

Abstract
Antidiuretic hormone and/or cAMP increase Na+ transport in the rat renal collecting duct and similar epithelia, including Madin-Darby canine kidney (MDCK) cell monolayers grown in culture. This study was undertaken to determine if that increment in Na+ transport could be explained quantitatively by an increased density of ENaC Na+ channels in the apical membrane. MDCK cells with no endogenous ENaC expression were retrovirally transfected with rat α-, β-, and γENaC subunits, each of which were labeled with the FLAG epitope in their extracellular loop as described previously (Firsov, D., L. Schild, I. Gautschi, A.-M. Mérillat, E. Schneeberger, and B.C. Rossier. 1996. Proc. Natl. Acad. Sci. USA. 93:15370–15375). The density of ENaC subunits was quantified by specific binding of 125I-labeled anti-FLAG antibody (M2) to the apical membrane, which was found to be a saturable function of M2 concentration with half-maximal binding at 4–8 nM. Transepithelial Na+ transport was measured as the amiloride-sensitive short-circuit current (AS-I sc) across MDCK cells grown on permeable supports. Specific M2 binding was positively correlated with AS-I sc measured in the same experiments. Stimulation with cAMP (20 μM 8-p-chlorothio-cAMP plus 200 μM IBMX) significantly increased AS-I sc from 11.2 ± 1.3 to 18.1 ± 1.3 μA/cm2. M2 binding (at 1.7 nM M2) increased in direct proportion to AS-I sc from 0.62 ± 0.13 to 1.16 ± 0.18 fmol/cm2. Based on the concentration dependence of M2 binding, the quantity of Na+ channels per unit of AS-I sc was calculated to be the same in the presence and absence of cAMP, 0.23 ± 0.04 and 0.21 ±0.05 fmol/μA, respectively. These values would be consistent with a single channel conductance of ∼5 pS (typically reported for ENaC channels) only if the open probability is <0.02, i.e., less than one-tenth of the typical value. We interpret the proportional increases in binding and AS-I sc to indicate that the increased density of ENaC subunits in the apical membrane can account completely for the I sc increase produced by cAMP.
Keywords: retroviral transfection, FLAG epitope, channel number, membrane trafficking, short-circuit current
INTRODUCTION
Antidiuretic hormone (ADH),* in addition to its eponymous action, also stimulates Na+ reabsorption in the skin and urinary bladder epithelia of amphibians, in the rat cortical collecting duct (CCD), and in many in vitro and cultured epithelia possessing transport characteristics similar to the mammalian collecting duct (Tomita et al., 1985; Reif et al., 1986; Garty and Benos, 1988; Schafer and Hawk, 1992; Schafer, 1994; Garty and Palmer, 1997). As demonstrated previously in anuran epithelia, the increase in intracellular cAMP generated by the binding of ADH to V2 receptors results in an increase in the amiloride-sensitive conductance of the apical membrane of the principal cells of the collecting duct, allowing increased Na+ entry into the cell, which ultimately results in increased Na+ reabsorption (Schafer and Troutman, 1990).
The Na+ channels involved in this response to ADH and cAMP are now known to be oligomeric complexes of three protein subunits, α-, β-, and γENaC, encoded by discrete genes (Canessa et al., 1994). All three subunits share a similar topology, consisting of two transmembrane regions, a large cysteine-rich extracellular loop, and short COOH- and NH2-terminal regions in the cytoplasm. When the three cloned subunits are expressed together in Xenopus oocytes, they form channels with characteristics comparable to those found in the apical membrane of intact epithelia such as the CCD, including: a single channel conductance of ∼5 pS, a high Na+ to K+ selectivity ratio, and inhibition by submicromolar concentrations of amiloride (Canessa et al., 1994).
Interestingly, not all epithelia expressing ENaC respond to ADH or cAMP with increased Na+ transport. For example, ADH-dependent cAMP production in the rabbit CCD produces either no change or a decrease in Na+ transport (Schafer and Hawk, 1992; Schafer, 1994, 2002). Even in the same species, cAMP or ADH may have a different effect in different tissues, for example, stimulating Na+ transport in the rat CCD (Tomita et al., 1985; Reif et al., 1986), but not in the rat colon (Bridges et al., 1984). This variability in the response to cAMP or ADH has been attributed to differences in the structure of the αENaC subunit (Schnizler et al., 2001), or to differences in the actions of autacoids such as prostaglandin E2 that modify the response to ADH (Schafer, 2002).
The increase in Na+ reabsorption that occurs with ADH or cAMP in the rat CCD and other responsive epithelia represents an increase in the activity of ENaC hetero-oligomers in the apical membrane. Two general mechanisms have been put forward to explain this increased channel activity. First, modification of ENaC subunits, occurring as one of the ultimate events of ADH action, might cause an increase in the open probability (P o) or an increase in the unit conductance of the multimeric channel complexes already present in the apical membrane with no change in their numbers. Alternatively, or in addition to its effects on individual channel kinetics, ADH might cause the insertion of assembled ENaC multimers into the apical membrane in a manner analogous to the ADH-stimulated insertion of aquaporin-2 water channels in the CCD (Wade, 1985).
The former mechanism of ADH action, i.e., an increase in P o of individual Na+ channels, has been supported by studies using patch clamping and reconstituted Na+ channels. Changes in the single channel properties might be a consequence of protein kinase A (PKA) activation by cAMP, and Shimkets et al. (1998) have shown that the COOH-terminal regions of β- and γENaC are phosphorylated by this signaling cascade. Using inside-out patches of A6 cell apical membranes, Prat et al. (1993) showed that application of PKA plus ATP activated Na+ channels at least in part by an increase in P o. They also showed, however, that this effect of PKA was dependent on the presence of “short” actin filaments and hypothesized that the action of PKA might be mediated by phosphorylation of G-actin rather than the channel itself. A role for actin in activating ENaC was also supported by Berdiev et al. (1996), who demonstrated that PKA activation of α-, β-, and γENaC subunits incorporated into planar lipid bilayers required the presence of short actin filaments. PKA has also been shown to increase the P o of several biochemically purified Na+ channel complexes incorporated into planar bilayers (Oh et al., 1993; Bradford et al., 1995; Ismailov and Benos, 1995; Senyk et al., 1995).
Although variable in magnitude, the increases in P o that occur in these patch-clamp and bilayer experiments have been taken to support the view that direct activation of channels by cAMP/PKA can contribute to the increased amiloride-sensitive Na+ transport produced by ADH. This conclusion must, however, be tempered by the limitations of the experimental systems used. As indicated by the studies in lipid bilayers and detached membrane patches (Prat et al., 1993; Berdiev et al., 1996), the activity of ENaC channels depends on the presence of components of the cytoskeleton. In a detached patch or in a reconstituted system in which normal interactions of ENaC subunits with the cytoskeleton are disrupted, activation by PKA may involve changes in the interaction between residual or exogenous actin and ENaC that do not occur in the intact cell. It is also impossible to quantify the density of channels in the membrane of the native cells from the numbers measured in detached patches. When multiple channels are present in a patch, not only is it technically very difficult to determine their precise number, but the channels may also not be uniformly distributed, resulting in the “hot spots” of channel activity as described by Marunaka and Eaton (1991) in vasotocin-treated A6 cells.
Both patch-clamp and biochemical approaches have been used in support of the alternative hypothesis that ADH increases Na+ transport by the insertion of additional channels into the apical membrane. Marunaka and Eaton (1991) used patch clamping in A6 cell monolayers to show that both vasotocin and cAMP increased the number of Na+ channels per patch with no change in single channel kinetics. Kleyman et al. (1994) addressed the effect of ADH on the total pool of Na+ channels in A6 apical membranes using an antiidiotypic antibody (anti–anti-amiloride) that has been shown to recognize αENaC (Kieber-Emmons et al., 1995). In these experiments, apical membrane proteins in intact A6 cells, with and without ADH treatment, were radio iodinated and the Na+ channels were subsequently immunoprecipitated from extracted proteins using the antiidiotypic antibody. Kleyman et al. (1994) reported there was approximately a doubling of the immunoprecipitated Na+ channels after treatment with the hormone, which they concluded would account for the increase in Na+ transport produced by ADH. In another approach to quantifying Na+ channel density in the apical membrane, Snyder (2000) used transient transfection to introduce human ENaC subunits, which had been modified to allow fluorescence labeling, into cultured epithelial cells of thyroid origin. He demonstrated that cAMP stimulation produced an acute increase in the fluorescent labeling of ENaC in the apical membrane that paralleled the increase in Na+ transport.
The measurement of the surface density of ENaC subunits in the intact epithelium has definite advantages. Not only are the subunits in their native environment, but the use of epithelial monolayers with >106 cells inherently provides the statistical averaging that is lost when examining individual patches or reconstituted transporters. However, although the two surface-labeling studies discussed above undeniably show an increase in the density of ENaC subunits in the apical membrane of A6 cells with ADH or cAMP, both are limited in their ability to relate the change in the surface density of the label quantitatively to the change in Na+ transport. In the experiments of Kleyman et al. (1994), the identity and the number of antigenic sites per ENaC subunit, as well as the affinity of the antiidiotypic antibody for these sites, are unknown. Similarly, in Snyder (2000) fluorescence could not be related quantitatively to channel density, and the cells examined by confocal microscopy were not representative of the average labeling of the whole epithelium, because not all cells expressed ENaC subunits in this transient transfection system.
These limitations were overcome in the surface-labeling experiments described by Firsov et al. (1996). These investigators modified rat α-, β-, and γENaC by introducing the octapeptide FLAG epitope (DYKDDDDY) into the early (NH2-terminal) region of the extracellular loop of each. When these subunits were expressed in Xenopus oocytes, the binding of 125I-labeled monoclonal antibody to FLAG allowed these investigators to determine the surface density of the ENaC subunits on a femtomole per oocyte basis. The macroscopic amiloride-sensitive Na+ current and the surface density of ENaC subunits measured in the same oocyte were linearly related over a wide range with a slope of 1.1 μA/fmol. Although Firsov et al. (1996) did not examine the effect of cAMP,1 they found that a mutation in the β subunit associated with Liddle's syndrome resulted in an increase in the surface density of ENaC subunits and an increase in amiloride-sensitive current.
Our objective in the present studies was to use FLAG antibody surface labeling to quantify any change in the surface density of ENaC in the apical membrane of mammalian epithelial cells with cAMP treatment and to relate it to transepithelial Na+ transport measured in the same epithelia. We used retroviral transfection to develop a line of MDCK cells expressing “flagged” rat ENaC subunits, i.e., subunits labeled in the extracellular loop-domain with FLAG as described by Firsov et al. (1996). We measured the density of total ENaC subunits on the apical membrane surface and compared it with the amiloride-sensitive short-circuit current (I sc) measured in the same cells in the presence and absence of cAMP stimulation. Our results indicate that the increase in I sc produced by cAMP can be accounted for entirely by a proportional increase in the surface density of ENaC.
MATERIALS AND METHODS
MDCK Cell Cultures
MDCK cells were obtained as a gift from Dr. D. Rotin (University of Toronto, Toronto, Canada) with the permission of Dr. B. Rossier (University of Lausanne, Lausanne, Switzerland), whose laboratory originally developed the clone. This cell line, which exhibited most of the characteristics of the type-1 MDCK cells, including a transepithelial resistance (R T) >1,000 Ω·cm2, was chosen because it had been shown to have a negligible amiloride-sensitive conductance (Ishikawa et al., 1998). The unmodified wild-type cells are referred to below as parental MDCK cells. We also obtained a different clone of type-1 MDCK cells (a gift of Dr. D. Balkovetz, Nephrology Research and Training Center, University of Alabama at Birmingham), to serve as a source of protein for some of the immunoblots described below. These are referred to as type-1 MDCK cells.
Cultures of MDCK cells were expanded in T-75 flasks in a humidified incubator at 37°C in the presence of 4% CO2. The medium was Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 50 mM HEPES (pH 7.4), 1% Pen/Strep-fungizone, and 10% FBS. The medium was exchanged as needed, usually daily, and the cells were split upon confluence. For the both electrophysiological and surface labeling experiments, the cells were seeded on inserts with permeable membranes: either 24-mm cyclopores (Catalog No. 35-3090; Falcon) or 24-mm transwells (Catalog No. 3412; Costar) at a density of ∼400,000 cells/insert, after which they were grown in the modified DMEM without selection antibiotics (see below).
Other Tissues
In some of the immunoblotting and RT-PCR studies described below, we also used protein and total RNA extracted from fresh samples of dog and rat renal cortex and inner medulla. These tissues were obtained from cadarveric animals that had been used in other studies in our own laboratory or in other laboratories at this university. All such studies were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Retroviral Transfection of Parental MDCK Cells
Three plasmids containing rat α-, β-, and γENaC subunits, into each of which the FLAG epitope had been introduced in the early portion of the extracellular loop as described by Firsov et al. (1996), were provided by Dr. B. Rossier (University of Lausanne, Lausanne, Switzerland). These flagged subunits are designated hereafter by a subscript F, e.g., αFENaC. These subunit cDNAs were provided in mammalian plasmids (a modification of pSD65), and new restriction sites appropriate for subcloning into the retroviral vectors (5′ XhoI and 3′ NotI) were added to each by PCR. The resulting cDNA constructs were bidirectionally sequenced in order to verify that the ENaC subunits had not been modified, and they were then subcloned into bicistronic retroviral vectors, which were generously provided by Dr. R. Boucher (University of North Carolina, Chapel Hill, NC). The three vectors, LXPIN, LXPIH, and LXPIP, contained antibiotic resistance genes for, respectively, neomycin (G418), hygromycin, and puromycin, in one cloning position, with the αF-, βF-, or γFENaC subunit in the other. Sequencing from the 5′ and 3′ termini verified the orientation and nucleotide fidelity of the subcloned ENaC subunits. Using the protocol of Comstock et al. (1997), PA317 packaging cells (American Type Culture Collection) were transfected with the retroviral vectors to produce three retrovirions: LαF PIN, LβF PIH, and LγF PIP, each containing the indicated flagged ENaC subunit and the respective antibiotic resistance gene.
Parental MDCK cells were first infected with one of the three retrovirions, and then selected with the corresponding antibiotic, followed by transfection and selection cycles with the other two virions. Selection media included (in combinations appropriate to the history of infection): G418 (800 μg/ml), hygromycin (300 μg/ml), and puromycin (5 μg/ml) DMEM. At the end of three rounds of infection and selection, colonies of cells that had been infected with all three subunits and exhibited the corresponding antibiotic resistances were isolated and expanded for screening while maintaining selection pressure with all three antibiotics.
Clones of the triply transfected MDCK cells (referred to as αFβFγF MDCK cells) were subsequently grown on permeable supports. After 4–5 d in culture, expression of the transfected ENaC subunits was increased by overnight “induction” with 1 μM dexamethasone plus 2 mM Na+ butyrate in the culture medium as described by Stutts et al. (1995). Functional expression of the transfected ENaC subunits was monitored by the amiloride-sensitive transepithelial voltage (V T) using an EVOM (World Precision Instruments). The clone with the most robust transepithelial amiloride–sensitive voltage was selected. RT-PCR was used to verify the presence of flagged (but not wild-type) ENaC subunits in the transfected cells, and to confirm the absence of wild-type ENaC subunit cDNA in the parental cell line. The suitability of the primers used for this purpose was confirmed by using RNA extracted from fresh dog kidney as a positive control. Expression of all three subunits in the αFβFγF MDCK cells was also confirmed by immunoblotting as described below.
Electrophysiological Measurements
Short-circuit currents (I sc) were measured by mounting 12-mm diameter cell culture inserts (Millicell PCF; Millipore) in Ussing-type chambers (Jims Instruments) enclosed in a circulating water-jacket maintained at 37°C and equipped with bubble-lifts driven by the 95% O2/5% CO2 gassing. The half-chambers were connected via agar bridges to potential-sensing and current-passing Ag-AgCl electrodes. All electrodes were connected to a voltage clamp (model VCC600; Physiological Instruments). I sc was continuously recorded on a strip-chart recorder.
In addition to the traditional Ussing-type chambers, I sc was also measured in the 24-mm diameter cyclopore and transwell culture inserts that were used to conduct binding experiments. For these experiments, we built a special “multiinsert apparatus” that allowed rapid sequential recording of I sc and R T, as well as the open-circuit V T. The inserts were held in a manifold constructed from a Lucite sheet with 24 equally spaced openings. The lower portions of the inserts protruded below the manifold into the basolateral medium, which was contained in an inexpensive plastic container and was gassed with a mixture of 95% O2/5% CO2. This container was placed in a larger plastic container that served as a water bath whose temperature was maintained at 37°C by an immersion heater and temperature regulator. The basolateral current–receiving electrode was a stainless-steel sheet, whereas the basolateral voltage–sensing electrode, an Ag-AgCl2 electrode, was connected to the basolateral medium by an agar bridge. These basolateral electrodes served as a common electrode pair for all 24 inserts mounted in the Lucite manifold. The apical electrodes consisted of a small stainless-steel disc for current passing and a central agar bridge connected to an Ag-AgCl2 electrode. The apical electrode pair was encased in a housing made from a rubber stopper, which rested on the top rim of the cell culture inserts with the current-passing electrode and the agar bridge extending into the apical solution. Using this housing, the apical electrode pair could be moved rapidly from insert to insert for sequential electrophysiological measurements with the VCC600 voltage-clamp. A MacLab model 8e (ADInstruments) interface and a Macintosh computer were used to control the clamp and perform a rapid sequence of short-circuit, open-circuit, and current-passing recordings, and the data were stored on the computer.
Two solutions were used in the Ussing chambers, as well as in the multiinsert apparatus: unmodified DMEM or a “chloride-free” solution containing (in mM): 140 Na+ aminobenzenesulfonate, 24 Na+ bicarbonate 6 Ca2+ gluconate, 3 Mg2+ gluconate, 5 K+ gluconate, and 10 D-glucose.
Surface Labeling of Flagged ENaC Subunits
Anti-FLAG antibody (Sigma-Aldrich; referred to below as M2) was radiolabeled with 125I in the UAB radiolabeling core facility at the Comprehensive Cancer Center using Iodo-Beads (Pierce Chemical Co.) per the manufacturer's instructions. Labeled M2 antibody was initially purified over an anion-exchange column, eluted, and subsequently placed in a 10,000 MWC Slide-A-Lyzer dialysis cassette (Pierce Chemical Co.) and dialyzed for at least 24 h against PBS. The amount of labeled antibody was determined by a microprotein assay (# 23235; Pierce Chemical Co.) and was diluted to the desired specific activity by the addition of unlabeled M2 antibody. The specific activity was typically ∼1.8 μCi/μg antibody, but it was modified as needed to give sample count rates that ranged from at least 20 times background to >10,000 cpm, depending on the antibody concentration used.
Equilibrium binding assays were performed on monolayers of αFβFγF MDCK cells grown on either cyclopore or transwell 24-mm cell culture inserts. After experimental measurements and treatments, the inserts were placed in an ice bath and briefly rinsed with ice-cold PBS, and then incubated for 30 min with ice-cold blocking solution (PBS + 5% FBS). The apical blocking solution was aspirated and replaced with 500 μl of ice-cold blocking solution containing the desired concentration of 125I-labeled M2 antibody and incubated for 1 h. To determine the nonspecific binding, M2 antibody was added to paired inserts together with a 100-fold excess (by weight) of FLAG peptide (Research Genetics). After a 1-h incubation, excess label was aspirated and monolayers were washed four times with 1 ml of ice-cold blocking solution. Label remaining bound to the apical surface of the monolayers was then removed by “acid-stripping” as per Wiley and Cunningham (1982). Briefly, 750 μl of ice-cold acid wash solution (0.5 M NaCl, 0.2 M Na+ acetate, pH 2.4) was aliquoted onto the apical membrane of monolayers, incubated on ice for 5 min, and then harvested for gamma counting. Two acid strips were performed per monolayer to confirm that ≥80% of the bound label was removed. The counts from both acid strips were combined for data analysis. For each experimental point, triplicate inserts were labeled with 125I-labeled M2 antibody in the absence of competing peptide and a paired triplicate in its presence. Specifically, bound counts were taken to be the difference in average counts obtained in the presence and absence of peptide.
Western Blotting and Immunoprecipitation of ENaC Subunits
For each Western blot, protein was harvested from two confluent 24-mm monolayers of αFβFγF or parental MDCK cells. Monolayers were washed extensively with ice-cold PBS, removed from the filter inserts with cell scrapers, and briefly centrifuged. After aspirating the PBS, 600 μl of ice-cold lysis buffer (10 mM triethanolamine, 250 mM sucrose, pH 7.6, plus PMSF [1 mg/10 ml] and leupeptin [1 μg/ml]) was added to the pellet, and the cells were homogenized with a glass mortar and pestle. SDS was then added (1% final concentration) to the homogenate, which was mixed with a 21-ga needle and syringe. The sample was transferred to a 1.5-ml tube and sheered with a 30-ga needle 3–4 times before centrifuging 15 min at 14,000 g. Approximately 90% of the supernatant was then recovered and concentrated using a Microcon 30 (Millipore) as per the manufacturer's instructions. Protein determinations were made with a MicroBCA Kit (Pierce Chemical Co.). Laemmli buffer containing mercaptoethanol was added to 10-μg samples of protein, which were briefly boiled and run on 10% polyacrylamide gels. Protein was electrophoretically transferred to a nitrocellulose membrane (Nitrobind; Osmonics) and processed for immunoblotting.
Antibodies specific to three rat ENaC subunits (Masilamani et al., 1999) were provided by Dr. M. Knepper (National Institutes of Health, Bethesda, MD). The membrane blots were blocked with Blotto for 1 h at room temperature and incubated overnight at 4°C with primary ENaC antibodies at the appropriate dilutions (anti-αENaC L766 1:2,500, anti-βENaC L558 1:1,500, anti-γENaC L550 1:2,000). They were then washed three times with T-TBS (TBS/0.05% Tween-20), incubated for 1 h at room temperature with secondary antibody diluted 1:5000 in Blotto, and washed again with T-TBS. Blots were visualized with the ECL detection system (Amersham Pharmacia Biotech) per the manufacturer's instructions. Autoradiograms of immunoblots were digitized on an Epson transparency scanner (Expression Model 1600). Densitometric comparisons of bands on the same blot were made using the NIH Image program and calibrated gel analysis subroutine (version 1.62; National Institutes of Health).
To verify that the expressed ENaC subunits were indeed flagged, αF, βF, and γF subunits were immunoprecipitated from protein harvested from two confluent 24-mm diameter monolayers of induced αFβFγF MDCK cells using M2 antibody (5 μl) and a protein-G immunoprecipitation kit (1719386; Roche) according to the manufacturer's instructions. After immunoprecipitation, samples were boiled briefly in Laemmli buffer and processed for Western blotting as described above. In other experiments, the subunit-specific antibodies were used to immunoprecipitate the proteins, and the immunoblots were probed with the M2 antibody.
Statistics and Nonlinear Regression of Binding Saturation Plots
StatView for Macintosh (version 4.5.1, SAS Institute Inc.) was used for standard statistical calculations. Numerical results are presented as averages ±SEM. ANOVA with Bonferroni-Dunn post-hoc testing was used for comparisons of paired and nonpaired averages as appropriate, with significance ascribed at P < 0.05.
Kaleidagraph for Macintosh (version 3.5.1; Synergy Software) was used for nonlinear least squares fitting of specific binding data to the Michaelis-Menten relationship. Quality of fit is indicated by the correlation coefficient (r) and associated P value.
RESULTS
Expression of ENaC Subunit Proteins in MDCK Cells
In preliminary experiments, we first tested the antibodies to the rat α-, β-, and γENaC subunits in immunoblots with protein extracted from rat kidney cortex, and we obtained the same bands reported by Masilamani et al. (1999) in their original description of the antibodies. Because MDCK cells were derived from dog kidney, we also tested the same rat antibodies against protein isolated from dog kidney and from type-1 MDCK cells from which our parental strain was derived. Unfortunately, only the antibody for the rat βENaC was found to also recognize the dog subunit as shown in Fig. 1. Using the βENaC antibody, a single band at ∼101 kD was obtained in proteins isolated from dog kidney cortex and inner medulla, type-1 MDCK cells, and triply-transfected (αFβFγF) MDCK cells. However, there was no band in the parental MDCK cells used for transfection in these studies (first lane in Fig. 1). (All MDCK cells used for protein extraction had been induced overnight with butyrate and dexamethasone, and equal protein loading on each blot was independently verified among all MDCK cell lanes.) We also performed RT-PCR using degenerate, ENaC subunit–specific primer pairs that were shown to amplify ENaC subunits from reverse-transcribed total mRNA samples from dog kidney cortex. When cDNA that had been prepared in the same manner from parental MDCK cells (induced overnight with butyrate and dexamethasone) was amplified with the same primers, only a faint band of the expected size for αENaC was obtained even with 40 cycles of amplification, but there was no amplification of β- or γENaC (unpublished data). The apparent low (or absence of) expression of significant βENaC protein or mRNA for any of the three subunits is consistent with the very low or absent amiloride-sensitive Na+ transport activity observed previously in this MDCK cell line (Ishikawa et al., 1998), and confirmed in our electrophysiological studies below.
Figure 1.

Western blot demonstrating the expression of the βENaC subunit in MDCK cells and dog kidney tissues. The lanes contained protein extracts from: parental MDCK cells (the line subsequently used for transfection of flagged ENaC subunits), another clone of Type-I MDCK cells, αFβFγF MDCK cells (the triply transfected cell line developed in this study), dog kidney cortex, and inner medulla. All three lines of MDCK cells were induced overnight with 2 mM butyrate plus 1 μM dexamethasone, and 20 μg of extracted protein was present in each lane. In the case of the dog kidney tissues, 50 μg of protein was present for each. The numbers on the left side indicate the molecular weights (kD) of proteins in the molecular weight calibration ladder.
As shown in Fig. 2 A, the subunit-specific antibodies to rat ENaCs showed the presence of all three ENaC subunits in the retrovirally transfected αFβFγF MDCK cells with and without induction, and the approximate molecular weights were: α, 94 kD; β, 98 kD; γ, 89 kD. It should be noted that, whereas only a single band was obtained in all immunoblots for αENaC and γENaC, in some blots for βENaC we obtained two bands (ranges: 93–102 kD and 105–116 kD) as shown in Fig. 2 B. For nine separate protein samples from induced αFβFγF MDCK cells, the βENaC doublet was present in six. In six of these blots in which protein samples from uninduced and induced αFβFγF MDCK cells were paired, three showed the doublet and it was present in both induced and uninduced samples. The source of the higher molecular weight member of the doublet is unknown, and it was not observed in protein isolates from rat kidney by Masilamani et al. (1999). The 110-kD band was variable among different protein isolates, ranging from barely detectable in comparison with the 98-kD band to almost as dense (as in Fig. 2 B). The molecular weight difference between the bands of the doublet was ten times larger than weight of the FLAG epitope (∼1.1 kD). This comparison and the absence of any β band in the parental cells (Fig. 1) rules out the possibility that the doublet was due to the presence of both wild-type and flagged βENaC in the transfected cells.
Figure 2.
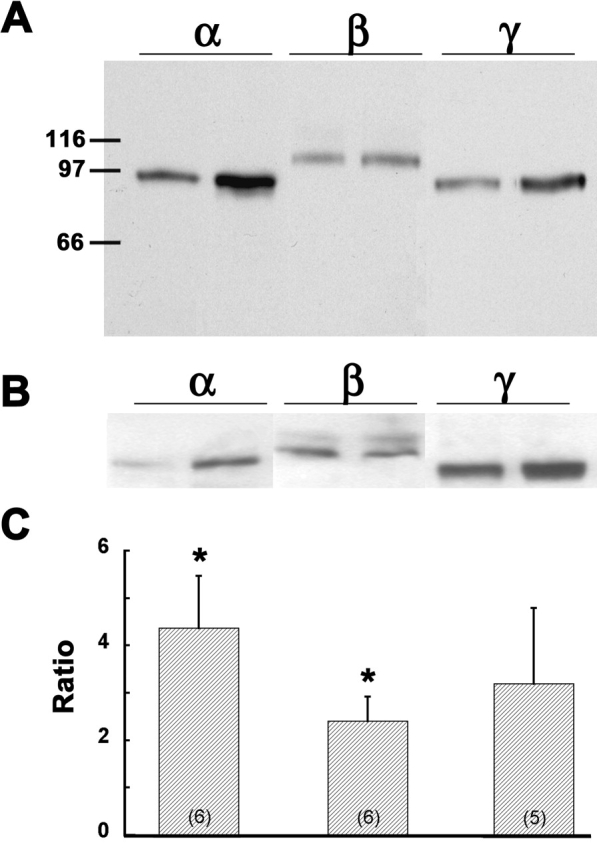
Expression of ENaC subunits in retrovirally transfected MDCK cells with and without induction. (A) Western blot of protein from triple-flagged (αFβFγF MDCK) cells. The lanes are grouped according to the subunit-specific antibody, and in each subunit group extracts are from αFβFγF MDCK cells without (first lane of pair) and with (second lane) overnight induction with 2 mM butyrate plus 1 μM dexamethasone. The numbers on the left side indicate the molecular weights (kD) of proteins in the molecular weight calibration ladder. (B) Another composite of blots prepared in the same way as panel A, but showing the doublet for βENaC (see text). (C) Ratios of the densitometry measurements for the α-, β-, and γENaC subunit bands in the presence of induction to that in the absence of induction. The number of separate protein extractions and immunoblots is indicated in parentheses in each bar. Asterisk indicates average ratio is significantly different from 1.0.
In the representative blot shown in Fig. 2 A, the levels of all three ENaC subunits appeared to be increased by overnight induction with butyrate and dexamethasone, whereas only αENaC appears to be significantly increased in Fig. 2 B. Densitometric ratios of paired induced and uninduced bands for each of the three ENaC subunits are presented in Fig. 2 C and show that there was significant induction only of αENaC (ratio of induced to uninduced 4.4 ± 1.1, P < 0.02) and βENaC (2.4 ± 0.5, P < 0.01), but not γENaC (3.2 ± 1.6, not significantly different from 1.0). However, as shown below, induced αFβFγF MDCK cells exhibited a much higher amiloride-sensitive I sc.
To verify that the ENaC subunits expressed in transfected MDCK cells contained the FLAG epitope, two series of immunoprecipitation experiments were performed. First, proteins were immunoprecipitated from αFβFγF MDCK cell lysates with the M2 antibody. The immunoprecipitated proteins were visualized on subsequent Western blots using the same subunit-specific antibodies as above. Bands of the appropriate molecular weights (Fig. 3 A) confirmed the presence of the FLAG in each of the ENaC subunits, but with the presence of a doublet for the β subunit. As further confirmation, a second series of immunoprecipitation experiments was performed in which each ENaC subunit was initially immunoprecipitated from αFβFγF MDCK cell lysates using the subunit-specific anti-ENaC antibodies. Immunoprecipitated proteins were processed for Western blotting and visualized with the M2 antibody shown in Fig. 3 B. Using this approach, the doublet for βF subunit was absent, although, as noted above, the presence of this band was variable. Together, the results (Figs. 2, A and B, and 3, A and B) demonstrate that all three flagged ENaC subunits are expressed in the αFβFγF MDCK cells.
Figure 3.

Immunoprecipitation of ENaC subunits from lysates of αFβFγF MDCK cells induced with butyrate and dexamethasone. (A) Monoclonal anti-FLAG (M2) antibody was used to immunoprecipitate ENaC subunits. Three aliquots of the immunoprecipitate were subsequently visualized in immunoblots using polyclonal, subunit-specific anti-ENaC antibodies as probes. (B) Three aliquots of cell lysates were separately immunoprecipitated with polyclonal, subunit-specific anti-α, -β, and -γ antibodies. M2 antibody was used as the probe in immunoblots.
Specific Binding of 125I-labeled M2 Antibody to αFβFγF MDCK cells
M2 antibody labeled with 125I was used to measure the apical membrane surface density of FLAG. In initial experiments we found appreciable binding of this antibody to the αFβFγF MDCK cells and culture inserts that persisted even in the presence of an excess of the unlabeled M2 antibody or the FLAG peptide. It was necessary to measure this nonspecific binding for each data point by adding a 100-fold excess (by weight) of FLAG peptide. (The ratio of specific to nonspecific binding, i.e., the signal-to-noise ratio, varied from 3:1 to 4:1, depending on the antibody concentration.) Thus, the specific binding of 125I-labeled M2 antibody was defined as the portion of the total binding displaced by the competing peptide.
Fig. 4 presents the results of seven experiments with αFβFγF MDCK cells in DMEM in which specific binding was measured as a function of the 125I-labeled M2 antibody concentration. The specific binding showed saturation with increasing antibody concentration as would be expected for a typical receptor ligand interaction; in this case, the binding of 125I-labeled M2 antibody to the FLAG present in the ectodomain of the ENaC subunits in the apical membrane. Because less than 1% of the M2 antibody added to the apical medium was bound to the membrane surface at equilibrium, we fit the data in Fig. 4 according to the Michaelis-Menten equation. Nonlinear least squares fitting gave estimates of k 0.5 and B max of 7.9 ± 1.9 nM and 7.4 ± 0.7 fmol/cm2, respectively.2 The k 0.5 thus obtained is not significantly different from the k 0.5 of ∼3 nM reported by Firsov et al. (1996) using the same FLAG-M2 antibody pairing in the Xenopus oocyte expression system.
Figure 4.

Specific binding of 125-I-labeled M2 antibody in αFβFγF MDCK cells as a function of antibody concentration. Cells were induced overnight with butyrate and dexamethasone, and the experiments were performed in DMEM. M2 antibody was added at seven different concentrations to the apical membrane only. The data were fit to the Michaelis-Menten relationship by nonlinear, weighted least squares (r = 0.95, P < 0.001). Estimates of maximal specific antibody binding in fmol/cm2 (B max) and affinity (k 0.5) were 7.4 ± 0.7 fmoles/cm2 and 7.9 ± 1.9 nM, respectively. Averages from seven experiments with 126 inserts.
We also performed three control-binding experiments on MDCK cells transfected with rat wild-type β- and γENaC subunits together with a rat αENaC subunit that was modified to contain an unrelated epitope (hemagglutinin, [HA]: YPYDVPDYA) in the same ectodomain region as the FLAG epitope in the construct of Firsov et al. (1996). Using a single 125I-labeled M2 antibody concentration (1.7 nM) revealed an insignificant amount of binding that could be displaced by the flag peptide (n = 3, 0.04 ± 0.01 fmol/cm2).
Finally, we conducted four experiments in which we measured the specific binding of 125I-labeled M2 antibody applied to the basolateral surface of the αFβFγF MDCK monolayers in the presence and absence of FLAG peptide. There was no significant specific binding, however, it would have been difficult to discern specific binding at the lower levels observed on the apical surface (i.e., <2 fmol/cm2) because of the high levels of nonspecific binding when the antibody was added to basolateral side. We attributed these higher nonspecific counts to antibody binding to the membrane support because we found similar levels of nonspecific binding to membrane inserts in the absence of cells. The absence of significant ENaC activity in the apical membrane was also supported by the absence of any change in V T or I sc when amiloride was added to the basolateral membrane (see below).
Electrophysiological Characteristics
Parental MDCK cells exhibited very little evidence of amiloride-sensitive Na+ transport. In three experiments with parental MDCK cells that were not induced, the average baseline open-circuit V T and I sc in DMEM were, respectively, −1.2 ± 0.3 mV and 0.8 ± 0.1 μA/cm2. In five experiments with parental cells that had been induced overnight with butyrate and dexamethasone, baseline V T and I sc were, respectively, −7.1 ± 2.1 mV and 3.7 ± 0.3 μA/cm2. After the latter cells were treated with 20 μM 8-CPT-cAMP plus 200 μM IBMX, I sc decreased by only 1.8 ± 0.3 μA/cm2 upon addition of 20 μM amiloride to the apical solution. In many other groups of parental cells, treated with both induction and cAMP, there was no change whatsoever in I sc upon the addition of amiloride to the apical solution. Thus, while there may be a small amiloride-sensitive I sc in some groups of parental cells after overnight induction, it was small in comparison with the transfected cells.
Table I documents the effects of 20 μM amiloride on the open-circuit V T and R T that develop across confluent monolayers of αFβFγF MDCK cells after overnight induction with both dexamethasone and butyrate. Addition of 20 μM amiloride to the apical medium significantly reduced V T from −27.7 ± 1.2 mV to −6.4 ± 0.3 mV (n = 95, P < 0.001), and significantly elevated R T from 2.2 ± 0.1 to 2.8 ± 0.1 kΩ·cm2 (n = 90, P < 0.001). We also examined the dose response of the transfected cells to amiloride. Based on the percent inhibition of I sc, the k i for amiloride in the absence and presence of cAMP treatment (20 μM 8-CPT-cAMP plus 200 μM IBMX) was, respectively, 0.48 ± 0.14 and 0.48 ± 0.18 μM. Addition of 10–100 μM amiloride to the basolateral membrane had no significant effect on either the basal V T, or on I sc in the presence or absence of cAMP treatment. These observations demonstrate the presence of functional amiloride-sensitive Na+ channels in the apical membranes of the αFβFγF MDCK cells.3
TABLE I.
Effect of Amiloride on Transepithelial Voltage and Resistance in αFβFγF MDCK Cells
| Treatmenta | V T b | R T c | Eq. I SC d |
|---|---|---|---|
| mV | kΩ·cm2 | μA/cm2 | |
| Basal | −27.7 ± 1.2 | 2.2 ± 0.1 | 12.5 |
| Amiloride | −6.4 ± 0.3e | 2.8 ± 0.1e | 2.2 |
After overnight induction with 2 mM butyrate and 1 µM dexamethasone, triple-flagged (αFβFγF) MDCK cells on either cyclopore or transwell membrane inserts were incubated in DMEM. Basal transepithelial voltage (V T, apical to basolateral) and resistance (R T) were measured during the first 20–30-min incubation with no additions to the medium. Amiloride (20 μM) was then added to the apical solution and the same parameters were measured when they reached a nadir 1–3 min after amiloride addition.
n = 95.
n = 90.
The “equivalent” short-circuit current (Eq. Isc) is calculated as V T/R T.
Difference between basal and amiloride values of V T and R T are significant (P < 0.001) by paired t test.
The effect of cAMP on transepithelial transport in αFβFγF MDCK cells was examined in experiments measuring I sc in Ussing chambers. Fig. 5 A compares the time course I sc of representative experiments with αFβFγF MDCK cells in DMEM and chloride-free media, and the average results of several experiments are presented in Fig. 5 B. In DMEM, basal I sc was significantly stimulated from 8.1 ± 1.0 to a peak value of 18.9 ± 2.2 μA/cm2 by the addition of 20 μM 8-CPT-cAMP plus 200 μM IBMX to the apical and basolateral media. There was rapid spike in I sc followed by a broader peak, which decayed over 30 min. Addition of 20 μM amiloride to the apical solution rapidly reduced I sc to ∼5 μA/cm2. The effect of amiloride was statistically significant and confirmed the presence of amiloride-sensitive, cAMP-stimulated Na+ transport in αFβFγF MDCK cells. As also shown in Fig. 5 A, in chloride-free medium the time course of 20 μM 8-CPT-cAMP plus 200 μM IBMX action was quite different. I sc rose from 7.4 ± 1.0 to 13.6 ± 1.2 μA/cm2 without the initial spike observed in DMEM. The cAMP-stimulated current also decayed more slowly in chloride-free medium. Again, addition of 20 μM amiloride to the apical side significantly reduced I sc to ∼3 μA/cm2.
Figure 5.

Effect of Cl− on the response of short-circuit current (I sc) to cAMP stimulation. (A) Time course of I sc response to cAMP treatment in DMEM and chloride-free medium. I sc was measured across monolayers of αFβFγF MDCK cells induced overnight with 2 mM butyrate and 1 μM dexamethasone. A mixture of 20 μM CPT-cAMP and 200 μM IBMX was added at the time indicated, followed by 20 μM amiloride. (B) Average I sc in experiments described in A. Six experiments were conducted in DMEM and 20 in chloride-free medium. Basal I sc was measured just before, and the cAMP I sc just after the addition of 20 μM CPT-cAMP plus 200 μM IBMX, at the point of maximal response, 5–10 min in DMEM and 10–20 min in chloride-free medium. The final measurement was made 1–3 min after the addition of 20 μM amiloride.
The biphasic response to cAMP in the presence of Cl− in the bathing solution has been described previously for MDCK and similar epithelia. It has been interpreted to reflect at least two components of I sc: active Cl− secretion and active Na+ transport via ENaC (Chalfant et al., 1993; Kleyman et al., 1994; Letz and Korbmacher, 1997; Morris et al., 1998), both of which are stimulated by cAMP. Under short-circuit conditions in these epithelia, cAMP produces a rapid initial increase in Cl− secretion followed by a slower activation of Na+ transport. Furthermore, as observed in Fig. 5 A, after cAMP stimulation in the presence of Cl−, I sc falls more rapidly than in its absence. Because our objective was to correlate any changes in the density of ENaC subunits in the apical membrane with the amiloride-sensitive I sc, we used a chloride-free medium in the remainder of these studies to reduce any variability that might be introduced by differences in the time course of the I sc response to cAMP.
Correlation between ENaC Surface Labeling and Isc
We used two different approaches to produce cell cultures that exhibited a wide range of ENaC expression and, thus, of surface binding. In both sets of experiments, the chloride-free medium was used. First, we prepared mixtures of varying proportions of parental (wild-type) to αFβFγF MDCK cells at the time of seeding onto cell culture inserts while keeping the total number of cells constant. For each experiment, a tray of six cell culture inserts was seeded for each mixture of cells. This arrangement typically resulted in a total of four trays, or 24 inserts for each experiment. After reaching confluence, the cells were induced with butyrate and dexamethasone and the following day the cell culture inserts were placed in the multiinsert apparatus for rapid, consecutive measurement of I sc in each of the 24 inserts. The amiloride-sensitive short-circuit current (AS-I sc) was calculated as the mean paired difference between I sc measured just before and that measured within 3 min after adding 20 μM amiloride to the apical solution. There was considerable variability in AS-I sc measured for a given cell mixture, which probably reflects a variable rate of growth of parental compared with αFβFγF MDCK cells among the different preparations of cells used to plate the inserts. Nevertheless, this approach provided inserts with the wide range of AS-I sc that was desired.
In the second protocol, we varied the expression of ENaC among the four trays used for each experiment by varying the induction method. In each experiment, we seeded αFβFγF MDCK cells onto cell culture inserts resulting in four trays, or 24 inserts, per experiment. The evening before the experiment, one tray was not induced, the second tray was induced with 2 mM butyrate only, the third with 1 μM dexamethasone only, and the fourth tray of cells was induced with the standard combination of dexamethasone and butyrate. As in the previous protocol, I sc was measured in the presence and absence of amiloride using the multi-insert apparatus. In nine experiments conducted in chloride-free medium, the average AS-I sc values in uninduced MDCK cells, and in cells induced with butyrate alone, dexamethasone alone, or both agents were, respectively, 2.7 ± 0.7, 8.1 ± 1.3, 11.8 ± 0.7, and 16.7 ± 1.6 μA/cm2 (each mean significantly different from the other three by ANOVA).
In each of the two sets of experiments described above, the specific binding 125I-labeled M2 antibody was measured at an antibody concentration of 16 nM. This intermediate concentration was chosen because the ratio of specific to nonspecific binding (i.e., the signal-to-noise ratio) decreased at higher antibody concentrations as specific binding sites became saturated. Specific binding was measured in the same inserts as I sc, and it was plotted against the AS-I sc as shown in Fig. 6. Linear regression demonstrated a significant correlation between specific binding and AS-I sc (n = 45, r = 0.82, P < 0.001). The slope of this relationship was 0.12 ± 0.03 fmol/μA, and the intercept (0.62 ± 0.33 fmol/cm2) was not significantly different from zero.
Figure 6.

Correlation between specific binding of 125I-labeled M2 antibody and amiloride-sensitive short-circuit current (I sc). The M2 antibody concentration in the binding experiments was 16 nM. Chloride-free medium was used for both the apical and basolateral solutions. Two methods were used to vary the range of I sc in the MDCK cultures: (a) the culture inserts were seeded with mixtures of αFβFγF and parental MDCK cells (square data points, with ratio of parental to αFβFγF MDCK cells indicated in the symbol key). (b) The αFβFγF MDCK cells either were given no induction or were induced with 2 mM butyrate and/or 1 μM dexamethasone, as indicated in the symbol key. The least-squares linear regression, indicated by the line, has a slope of 0.12 ± 0.03 fmol/μA (P < 0.001) and intercept of 0.62 ± 0.33 (NS). The correlation coefficient r is 0.82 (P < 0.001).
Effect of cAMP on Surface Labeling and Isc
Fig. 7 presents the cumulative results of paired experiments examining the specific binding of 125I-labeled M2 antibody to the apical surface of αFβFγF MDCK cells as a function of M2 antibody concentration, in the presence and absence of cAMP stimulation. After the standard overnight induction with dexamethasone and butyrate, the cell culture medium was replaced with chloride-free medium. After a 30-min equilibration period, 20 μM 8-CPT-cAMP plus 200 μM IBMX (hereafter referred to as cAMP treatment), or vehicle for control monolayers, was added to the apical and basolateral media. 30 min after the addition of the reagents, the trays of inserts were processed for the binding assay as described previously. For each of six experiments (432 total inserts), paired binding assays were performed at six different 125I-M2 antibody concentrations. In other words, for each experiment 72 total inserts were used in the following manner. Six trays, each containing six cell culture inserts, were used for the control group with one 125I-M2 antibody concentration (three inserts with and three without competing FLAG peptide) per tray. The experimental group was paired in the same manner; six trays each containing six cell culture inserts with a single 125I-M2 antibody concentration per tray. Specific counts were converted to fmol/cm2 of specifically bound (displaceable by FLAG peptide) M2 antibody and plotted as a function of the M2 antibody concentration (Fig. 7).
Figure 7.

Specific binding of 125I-labeled M2 antibody in αFβFγF MDCK cells as a function of antibody concentration in the presence and absence of cAMP treatment. In each of six experiments, 72 culture inserts were incubated in chloride-free medium for 30–40 min at 37°C. At this point, 20 μM CPT-cAMP plus 200 μM IBMX was added to the apical solution of one-half of the inserts (+cAMP) and only the vehicle to the other half (control). 30 min after cAMP treatment, the inserts where chilled in an ice bath and surface labeling was conducted as described in materials and methods. Specifically bound 125I-labeled M2 antibody is plotted as a function of the 125I-labeled M2 antibody concentration added to the apical membrane. The data were fit to the Michaelis-Menten relationship by nonlinear regression (control: r = 0.99, P < 0.001; +cAMP: r = 0.97, P < 0.001). Asterisk denotes specific binding was significantly higher in cAMP-treated than controls.
In every instance except one (a single pair of triplicate inserts out of 36 total pairs) specific binding was lower in control cells than in those treated with cAMP. As shown in Fig. 7, paired comparisons showed significantly higher binding in the cAMP-treated cells at five of the six M2 antibody concentrations examined. The data in Fig. 7 were fit with Michaelis-Menten curves as described previously, and k 0.5 and B max from the nonlinear fitting were: 4.8 ± 2.1 nM and 2.5 ± 0.5 fmol/cm2, respectively, for the control group, and 4.5 ± 1.6 nM and 3.4 ± 0.6 fmol/cm2, respectively, for the cAMP-stimulated group.4 The mean paired difference of the estimates of B max with versus without cAMP stimulation was statistically significant (n = 6, P < 0.001), with B max in cAMP-treated cells 40% higher than in control cells, whereas treatment with cAMP had no significant effect on k 0.5. These data indicate that cAMP treatment increased the apical membrane surface density of ENaC.
To correlate the cAMP-stimulated increase in the surface density of ENaC subunits with the increase in amiloride-sensitive I sc, additional experiments were conducted in which AS-I sc measurements in the absence and presence of cAMP stimulation were performed contemporaneously with binding studies using inserts seeded at the same time and grown under the same conditions. In this series of experiments, a single concentration of M2 antibody (1.7 nM) was used for the binding assay because of the greater sensitivity (larger ratio of specific to nonspecific counts) at antibody concentrations at or below k 0.5. In the seven experiments summarized in Table II, the amiloride-sensitive I sc increased from 11.2 ± 1.3 to 18.1 ± 1.3 μA/cm2 after cAMP treatment, and the specific binding of M2 antibody increased from 0.623 ± 0.126 to 1.159 ± 0.183 fmol/cm2 (n = 9) after addition of cAMP plus IBMX.
TABLE II.
Comparison of M2 Antibody Binding to Amiloride-sensitive Short-circuit Current in αFβFγF MDCK Cells with and without cAMP Treatment
| Bound M2 Aba | AS-I sc b | Est. B max | Est. B max/AS-I SC | |
|---|---|---|---|---|
| fmoles/cm2 | μA/cm2 | fmoles/cm2 | fmol/μA | |
| Control | 0.62 ± 0.13 | 11.2 ± 1.3 | 2.37 ± 0.48 | 0.21 ± 0.05 |
| +cAMP | 1.16 ± 0.18c | 18.1 ± 1.3c | 4.49 ± 0.70c | 0.23 ± 0.04 |
After overnight incubation with 2 mM butyrate and 1 μM dexamethasone triple-flagged (αFβFγF) MDCK cells grown on transwell membrane inserts were incubated in chloride-free medium in the absence (control) and presence of 20 μM 8-CPT-cAMP plus 200 μM IBMX (+cAMP). The amiloride-sensitive short-circuit current AS-I sc was measured in Ussing chambers. In contemporaneous experiments with inserts from the same platings, 125I-labeled M2 antibody was added to the apical medium at 1.7 nM, and the specific binding (second column) was measured as described in the text. For comparison with AS-I sc, B max was estimated (Est. B max) using the value of k 0.5 determined from the data in Fig. 7. The last column is the ratio of the estimated B max to AS-I sc.
n = 9.
n = 7.
Mean paired difference between control and +cAMP groups is significant with P < 0.001.
DISCUSSION
The data presented above relate to two main issues that are addressed below. First, the fundamental properties of the MDCK cell line retrovirally transfected with flagged ENaC subunits are discussed in the context of its applicability as a model epithelium for the study of ENaC regulation. Second, this model is used to examine the acute effects of cAMP on ENaC expression in the apical membrane of a high resistance mammalian epithelium and to correlate this expression with cAMP-stimulated increases in I sc.
ENaC Protein Expression and Amiloride-sensitive Na+ Transport in Transfected MDCK Cells
The parental line of MDCK cells that was used for retroviral transfection had no significant expression of any of the three ENaC subunits at the mRNA level and no expression of the endogenous (dog) βENaC protein (Fig. 1), whereas the triply transfected αFβFγF MDCK cells expressed all three exogenous rat ENaC proteins (Fig. 2). The immunoprecipitation experiments in Fig. 3 further demonstrate that the FLAG epitope was present in all three subunits. The αFβFγF MDCK cells also exhibited a significantly higher V T than observed in the parental cells, and that V T was inhibited >75% by amiloride (Table I) added to the apical but not to the basolateral membrane. These results demonstrated that the ENaC subunits expressed in the αFβFγF MDCK cells were assembled and trafficked preferentially to the apical membrane, and that they were mediating amiloride-sensitive Na+ transport.
The results of experiments in which I sc was measured in αFβFγF MDCK cells was also consistent with previous studies in MDCK cell lines that have high endogenous rates of amiloride-sensitive reabsorption as well as similar cultured lines, such as A6 cells (Bindels et al., 1988; Chalfant et al., 1993; Kleyman et al., 1994; Blazer-Yost and Helman, 1997; Morris et al., 1998). In particular, in DMEM the time course of the transport response to cAMP (Fig. 5 A) was the same as that observed in these previous studies. There was an initial rapid increase in I sc followed by a decline, which has been attributed to stimulation of Cl− secretion (under the voltage-clamp conditions) by cAMP, then a lesser secondary maximum, which has been attributed to stimulation of Na+ transport via ENaC and which subsequently decays (Chalfant et al., 1993; Letz and Korbmacher, 1997; Morris et al., 1998). The initial transient was absent in a chloride-free medium, and I sc peaked 10–15 min after cAMP treatment, followed by a slow decay, remaining elevated above baseline even 30 min after the addition. It is tempting to speculate that this more sustained current reflects the loss of an inhibitory influence of CFTR-mediated Cl− transport on Na+ transport (Schwiebert et al., 1995); however, Stutts et al. (1995) showed that the inhibitory effect of CFTR on ENaC channels in MDCK cells transfected with both transporters occurred even in a chloride-free medium. Regardless of the mechanism involved in the effect of Cl−, we used the chloride-free medium for most of the experiments in this study in order to avoid the complications presented by the more labile I sc in DMEM and to minimize the possible contribution of Cl− conductance changes on I sc.
Comparison of the Effects of cAMP on Isc and ENaC Subunit Density in the Apical Membrane
Labeling of the apical membrane of the αFβFγF MDCK cells with the M2 antibody was shown to be saturable with an affinity in the nanomolar range. The k 0.5 ranged from 3 to 8 nM (Figs. 4 and 7), which was comparable to the k 0.5 of ∼3 nM reported by Firsov et al. (1996) for M2 antibody binding to Xenopus oocytes transfected with the same flagged ENaC subunits. Most of the 125I-labeled M2 antibody binding could be displaced competitively either by FLAG or unlabeled antibody, but 20–30% of the counts remained even in the presence of a 100-fold excess (by weight) of FLAG. These residual counts were attributed to nonspecific binding to the cells and the culture inserts, and in all cases the specific binding was calculated as the paired difference between binding in the presence and absence of FLAG. Specific binding of 125I-labeled M2, defined in the manner just described, was demonstrated to be directly proportional to the amiloride-sensitive I sc in the αFβFγF MDCK cells (Fig. 6).
The effects of cAMP on the binding of M2 antibody to the apical membrane of αFβFγF MDCK cells were examined in two separate series of experiments in chloride-free medium. First, the results shown in Fig. 7 demonstrated that the specific binding of 125I-labeled M2 antibody was greater in cAMP-stimulated cells than in controls over a range of antibody concentrations with no change in the binding affinity. Furthermore, B max, obtained by nonlinear fitting to the Michaelis-Menten equation, was 40% higher than in controls (P < 0.001).
Second, in the experiments summarized by Table II, I sc measurements and binding assays were conducted in trays of cultured cells on inserts plated at the same time and treated in the same manner. The binding assays in this series of experiments were performed using a single concentration (1.7 nM) of 125I-labeled M2 antibody. The specific surface binding of radioligand was significantly higher by 86 ± 19% in αFβFγF MDCK cells treated with cAMP compared with untreated controls (Table II). Extrapolation of these binding values obtained with 1.7 nM M2 antibody gives B max values comparable to the B max values obtained from the previous saturation binding experiments (Table II). In the parallel electrophysiological experiments, cAMP stimulation also significantly increased the amiloride-sensitive I sc by 61 ± 10% (Table II). The difference between the fractional increases in cAMP-stimulated I sc and M2 antibody binding (−25 ± 21%) was not statistically significant. Put another way, the increases in the surface density of ENaC subunits in the apical membrane and the amiloride-sensitive I sc were proportional.
One might argue, however, that the increase in M2 Ab binding produced by cAMP treatment was due to some allosteric rearrangement of ENaC subunits, which were already present in the apical membrane, that made more FLAG binding sites accessible to the antibody. If this putative molecular rearrangement were accompanied by an increase in channel P o, our results would not necessarily be inconsistent with other findings that PKA, the intermediate mediator of cAMP effects, can increase P o of Na+ channels in planar lipid bilayers (Berdiev et al., 1996; Jovov et al., 1999) or detached membrane patches (Prat et al., 1993). However, there are no data in support of any molecular rearrangement. Furthermore, it seems unlikely that the number of sites newly exposed by cAMP action would be in direct proportion to the increase in AS-I sc, or that there would be no change in the apparent affinity of binding, as we have found in our studies. For these reasons, we feel that our data are more easily explained by an increase in the surface density of ENaC channels.
Quantitative Comparison of the ENaC Subunit Density with Single Channel Characteristics
Because there is one FLAG epitope per ENaC subunit, the M2 binding data permit an estimate of the molar density of ENaC subunits in the apical membrane assuming one bound antibody per subunit as shown by Firsov et al. (1996). Thus, the B max estimates from Fig. 7 and Table II give a range of 2.4–2.5 fmoles of ENaC subunits/cm2 of apical membrane in the control cells, and 3.4–4.5 fmol/cm2 in the cAMP-treated cells. Based on a cell density of ∼2.5·106 cells/cm2 in the confluent cultures, the subunit density would be on the order of 500–600 per cell under control conditions, and 800–1,100 per cell in the presence of cAMP.
The analysis presented in Table II shows that ∼0.22 fmoles of ENaC subunits are present per microampere of AS-I sc both in the presence and absence of cAMP. Thus, ∼130 subunits are present in the membrane for every picoampere of AS-I sc, This result can be compared with the number of subunits predicted from the single channel kinetic data obtained from patch clamp and fluctuation analyses. Ishikawa et al. (1998) obtained a single channel conductance of 4.7 pS when unmodified rat ENaC subunits were transfected into the same MDCK cell line used in our studies. This conductance agrees well with the range of 3.7–4.9 pS observed in a variety of epithelia possessing the highly selective Na+ channel (Garty and Palmer, 1997). Assuming an apical membrane voltage of −80 mV under short-circuit conditions, a 4.7 pS conductance would predict a single channel current of 0.38 pA. Using the average P o of 0.5 typically reported for the highly selective Na+ channel (Garty and Palmer, 1997), our estimate of 130 subunits/pA indicates that ∼25 subunits are present in the membrane for each channel.
This estimate of the subunit number per channel exceeds that expected based on estimates of the channel composition. The majority of studies indicate that the channel is a heterotetrameric assembly of ENaC subunits, but stoichiometries involving up to nine subunits per channel have been proposed (Firsov et al., 1998; Kosari et al., 1998; Snyder et al., 1998). Although our calculated number of subunits per channel may be compromised by the accuracy of the B max values and the assumptions made, Firsov et al. (1996) also found that the membrane density of ENaC subunits transfected into Xenopus oocytes exceeded that expected from the amiloride-sensitive current. There are two possible explanations for the apparent excess number of subunits. A fraction of the subunits in the membrane may be unassembled, i.e., not associated with a functional channel. Firsov et al. (1996) discounted this possibility as well as the presence of a pool of assembled but electrically silent channels in their oocyte expression studies based on the fact that the relationship between current and binding was linear and had a zero intercept. Although the Y-intercept of our correlation plot (Fig. 5) is not significantly different from zero, the error of this estimate cannot exclude the possibility that 20–30% of the specific binding was not associated with transporting channels.
An alternative explanation for the apparent excess of subunits is that the P o of the channels is <0.5. Firsov et al. (1996) proposed that the ENaC channels expressed in oocytes had a P o in the range of 0.004–0.014. In the present experiments, assuming a heterotetrameric stoichiometry, P o would have to be <0.02 for the subunit density to be consistent with the macroscopic currents measured. As argued by Firsov et al. (1996), the long open and closed times that characterize ENaC channels make the determination of the number of channels in a patch difficult and bias the P o determination to higher values. In their review of such studies, Garty and Palmer (1997) noted that the average P o of 0.5 is misleading because individual measurements exhibit a bimodal distribution with the majority of the measurements either >0.7 or <0.3.
An interesting sidelight to this issue is raised by our binding results in DMEM, in which the AS-I sc is calculated to be 10.3 μA/cm2 (Table I), whereas B max is 7.4 fmol/cm2. The ratio of binding to AS-I sc is thus ∼0.7 fmol/μA or >400 subunits/pA, which is threefold higher than calculated above for experiments in chloride-free medium. In other words, it appears that in DMEM the number of electrically silent subunits is greater or the P o lower than in the absence of chloride. However, these calculations should be regarded with caution for two reasons. First, the electrophysiological parameters were not measured in the same experiments as the binding for the experiments in DMEM. Second, we did not measure I sc but rather V T and R T in the electrophysiological experiments, and thus we could compute only an “equivalent I sc” (V T/R T) under open circuit conditions (Table I). These values would be expected to underestimate the true I sc, but certainly not by an amount sufficient to explain a threefold higher estimate of the subunits per μA. Thus, the possibility that the channel density per μA is greater in the presence of chloride is deserving of examination in further studies.
Conclusions
The results summarized above lead us to conclude that increases Na+ transport produced by cAMP are due to a proportional increase in the number of ENaC channels in the apical membrane, which is in agreement with the previous studies using less quantitative methods of determining the ENaC surface density as described in the introduction (Marunaka and Eaton, 1991; Kleyman et al., 1994; Snyder, 2000). Thus, our findings support the view that membrane trafficking is responsible for the regulation of ENaC activity that is mediated by cAMP.
Vesicular trafficking as a mechanism of ENaC regulation has received recent support from observations that all ENaC is present in cytoplasmic vesicles in subapical membrane region and that trafficking events are stimulated by cAMP. Using confocal and electron microscopy in immunohistochemical studies of the rat kidney, Hager et al. (2001) have shown that all three ENaC subunits are present in the apical membrane and cytoplasmic membrane vesicles of principal cells in the cortical collecting duct, and that cytoplasmic αENaC was found exclusively in the apical membrane region, where it colocalized with the aquaporin-2 water channel. Butterworth et al. (2001) used a membrane-specific fluorescent probe and confocal microscopy in A6 cells to show that elevation of intracellular cAMP increases the rate of endo- and exocytosis, with the increase exocytosis dominating.
Other studies have shown that the half-life of ENaC channels in the apical membrane is on the order of one to several hours (Weisz et al., 2000; Rotin et al., 2001), implying that ultimately ENaC activity must be maintained by trafficking. The pulse-chase experiments of Weisz et al. (2000) have also shown that aldosterone and ADH increase the membrane surface expression of βENaC in A6 cells, but not that of α- or γENaC, which contrasts with the apical localization of αENaC in the experiments of Hager et al. (2001). Our results, however, do not support the view that trafficking of a single subunit is responsible for cAMP regulation of ENaC because they show an increase in total (α, β, and γ) ENaC in the apical membrane. As proposed by Firsov et al. (1996), it seems more likely that a preassembled complex of all three ENaC subunits is trafficked to the apical membrane.
Acknowledgments
We thank Dr. L. Li and Ms. M.L. Watkins for their consistently superb technical assistance. The authors are particularly grateful to Drs. R. Boucher and J. Olsen for the training in retroviral transfection that R.G. Morris received in their laboratories with the help of Ms. N. Burch, and for providing the retroviral plasmids. Special thanks are also given to several members of the faculty at the University of Alabama at Birmingham: Drs. J.K. Bubien and Z.-H. Zhou conducted preliminary patch clamp experiments of ENaC expressed in our MDCK cells; Drs. C. Venglarik and J. Collawn shared specialized equipment in their laboratory as well as their expertise; Drs. D. Benos, J. Collawn, K. Kirk, and M. Quick were members of the dissertation committee for RGM and provided assistance with many aspects of the project.
This study was supported by National Institutes of Health grant DK-25519-21. This work was part of a dissertation submitted in partial fulfillment of the requirements for the Ph.D. degree awarded to R.G. Morris by the School of Graduate Studies at the University of Alabama at Birmingham.
Ryan G. Morris's present address is National Institutes of Health, National Heart, Lung, and Blood Institute, Laboratory of Kidney and Electrolyte Metabolism, Bethesda, MD 20892.
Portions of this work were previously published in abstract form (Morris, R.G., and J.A. Schafer. 2000. J. Am. Soc. Nephrol. 11:34A; and Morris, R.G., and J.A. Schafer. 2001. J. Am. Soc. Nephrol. 12:37A).
Footnotes
Abbreviations used in this paper: ADH, antidiuretic hormone; AS-I sc, amiloride-sensitive short-circuit current; B max, maximal specific binding; CCD, cortical collecting duct; CPT, 8-p-chlorophenylthio; DMEM, Dulbecco's modified Eagle's medium; ENaC, epithelial (amiloride-sensitive) Na+ channel; FLAG, octapeptide epitope DYKDDDDY; IBMX, isobutylmethylxanthine; I sc, short-circuit current; MDCK, Madin-Darby canine kidney; PKA, protein kinase A; R T, transepithelial resistance; V T, transepithelial voltage.
In the course of the present experiments, we also measured whole cell amiloride-sensitive currents in Xenopus oocytes expressing wild-type and FLAG-labeled rat ENaC subunits. Using a variety of cAMP analogs, we were unable to show any stimulatory effect, even at high concentrations in the presence of IBMX. Most likely the lack of effect is due to the high rate of endogenous cAMP production that we confirmed in these oocytes (unpublished data).
Plots of specific binding as a function of antibody concentration are presented and analyzed using the Michaelis-Menten equation as described in materials and methods. This approach is used in preference to a Scatchard plot (bound/free versus free antibody concentration) because the Michaelis-Menten plot shows the actual data obtained and its variation, and because the nonlinear regression of the data according to the Michaelis-Menten relationship avoids the bias introduced into least-squares fitting by the linearized Scatchard relationship. Nevertheless, the results of Scatchard analysis were quite comparable with those obtained from nonlinear regression using the Michaelis-Menten equation. In the case of Fig. 4, linear regression of the Scatchard plot gave k 0.5 and B max estimates of, respectively, 9.2 ± 2.2 nM and 8.0 ± 0.8 fmol/cm2 (r = 0.90, P < 0.001).
Drs. J.K. Bubien and Z.-H. Zhou in our department were kind enough to conduct preliminary patch clamp studies of the cells used in these studies using methods they have described previously (Bubien et al., 2001; Zhou and Bubien, 2001). In detached patches from αFβFγF MDCK cells, they observed one to three channels per patch with conductances in the range of 5–10 pS, NP o products of 0.5–1.5, and the long open and closed intervals characteristic of ENaC channels with or without the FLAG label (Firsov et al., 1996; Garty and Palmer, 1997). In one inside-out patch from an αFβFγF MDCK cell, the I-V relationship was linear in the range of ±80 mV with a slope conductance of 8.6 pS. In whole-cell patches of untreated αFβFγF MDCK cells, there was a basal level of ENaC activity that was markedly increased by cAMP. In whole-cell patch clamps of parental cells, there was no evidence of ENaC-like channel activity in the presence or absence of cAMP treatment (Bubien, J.K., and A.L. Birmingham, personal communication).
Scatchard analyses of these data gave k 0.5 estimates of: 4.6 ± 0.8 (control) and 3.5 ± 0.6 (+cAMP), and B max estimates of 1.9 ± 0.5 (control) and 2.9 ± 0.5 (+cAMP).
References
- Berdiev, B.K., A.G. Prat, H.F. Cantiello, D.A. Ausiello, C.M. Fuller, B. Jovov, D.J. Benos, and I.I. Ismailov. 1996. Regulation of epithelial sodium channels by short actin filaments. J. Biol. Chem. 271:17704–17710. [DOI] [PubMed] [Google Scholar]
- Bindels, R.J.M., J.A. Schafer, and M.C. Reif. 1988. Stimulation of sodium transport by aldosterone and arginine vasotocin in A6 cells. Biochim. Biophys. Acta. 972:320–330. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost, B.L., and S.I. Helman. 1997. The amiloride-sensitive epithelial Na+ channel: binding sites and channel densities. Am. J. Physiol. 272:C761–C769. [DOI] [PubMed] [Google Scholar]
- Bradford, A.L., I.I. Ismailov, J.M. Achard, D.G. Warnock, J.K. Bubien, and D.J. Benos. 1995. Immunopurification and functional reconstitution of a Na+ channel complex from rat lymphocytes. Am. J. Physiol. 269:C601–C611. [DOI] [PubMed] [Google Scholar]
- Bridges, R.J., W. Rummel, and P. Wollenberg. 1984. Effects of vasopressin on electrolyte transport across isolated colon from normal and dexamethasone-treated rats. J. Physiol. 355:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubien, J.K., B. Watson, M.A. Khan, A.L. Langloh, C.M. Fuller, B. Berdiev, A. Tousson, and D.J. Benos. 2001. Expression and regulation of normal and polymorphic epithelial sodium channel by human lymphocytes. J. Biol. Chem. 276:8557–8566. [DOI] [PubMed] [Google Scholar]
- Butterworth, M.B., S.I. Helman, and W.J. Els. 2001. cAMP-sensitive endocytic trafficking in A6 epithelia. Am. J. Physiol. 280:C752–C762. [DOI] [PubMed] [Google Scholar]
- Canessa, C.M., L. Shild, G. Buell, B. Thorens, I. Gautschi, J.-D. Horisberger, and B.C. Rossier. 1994. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 367:463–467. [DOI] [PubMed] [Google Scholar]
- Chalfant, M.L., B. Coupaye-Gerard, and T.R. Kleyman. 1993. Distinct regulation of Na+ reabsorption and Cl- secretion by arginine vasopressin in the amphibian cell line A6. Am. J. Physiol. 264:C1480–C1488. [DOI] [PubMed] [Google Scholar]
- Comstock, K.E., N.F. Watson, and J.C. Olsen. 1997. Design of retroviral expression vectors. Methods Mol. Biol. 62:207–222. [DOI] [PubMed] [Google Scholar]
- Firsov, D., I. Gautschi, A.M. Merillat, B.C. Rossier, and L. Schild. 1998. The heterotetrameric architecture of the epithelial sodium channel (ENaC). EMBO J. 17:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firsov, D., L. Schild, I. Gautschi, A.-M. Mérillat, E. Schneeberger, and B.C. Rossier. 1996. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. USA. 93:15370–15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garty, H., and D.J. Benos. 1988. Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiol. Rev. 68:309–373. [DOI] [PubMed] [Google Scholar]
- Garty, H., and L.G. Palmer. 1997. Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 77:359–396. [DOI] [PubMed] [Google Scholar]
- Hager, H., T.H. Kwon, A.K. Vinnikova, S. Masilamani, H.L. Brooks, J. Frokiaer, M.A. Knepper, and S. Nielsen. 2001. Immunocytochemical and immunoelectron microscopic localization of α-, β-, and γ-ENaC in rat kidney. Am. J. Physiol. 280:F1093–F1106. [DOI] [PubMed] [Google Scholar]
- Ishikawa, T., Y. Marunaka, and D. Rotin. 1998. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+. J. Gen. Physiol. 111:825–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismailov, I.I., and D.J. Benos. 1995. Effects of phosphorylation on ion channel function. Kidney Int. 48:1167–1179. [DOI] [PubMed] [Google Scholar]
- Jovov, B., A. Tousson, H.L. Ji, D. Keeton, V. Shlyonsky, P.J. Ripoll, C.M. Fuller, and D.J. Benos. 1999. Regulation of epithelial Na+ channels by actin in planar lipid bilayers and in the Xenopus oocyte expression system. J. Biol. Chem. 274:37845–37854. [DOI] [PubMed] [Google Scholar]
- Kieber-Emmons, T., C.M. Lin, K.V. Prammer, A. Villalobos, F. Kosari, and T.R. Kleyman. 1995. Defining topological similarities among ion transport proteins with anti-amiloride antibodies. Kidney Int. 48:956–964. [DOI] [PubMed] [Google Scholar]
- Kleyman, T.R., S.A. Ernst, and B. Coupaye-Gerard. 1994. Arginine vasopressin and forskolin regulate apical cell surface expression of epithelial Na+ channels in A6 cells. Am. J. Physiol. 26:F506–F511. [DOI] [PubMed] [Google Scholar]
- Kosari, F., S. Sheng, J. Li, D.O. Mak, J.K. Foskett, and T.R. Kleyman. 1998. Subunit stoichiometry of the epithelial sodium channel. J. Biol. Chem. 273:13469–13474. [DOI] [PubMed] [Google Scholar]
- Letz, B., and C. Korbmacher. 1997. cAMP stimulates CFTR-like Cl− channels and inhibits amiloride-sensitive Na+ channels in mouse CCD cells. Am. J. Physiol. 272:C657–C666. [DOI] [PubMed] [Google Scholar]
- Marunaka, Y., and D.C. Eaton. 1991. Effects of vasopressin and cAMP on single amiloride-blockable Na channels. Am. J. Physiol. 260:C1071–C1084. [DOI] [PubMed] [Google Scholar]
- Masilamani, S., G.-H. Kim, C. Mitchell, J.B. Wade, and M.A. Knepper. 1999. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J. Clin. Invest. 104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, R.G., A. Tousson, D.J. Benos, and J.A. Schafer. 1998. Microtubule disruption inhibits an AVT-stimulated chloride secretion but not sodium reabsorption in A6 cells. Am. J. Physiol. 274:F300–F314. [DOI] [PubMed] [Google Scholar]
- Oh, Y., P.R. Smith, A.L. Bradford, D. Keeton, and D.J. Benos. 1993. Regulation by phosphorylation of purified epithelial Na+ channels in planar lipid bilayers. Am. J. Physiol. 265:C85–C91. [DOI] [PubMed] [Google Scholar]
- Prat, A.G., D.A. Ausiello, and H.F. Cantiello. 1993. Vasopressin and protein kinase A activate G protein-sensitive epithelial Na+ channels. Am. J. Physiol. 265:C218–C223. [DOI] [PubMed] [Google Scholar]
- Reif, M.C., S.L. Troutman, and J.A. Schafer. 1986. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J. Clin. Invest. 77:1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotin, D., V. Kanelis, and L. Schild. 2001. Trafficking and cell surface stability of ENaC. Am. J. Physiol. 281:F391–F399. [DOI] [PubMed] [Google Scholar]
- Schafer, J.A. 1994. Salt and water homeostasis—is it just a matter of good bookkeeping? J. Am. Soc. Nephrol. 4:1933–1950. [DOI] [PubMed] [Google Scholar]
- Schafer, J.A. 2002. Abnormal regulation of ENaC: syndromes of salt retention and salt wasting by the collecting duct. Am. J. Physiol. Renal Physiol. In press. [DOI] [PubMed] [Google Scholar]
- Schafer, J.A., and C.T. Hawk. 1992. Regulation of Na+ channels in the cortical collecting duct by AVP and mineralocorticoids. Kidney Int. 41:255–268. [DOI] [PubMed] [Google Scholar]
- Schafer, J.A., and S.L. Troutman. 1990. cAMP mediates the increase in apical membrane Na+ conductance produced in the rat CCD by vasopressin. Am. J. Physiol. Renal Physiol. 259:F823–F831. [DOI] [PubMed] [Google Scholar]
- Schnizler, M., S. Schaffert, and W. Clauss. 2001. Regulation of cAMP-sensitive colonic epithelial Na+ channel in oocyte expression system. J. Comp. Physiol. [B]. 171:369–375. [DOI] [PubMed] [Google Scholar]
- Schwiebert, E.M., M.E. Egan, T.H. Hwang, S.B. Fulmer, S.S. Allen, G.R. Cutting, and W.B. Guggino. 1995. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 81:1063–1073. [DOI] [PubMed] [Google Scholar]
- Senyk, O., I. Ismailov, A.L. Bradford, R.R. Baker, S. Matalon, and D.J. Benos. 1995. Reconstitution of immunopurified alveolar type II cell Na+ channel protein into planar lipid bilayers. Am. J. Physiol. 268:C1148–C1156. [DOI] [PubMed] [Google Scholar]
- Shimkets, R.A., R. Lifton, and C.M. Canessa. 1998. In vivo phosphorylation of the epithelial sodium channel. Proc. Natl. Acad. Sci. USA. 95:3301–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, P.M. 2000. Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na+ channel to the cell surface. J. Clin. Invest. 105:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, P.M., C. Cheng, L.S. Prince, J.C. Rogers, and M.J. Welsh. 1998. Electrophysiological and biochemical evidence that DEG/ENaC cation channels are composed of nine subunits. J. Biol. Chem. 273:681–684. [DOI] [PubMed] [Google Scholar]
- Stutts, M.J., C.M. Canessa, J.C. Olsen, M. Hamrick, J.A. Cohn, B.C. Rossier, and R.C. Boucher. 1995. CFTR as a cAMP-dependent regulator of sodium channels. Science. 269:847–850. [DOI] [PubMed] [Google Scholar]
- Tomita, K., J.J. Pisano, and M.A. Knepper. 1985. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J. Clin. Invest. 76:132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, J.B. 1985. Membrane structural studies of the action of vasopressin. Fed. Proc. 44:2687–2692. [PubMed] [Google Scholar]
- Weisz, O.A., J.M. Wang, R.S. Edinger, and J.P. Johnson. 2000. Non-coordinate regulation of endogenous epithelial sodium channel (ENaC) subunit expression at the apical membrane of A6 cells in response to various transporting conditions. J. Biol. Chem. 275:39886–39893. [DOI] [PubMed] [Google Scholar]
- Wiley, H.S., and D.D. Cunningham. 1982. The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 257:4222–4229. [PubMed] [Google Scholar]
- Zhou, Z.-H., and J.K. Bubien. 2001. Nongenomic regulation of ENaC by aldosterone. Am. J. Physiol. C1118–C1130. [DOI] [PubMed] [Google Scholar]