terminal domain. J. Biol. Chem. 276:10963–10970. [PubMed] [Google Scholar]
- Ghanshani, S., H. Wulff, M.J. Miller, H. Rohm, A. Neben, G.A. Gutman, M.D. Cahalan, and K.G. Chandy. 2000. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 275:37137–37149. [DOI] [PubMed] [Google Scholar]
- Grissmer, S., A.N. Nguyen, and M.D. Cahalan. 1993. Calcium-activated potassium channels in resting and activated human T lymphocytes. Expression levels, calcium dependence, ion selectivity, and pharmacology. J. Gen. Physiol. 102:601–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., Y. Liu, Y. Xu, and G. Yellen. 1996. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 35:797–804. [DOI] [PubMed] [Google Scholar]
- Ishii, T.M., C. Silvia, B. Hirschberg, C.T. Bond, and J.P. Adelman. 1997. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA. 94:11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, B.S., N. Odum, N.K. Jorgensen, P. Christophersen, and S.P. Olesen. 1999. Inhibition of T cell proliferation by selective block of Ca(2+)-activated K(+) channels. Proc. Natl. Acad. Sci. USA. 96:10917–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Joiner, W.J., L.-Y. Wang, M.D. Tang, and L.K. Kaczmarek. 1997. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA. 94:11013–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna, R., M.C. Chang, W.J. Joiner, L.K. Kaczmarek, and L.C. Schlichter. 1999. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J. Biol. Chem. 274:14838–14849. [DOI] [PubMed] [Google Scholar]
- Klein, H., L. Garneau, M. Coady, G. Lemay, J.Y. Lapointe, and R. Sauvé. 1999. Molecular characterization of an inwardly rectifying K+ channel from HeLa cells. J. Membr. Biol. 167:43–52. [DOI] [PubMed] [Google Scholar]
- Kohler, M., B. Hirschberg, C.T. Bond, J.M. Kinzie, N.V. Marrion, J. Maylie, and J.P. Adelman. 1996. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 273:1709–1714. [DOI] [PubMed] [Google Scholar]
- Kuner, T., L.P. Wollmuth, A. Karlin, P.H. Seeburg, and B. Sakmann. 1996. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 17:343–352. [DOI] [PubMed] [Google Scholar]
- Langille, B.L., M.A. Reidy, and R.L. Kline. 1986. Injury and repair of endothelium at sites of flow disturbances near abdominal aortic coarctations in rabbits. Arteriosclerosis. 6:146–154. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Logsdon, N.J., J. Kang, J.A. Togo, E.P. Christian, and J. Aiyar. 1997. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 272:32723–32726. [DOI] [PubMed] [Google Scholar]
- Marchenko, S.M., and S.O. Sage. 1996. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 492:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morier, N., and R. Sauvé. 1994. Analysis of a novel double-barreled anion channel from rat liver rough endoplasmic reticulum. Biophys. J. 67:590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neylon, C.B., R.J. Lang, Y. Fu, A. Bobik, and P.H. Reinhart. 1999. Molecular cloning and characterization of the intermediate-conductance Ca(2+)-activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle cell function. Circ. Res. 85:e33–e43. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1996. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 70:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1997. Maximum likelihood estimation of aggregated Markov processes. Proc. R. Soc. Lond. B. Biol. Sci. 264:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane, S.G. 2000. The growth regulatory fibroblast IK channel is the prominent electrophysiological feature of rat prostatic cancer cells. Biochem. Biophys. Res. Commun. 269:457–463. [DOI] [PubMed] [Google Scholar]
- Rauer, H., M.D. Lanigan, M.W. Pennington, J. Aiyar, S. Ghanshani, M.D. Cahalan, R.S. Norton, and K.G. Chandy. 2000. Structure-guided transformation of charybdotoxin yields an analog that selectively targets Ca(2+)-activated over voltage-gated K(+) channels. J. Biol. Chem. 275:1201–1208. [DOI] [PubMed] [Google Scholar]
- Rittenhouse, A.R., D.H. Vandorpe, C. Brugnara, and S.L. Alper. 1997. The antifungal imidazole clotrimazole and its major in vivo metabolite are potent blockers of the calcium-activated channel in murine erythroleukemia cells. J. Membr. Biol. 157:177–191. [DOI] [PubMed] [Google Scholar]
- Roux, B., S. Bernèche, and W. Im. 2000. Ion channels, permeation, and electrostatics: insight into the function of KcsA. Biochemistry. 39:13295–13306. [DOI] [PubMed] [Google Scholar]
- Roux, B., and R. MacKinnon. 1999. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 285:100–102. [DOI] [PubMed] [Google Scholar]
- Sali, A., and T.L. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. [DOI] [PubMed] [Google Scholar]
- Sauvé, R., L. Parent, C. Simoneau, and G. Roy. 1988. External ATP triggers a biphasic activation process of a calcium-dependent K+ channel in cultured bovine aortic endothelial cells. Pflugers Arch. 41:469–481. [DOI] [PubMed] [Google Scholar]
- Sauvé, R., C. Simoneau, R. Monette, and G. Roy. 1986. Single-channel analysis of the potassium permeability in HeLa cancer cells: Evidence for a calcium-activated potassium channel of small unitary conductance. J. Membr. Biol. 92:269–282. [DOI] [PubMed] [Google Scholar]
- Schmid-Antomarchi, H., A. Schmid-Alliana, G. Romey, M.A. Ventura, V. Breittmayer, M.A. Millet, H. Husson, B. Moghrabi, M. Lazdunski, and B. Rossi. 1997. Extracellular ATP and UTP control the generation of reactive oxygen intermediates in human macrophages through the opening of a charybdotoxin-sensitive Ca2+-dependent K+ channel. J. Immunol. 159:6209–6215. [PubMed] [Google Scholar]
- Schreiber, M., and L. Salkoff. 1997. A novel calcium-sensing domain in the BK channel. Biophys. J. 73:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, M.A., A.F. Rivard, H.P. Bachinger, and J.P. Adelman. 2001. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 410:1120–1124. [DOI] [PubMed] [Google Scholar]
- Simoneau, C., D. Thuringer, S. Cai, L. Garneau, G. Blaise, and R. Sauvé. 1996. Effects of halothane and isoflurane on bradykinin-evoked Ca2+ influx in bovine aortic endothelial cells. Anesthesiology. 85:366–379. [DOI] [PubMed] [Google Scholar]
- Singh, S., C.A. Syme, A.K. Singh, D.C. Devor, R.J. Bridges, L.C. Lambert, A. DeLuca, and R.A. Frizzell. 2001. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J. Gen. Physiol. 296:600–611. [PubMed] [Google Scholar]
- Vandorpe, D.H., B.E. Shmukler, L. Jiang, B. Lim, J. Maylie, J.P. Adelman, M.D. Cappellini, C. Brugnara, and S.L. Alper. 1998. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J. Biol. Chem. 273:21542–21553. [DOI] [PubMed] [Google Scholar]
- Vergara, C., R. Latorre, N.V. Marrion, and J.P. Adelman. 1998. Calcium-activated potassium channel. Curr. Opin. Neurobiol. 8:321–329. [DOI] [PubMed] [Google Scholar]
- Warth, R., K. Hamm, M. Bleich, K. Kunzelmann, H.T. Von, R. Schreiber, E. Ullrich, M. Mengel, N. Trautmann, P. Kindle, A. Schwab, and R. Greger. 1999. Molecular and functional characterization of the small Ca(2+)-regulated K+ channel (rSK4) of colonic crypts. Pflugers Arch. 438:437–444. [DOI] [PubMed] [Google Scholar]
- Wulff, H., G.A. Gutman, M.D. Cahalan, and K.G. Chandy. 2001. Delineation of the clotrimazole/tram-34 binding site on the intermediate conductance calcium-activated potassium channel, ikca1. J. Biol. Chem. 276:32040–32045. [DOI] [PubMed] [Google Scholar]
- Xia, X.M., B. Fakler, A. Rivard, G. Wayman, T. Johnson-Pais, J.E. Keen, T. Ishii, B. Hirschberg, C.T. Bond, S. Lutsenko, et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 395:503–507. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y., K. Imaeda, and H. Suzuki. 1999. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 514:505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]