

Despite large changes in water and salt intake, the kidney is able to maintain the extracellular osmolarity and volume within narrow margins (Verrey et al., 2000). Such fine control requires specific factors or hormones; among them, aldosterone and antidiuretic hormone (ADH) (vasopressin in mammals) play the key role. The main effector for the sodium balance is the epithelial sodium channel expressed in the apical membrane of the principal cell located in the distal nephron (distal convoluted tubule and collecting duct). The effect of aldosterone on sodium transport has been highly conserved throughout the evolution of vertebrates, whereas that of vasopressin varies from species to species. In the urinary bladder of the toad (Girardet et al., 1986) and in the cortical collecting duct (CCD) of the rat (Reif et al., 1986), ADH increases sodium transport through a cAMP-dependent mechanism, via rapid (within minutes) nongenomic activation of ENaC and, later (within hours), by genomic activation (or repression) of a number of genes which have been identified recently (Robert-Nicoud et al., 2001). The natriferic effects of cAMP and aldosterone are synergistic (Girardet et al., 1986).
The molecular mechanism by which cAMP modulates ENaC activity to rapidly increase net sodium reabsorption remains a matter of controversies. Setting aside an increase in the unit conductance of ENaC, which has never been observed, one possible mechanism is that cAMP mediates an increase in open probability (Po) with no change in the number of molecules expressed in the apical membrane of the principal cell. An alternative possibility is that the hormonal effect is due to an increased insertion of new channels at the cell membrane and/or a decreased retrieval from the surface, leading to an increased number (N) of channels at the plasma membrane. The precise quantification of ENaC protein (N) present in the membrane and the measurement of its Po have been difficult for two main reasons: first, the overall density of ENaC molecules in the apical membrane of principal cells is very low, maybe <30–50 channels per cell. Under a normal salt diet and water repletion, ENaC protein is undetectable in the apical membrane of the CCD principal cell by classical immunohistochemical technique (Masilamani et al., 1999; Loffing et al., 2000) or by patch clamp (Pacha et al., 1993). This does not mean, however, that active ENaCs are not present in the apical membrane, but this small pool may be just undetectable with the available methodology. The immunohistochemical and physiological localization of ENaC at the apical membrane is seen only when the animal is put under low salt diet for a couple of days or weeks. Long term salt restriction (Pacha et al., 1993; Masilamani et al., 1999; Loffing et al., 2000) or short-term (15 h) Na deprivation (Frindt et al., 2001) lead to a relative hypovolemia, providing the physiological stimulus for aldosterone secretion by the adrenals.
Second, ENaC displays some unique gating properties, as assessed by patch clamp experiments. Hormone-induced changes in Po could theoretically be detected by patch clamp as for other ionic channels. But, as discussed by Palmer and Garty (2000), the open probability (Po) is highly variable; even two channels in the same patch may have widely different Po values. ENaC may exist in different gating modes. Therefore, ENaC could switch from one mode to another under the influence of a variety of factors or hormones. In addition, changes in opening and closing rates could account for the great variability of the observed Po (Palmer and Garty, 2000). Firsov et al. (1996) reported a quantitative and sensitive method to assess ENaC activity in the membrane of the Xenopus oocyte expressing heterologous ENaC channels (Firsov et al., 1996, 1997). To measure N, a binding assay was developed to quantitate the total number of channel molecules at the cell surface independently of their function. This was achieved by inserting a flag epitope in the ectodomain of each ENaC subunit, a domain of the molecule which is “physiologically” inactive, so that the biophysical and biochemical properties of ENaC (single channel conductance, Po, biosynthesis, membrane trafficking, and retrieval) were not affected. Using radio-iodinated monoclonal antibodies, a one to one stoechiometry of antibody binding to the channel subunit was demonstrated, which made it possible to determine precisely the number of molecules, silent or active, expressed at the cell surface. The binding assay was performed at 0°C, so that internalization was prevented. Then, the oocyte was rapidly washed and the temperature raised at 20°C, to measure amiloride-sensitive sodium transport mediated by ENaC (INa). According to the equation:
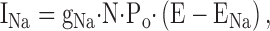 |
and setting E − ENa constant by clamping the membrane potential at −100 mV, it was possible to measure INa and N. Measuring gNa in independent patch clamp experiments and making some assumption on the stoichiometry of channel subunits, it was possible to deduce Po. Unlike the measurement of Po in the patch clamp experiment, which measures a restricted number of channels on a small membrane area for a limited duration, and thus may overlook channels with long closed state, this method averages the Po of all channels (with different gating modes and/or distinct opening and closing rates) expressed by the membrane of the whole oocyte. Therefore, Firsov et al. (1996) proposed that it would be prudent to term this average value the whole cell Po (wcPo), so as to distinguish it from the classical measurement of Po. This technique allowed Firsov et al. (1996) to conclude that in the oocyte membrane, channels were gating with a low wcPo in the presence of low intracellular sodium (lwcPo < 0.02) and even lower in the presence of a high intracellular sodium (wcPo < 0.004). The linearity of the relationship (with an X/Y axis intercept not different from 0) between INa and the number of channel subunits present at the cell surface of the same oocyte suggests that the increasing level of INa in individual oocytes was not due to the recruitment of nonconducting channels from a pool of inactive channels preexisting in the plasma membrane, but rather to the insertion of new channel molecules at the plasma membrane. Morris and Schafer (2002)(in this issue) have now used this technique to estimate the effect of cAMP on ENaC in an epithelial cell line (MDCK). In a real technical “tour de force,” they were able to obtain stable transfected cell lines expressing the three flagged subunits at the apical membrane, thus establishing an amiloride-sensitive transepithelial sodium transport (AS-I SC), which is absent in the untransfected MDCK cell line. This allowed the authors to perform a precise quantification of the number of channel subunits expressed at the apical membrane of MDCK cells and to relate that number to the amiloride-sensitive transepithelial sodium transport measured by the short-circuit current method. They found that cAMP was able to increase the amiloride-sensitive AS-I SC by about twofold in direct proportion with the increase in number of channels at the apical membrane, indicating that cAMP increases sodium transport, mainly by increasing the number of channels at the cell surface. Assuming that the hormone does not change the single channel conductance (typically 5 pS, in the presence of sodium), the authors conclude that the whole cell Po is in the range of 0.02 (if the channel is an heterotetramer), a value much lower than the typical value found in patch clamp experiments. This value in transfected epithelial cells agrees, however, with the value obtained in ENaC heterologously expressed oocytes (Firsov et al., 1996, 1997), showing that a number of silent channels or channels with very low Po is significant in the two cell systems. In an elegant series of experiments (Fig. 6), Morris and Schafer (2002) were able to vary the range of AS-I SC in the MDCK culture, using different mixtures of transfected or parental MDCK cells or inducing the transfected cells by butyrate and/or dexamethasone. Despite the difficulty associated with quantifying such a low number of channels in the apical membrane they observed, a linear relationship between AS-I SC and ENaC specific binding. At zero transepithelial current, a small residual binding was observed, suggesting that a small pool of truly silent channels could be present in the apical membrane of the transfected cells, but this residual binding was not statistically significantly different from 0. The results obtained in the oocyte and the epithelial cell line are therefore in good agreement.
If cAMP (and presumably vasopressin) controls N rather than Po, what about aldosterone? Recent findings indicate that aldosterone has also a major effect on N. The aldosterone-dependent signaling cascade just begins to be understood. Upon binding to the mineralocorticoid receptor, the ligand–receptor complex is translocated into the nucleus where it binds to the promoter of a large number of genes to control their transcription. The serum- and glucocorticoid-regulated kinase (Sgk1) is one of the early induced genes which is upregulated within 30 min of aldosterone addition (Chen et al., 1999; Naray-Fejes-Toth et al., 1999), promoting apical localization of ENaC in the distal nephron (Loffing et al., 2001). In the oocyte expression system, Sgk1 increases cell surface expression of ENaC (Alvarez de la Rosa et al., 1999) and phosphorylates Nedd4-2 (Debonneville et al., 2001; Snyder et al., 2002). The ubiquitin ligase Nedd4-2 normally binds tightly to ENaC by a well defined protein–protein interaction. Unphosphorylated Nedd4-2 is shown to have a higher affinity for ENaC, leading to its ubiquitination and rapid retrieval from the surface. Upon Sgk1-dependent phosphorylation of Nedd4-2, the rate of retrieval is slowed down, leading to an increased expression of ENaC at the cell surface. The effects of aldosterone or low salt diet on the apical localization of ENaC in vivo (Loffing et al., 2001) are consistent with the in vitro findings. In a recent issue of The Journal of General Physiology, Alvarez de la Rosa et al. (2002) reported that aldosterone induced a fourfold increase in the abundance of the three subunits in the apical membrane of an amphibian kidney cell line (A6). This effect could not be explained by changes in Po, kinetics of single channels, or in the rate of degradation of ENaC subunit, at variance with the prediction made from the effect of Sgk1 described above. The observed effect could be explained by insertion of channels from an endocytic pool or from de novo synthesis. Altogether, the data obtained recently would be consistent with previous findings on the measurement of N·Po in the rat kidney (Pacha et al., 1993) and in A6 cells using a modified method of blocker-induced noise analysis (Helman et al., 1998).
So, should we conclude that vasopressin and/or aldosterone have only one mode of action by increasing N by two distinct (cAMP and Sgk1, respectively) but synergistic pathways? This seems unlikely to me, since a dual control of N and Po would dramatically increase the dynamic range of ENaC regulation by each of the two hormones. A number of cytoplasmic factors (Na, pH, Ca, PKA, actin filament) or an external factor (self inhibition by Na) have been shown to affect Po in patch clamp or in bilayer experiments (for review see Palmer and Garty, 2000). The relevance of these effects on the regulation of ENaC in intact native cells remains to be established. Thus, several important questions remain to be addressed, but their resolution will require the development of new tools and methods. One of the most difficult challenges will be to measure the absolute number of channel molecules in the apical membrane of the native principal cell. It will also be necessary to understand and dissect the exocytic/endocytic pathways for ENaC, both in vitro and in vivo. Thanks to the Liddle mutation in the COOH terminus of β or γ subunit of ENaC, we know much more about the mechanism of membrane retrieval (Rotin et al., 2000) than that of insertion. Recent findings indicate that ENaC must be trafficked to the apical membrane by an unusual route, since the cell surface pool is core-glycosylated but remains fully EndoH-sensitive in transfected cells (Hanwell et al., 2002) or partially EndoH-sensitive in A6 cells (Alvarez de la Rosa et al., 2002). Our understanding of the mechanism of gating is also in its infancy. A loss of function mutation in the NH2 terminus of the β subunit (Chang et al., 1996) helped to define the sequence involved in gating, but the mechanism(s) are not yet understood (Grunder et al., 1997). The identification of the degenerin site in the ectodomain in C. elegans (Tavernarakis and Driscoll, 1997) led to the identification of a corresponding site in ENaC, which regulates Po (Snyder et al., 1999). Finally, it has been shown that channel-activating proteases (CAP) are important membrane-bound serine proteases, able to activate ENaC in the extracellular compartment (Vallet et al., 1997, 2002). CAP do not modify the cell surface–expressed ENaC and thus modulates wcPo, but the molecular mechanism of this effect is not understood and its physiological relevance in vivo not yet investigated in detail. Finally, one would like to know more about the function of the postulated small pool of ENaC molecules which may operate under a normal salt diet and respond to physiological stimuli for aldosterone secretion, for instance, during the circadian variation of volemia. Under these physiological stimuli where plasma aldosterone vary within low values (0.01–0.5 nM), ENaC activity was shown to be strictly regulated both in the kidney (Wang et al., 2000; Frindt et al., 2001) and in the colon (Wang et al., 2000). The sensitivity of the tools presently available, both at the protein and electrophysiological levels, is inadequate to tackle this aspect of ENaC function. I anticipate that both N and Po will be found to be under hormonal control. But these questions are nice challenges for the future.
Acknowledgments
We thank Laurent Schild and Jean-Daniel Horisberger for critically reading the manuscript.
This work was supported by a grant from the Swiss National Foundation (#31-061966.00) to B. Rossier.
References
- Alvarez de la Rosa, D.A., P. Zhang, A. Naray-Fejes-Toth, G. Fejes-Toth, and C.M. Canessa. 1999. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J. Biol. Chem. 274:37834–37839. [DOI] [PubMed] [Google Scholar]
- Alvarez de la Rosa, D., H. Li, and C.M. Canessa. 2002. Effects of aldosterone on biosynthesis, traffic, and functional expression of epithelilal sodium channels in A6 cells. J. Gen. Physiol. 119:427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, S.S., S. Grunder, A. Hanukoglu, A. Rosler, P.M. Mathew, I. Hanukoglu, L. Schild, Y. Lu, R.A. Shimkets, C. Nelson-Williams, et al. 1996. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkaliemic acidosis, pseudohypoaldosteronism type 1. Nat. Genet. 12:248–253. [DOI] [PubMed] [Google Scholar]
- Chen, S.Y., A. Bhargava, L. Mastroberardino, O.C. Meijer, J. Wang, P. Buse, G.L. Firestone, F. Verrey, and D. Pearce. 1999. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. USA. 96:2514–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debonneville, C., S.Y. Flores, E. Kamynina, P.J. Plant, C. Tauxe, M.A. Thomas, C. Münster, A. Chraïbi, J.H. Pratt, J.-D. Horisberger, et al. 2001. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 20:7052–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firsov, D., S. Gruender, L. Schild, and B.C. Rossier. 1997. Quantitation of cell surface expressed epithelial sodium channel: a novel approach to determine the channel open probability in a whole cell. Nova Acta Leopold NF75. 302:13–22. [Google Scholar]
- Firsov, D., L. Schild, I. Gautschi, A.M. Merillat, E. Schneeberger, and B.C. Rossier. 1996. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. USA. 93:15370–15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt, G., S. Masilamani, M.A. Knepper, and L.G. Palmer. 2001. Activation of epithelial Na channels during short-term Na deprivation. Am. J. Physiol. 280:F112–F118. [DOI] [PubMed] [Google Scholar]
- Girardet, M., K. Geering, H.-P. Gaeggeler, and B.C. Rossier. 1986. Control of transepithelial Na transport and Na-K-ATPase by oxytocin and aldosterone. Am. J. Physiol. 251:F662–F670. [DOI] [PubMed] [Google Scholar]
- Grunder, S., D. Firsov, S.S. Chang, N. Fowler Jaeger, I. Gautschi, L. Schild, R.P. Lifton, and B.C. Rossier. 1997. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 16:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell, D., T. Ishikawa, R. Saleki, and D. Rotin. 2002. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial Madin-Darbin canine kidney cells. J. Biol. Chem. 277:9772–9779. [DOI] [PubMed] [Google Scholar]
- Helman, S.I., X. Liu, K. Baldwin, B.L. Blazer-Yost, and W.J. Els. 1998. Time-dependent stimulation by aldosterone of blocker-sensitive ENaCs in A6 epithelia. Am. J. Physiol. 274:C947–C957. [DOI] [PubMed] [Google Scholar]
- Loffing, J., L. Pietri, F. Aregger, M. Bloch-Faure, U. Ziegler, P. Meneton, B.C. Rossier, and B. Kaissling. 2000. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am. J. Physiol. 279:F252–F258. [DOI] [PubMed] [Google Scholar]
- Loffing, J., M. Zecevic, E. Feraille, B. Kaissling, C. Asher, B.C. Rossier, G.L. Firestone, D. Pearce, and F. Verrey. 2001. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am. J. Physiol. 280:F675–F682. [DOI] [PubMed] [Google Scholar]
- Masilamani, S., G.H. Kim, C. Mitchell, J.B. Wade, and M.A. Knepper. 1999. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J. Clin. Invest. 104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, R.G., and J.A. Schafer. 2002. cAMP increases density of ENaC subunits in the apical membrane of MDCK cells in direct proportion to amiloride-sensitive Na+ transport. J. Gen. Physiol. 120:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naray-Fejes-Toth, A., C. Canessa, E.S. Cleaveland, G. Aldrich, and G. Fejes-Toth. 1999. Sgk is an aldosterone-induced kinase in the renal collecting duct - Effects on epithelial Na+ channels. J. Biol. Chem. 274:16973–16978. [DOI] [PubMed] [Google Scholar]
- Pacha, J., G. Frindt, L. Antonian, R.B. Silver, and L.G. Palmer. 1993. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J. Gen. Physiol. 102:25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, L.G., and H. Garty. 2000. Epithelial Na channels. The Kidney, Physiology and Physiopathology. Vol. 1, 3rd edition. D.W. Seldin and G. Giebisch, eds. Lippincott Williams and WilkinsPhiladelphia. 53:251–276. [Google Scholar]
- Reif, M.C., S.L. Troutman, and J.A. Schafer. 1986. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J. Clin. Invest. 77:1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Nicoud, M., M. Flahaut, J.M. Elalouf, M. Nicod, M. Salinas, M. Bens, A. Doucet, P. Wincker, F. Artiguenave, J.D. Horisberger, et al. 2001. Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc. Natl. Acad. Sci. USA. 98:2712–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotin, D., O. Staub, and R. Haguenauer-Tsapis. 2000. Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J. Membr. Biol. 176:1–17. [DOI] [PubMed] [Google Scholar]
- Snyder, P.M., D.R. Olson, and D.B. Bucher. 1999. A pore segment in DEG/ENaC Na(+) channels. J. Biol. Chem. 274:28484–28490. [DOI] [PubMed] [Google Scholar]
- Snyder, P.M., D.R. Olson, and B.C. Thomas. 2002. SGK modulates Nedd4-2-mediated inhibition of ENaC. J. Biol. Chem. 277:5–8. [DOI] [PubMed] [Google Scholar]
- Tavernarakis, N., and M. Driscoll. 1997. Molecular modeling of mechanotransduction in the nematode Caenorhabditis elegans. Annu. Rev. Physiol. 59:659–689. [DOI] [PubMed] [Google Scholar]
- Vallet, V., A. Chraibi, H.P. Gaeggeler, J.D. Horisberger, and B.C. Rossier. 1997. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 389:607–610. [DOI] [PubMed] [Google Scholar]
- Vallet, V., C. Pfister, J. Loffing, and B.C. Rossier. 2002. Cell surface expression of the channel activating protease xCAP1 is required for activation of ENaC in the Xenopus oocyte. J. Am. Soc. Nephrol. In press. [DOI] [PubMed] [Google Scholar]
- Verrey, F., E. Hummler, L. Schild, and B.C. Rossier. 2000. Control of sodium transport by aldosterone. The Kidney, Physiology and Physiopathology. Vol. 1, 3rd edition. D.W. Seldin and G. Giebisch, eds. Lippincott Williams and WilkinsPhiladelphia. 53:1441–1471. [Google Scholar]
- Wang, Q., J.D. Horisberger, M. Maillard, H.R. Brunner, B.C. Rossier, and M. Burnier. 2000. Salt- and angiotensin II-dependent variations in amiloride-sensitive rectal potential difference in mice. Clin. Exp. Pharmacol. Physiol. 27:60–66. [DOI] [PubMed] [Google Scholar]