

Abstract
Aims
Two studies were conducted to: (i) quantify the amount of drug-related radioactivity in blood, plasma, urine and faeces following a 14C-labelled dose of maraviroc; and (ii) investigate the pharmacokinetics, safety and tolerability of intravenous (i.v.) maraviroc and determine the absolute bioavailability of oral maraviroc. Metabolite profiling was also conducted. Data from both of these studies were used to construct a mass-balance model for maraviroc.
Methods
Study 1 was an open-label study in three healthy male subjects. All subjects received a single 300-mg oral solution dose of 14C-labelled maraviroc. Study 2 included two cohorts of subjects. Cohort 1 involved a double-blind (third party open), four-way crossover study where eight subjects received escalating i.v. doses of maraviroc (3, 10 and 30 mg) with placebo insertion. Cohort 2 involved an open, two-way crossover study where 12 subjects received 30 mg maraviroc by i.v. infusion and 100 mg maraviroc orally in random order. In study 1, blood samples and all urine and faeces were collected up to at least 120 h postdose. In study 2, blood samples were taken at intervals up to 48 h postdose. Urine was also collected up to 24 h postdose in cohort 1 only.
Results
After oral administration in study 1, maraviroc was rapidly absorbed with a plasma Tmax reached by 2 h postdose for all three subjects. The maximum concentrations of radioactivity also occurred within 2 h for all subjects. There was a higher amount of radioactivity in plasma than in blood (blood/plasma ratio ∼0.6 for AUCt and Cmax). Unchanged maraviroc was the major circulating component in plasma, accounting for ∼42% of the circulating radioactivity. Following a 300-mg 14C-labelled maraviroc dose, means of 76.4% and 19.6% of radioactivity were recovered in the faeces and urine, respectively. The mean total recovery of dosed radioactivity was 96%, with the majority of radioactivity being recovered within 96 h postdose. Profiling of the urine and faeces showed similar and extensive metabolism in all subjects. Unchanged maraviroc was the major excreted component (33%). The major metabolic pathways were determined and involved oxidation and N-dealkylation. Intravenous doses of maraviroc (3–30 mg) were well tolerated in study 2, and drug exposure was approximately proportional to dose within the studied range. Approximately 23% of total clearance (44 l h−1) was accounted for by renal clearance (10.2 l h−1). Mean volume of distribution at steady state was 194 l. Absolute bioavailability of a 100-mg oral tablet dose, by comparison with a 30-mg i.v. dose, was calculated to be 23.1%.
Conclusions
Maraviroc is rapidly absorbed and extensively metabolized, although unchanged maraviroc is the major circulating component in plasma and is the major excreted component after oral dosing. The pharmacokinetics of maraviroc after i.v. administration is approximately proportional over the dose range studied. Renal clearance contributes 23% of total clearance. The absolute bioavailability of 100 mg oral maraviroc is 23%.
Keywords: absorption, bioavailability, excretion, maraviroc, metabolism
Introduction
Maraviroc (4, 4-difluoro-N-{(1S)-3-[exo-3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1 phenylpropyl} cyclohexanecarboxamide) is a novel CCR5 antagonist recently approved for the treatment of human immunodeficiency virus (HIV) infection in treatment-experienced patients infected with CCR5-tropic HIV-1 [1]. The compound has a molecular weight of 514, is moderately lipophilic (log D7.4 = 2.1) and weakly basic with a pKa = 7.3. Pharmacokinetic and metabolism studies in animals have demonstrated species differences in bioavailability and absorption rates [2]. Urinary clearance of maraviroc accounts for a minor proportion of total clearance in mice, rats and dogs, with faecal elimination accounting for 72–94% of total excretion.
Findings from studies in Caco-2 cells and in P-glycoprotein (Pgp) homozygous null mice [2] have suggested that Pgp is involved in maraviroc transmembrane transport, potentially influencing both absorption in the intestine and biliary excretion. In vitro data demonstrate that cytochrome P450 3A4 is primarily responsible for maraviroc metabolism [3].
Clinical studies with maraviroc have shown that it has nondose-proportional oral pharmacokinetics [4, 5]. Two clinical studies were designed to investigate further the pharmacokinetics and excretion of maraviroc in healthy human subjects. The first study aimed to quantify the amount of drug-related radioactivity in blood, plasma, urine and faeces following a 14C-labelled dose of 300 mg maraviroc and to elucidate the metabolite profile of maraviroc. The second study investigated the pharmacokinetics, safety and toleration of intravenous (i.v.) maraviroc and determined the absolute bioavailability of 100 mg oral maraviroc. In addition, data from both of these studies, as well as predictions of absorption from other Phase I studies, were used to construct a mass-balance model to describe the disposition of maraviroc.
Methods
Subjects
All subjects provided written informed consent before participating in the studies. Subjects in study 1 were healthy men aged 45–65 years, weighing between 60 and 100 kg and with a body mass index between 18 and 28 kg m−2. Subjects in study 2 were healthy men aged 18–45 years, weighing between 60 and 100 kg with a body mass index between 18 and 28 kg m−2. Subjects were excluded from both studies if they had any history of clinically significant allergies, especially drug hypersensitivity; had any clinically significant abnormality; drank >28 units of alcohol per week; smoked more than five cigarettes per day; had evidence of drug abuse; had tested positive for HIV, hepatitis B or hepatitis C; had donated blood during the 2-month period preceding the study; had taken any experimental drug in the 4-month period preceding the study or any prescribed or over-the-counter medication (except paracetamol up to 3.0 g day−1) in the 3-week period preceding the study. Subjects in study 1, who were occupationally exposed to radioactivity, or had x-ray examinations or participated in a radiolabelled drug study in the preceding 12-month period, were also excluded. Both studies were conducted in compliance with the ethical principles set forth in the Declaration of Helsinki (1989) and with the local laws and regulations governing the use of new therapeutic agents.
Study designs
Study 1 was a single-centre, open-label study to investigate the absorption, metabolism and excretion of 14C maraviroc in healthy subjects. Subjects were admitted to the study centre on the evening before maraviroc dosing for physical examination, breath alcohol testing, and drug abuse screening, and to provide a predose faecal sample. Subjects fasted from 22.00 h on the evening before dosing until 4 h postdose on day 1, at which time they ate a standard lunch. Subjects had a light snack and a standard evening meal 8 and 10 h postdose, respectively. For each subject, 300 mg 14C maraviroc powder was dissolved in 10 ml of 0.01 M hydrochloric acid and 90 ml of water, to produce a solution (100 ml) of 3 mg ml−1 14C maraviroc. Doses contained nominally 48 μCi of 14C maraviroc.
Study 2 was a two-cohort, crossover design study to investigate the pharmacokinetics, safety and tolerability of i.v. maraviroc (cohort 1) and to determine the absolute bioavailability (cohort 2) of the 100-mg oral tablet dose. The 100-mg dose was selected, because at the time of this study it was predicted to be a clinically relevant dose [6]. Cohort 1 involved a double-blind (third-party open), four-way crossover design study in which eight subjects received escalating i.v. doses of maraviroc (3 mg, 10 mg, 30 mg) with placebo insertion. Cohort 2 involved an open, two-way crossover design study in which 12 subjects received maraviroc 30 mg by i.v. infusion and maraviroc 100 mg oral tablet in random order. In cohort 1, subjects attended the study centre six times: screening, study visits 1–4, and follow-up. In cohort 2, subjects attended the study centre four times: screening, study visits 1 and 2, and follow-up. Treatment periods in study 2 were separated by at least 7 days. All subjects were admitted to the study centre on the day prior to the first dosing period and remained there until 24 h postdose. Subjects fasted (except water) from 22.00 h the evening before each dose until at least 4 h postdose the following morning. Water was also restricted from 1 h predose to 1 h postdose. Subjects received maraviroc between 08.00 h and 10.00 h on the day of dosing. I.v. infusions were administered over 1 h and tablets were taken with 250 ml of water. Maraviroc was supplied by Pfizer as 100-ml vials (2.5 mg ml−1) for i.v. infusion and as 100-mg tablets for oral administration.
Pharmacokinetic assessments
Sample collection
In study 1, blood samples were collected predose and at intervals up to 120 h postdose for analysis of total radioactivity, maraviroc, and its metabolites. Samples were collected into beadless heparinized tubes; plasma was isolated by centrifugation and stored at −20°C within 1 h of collection. Aliquots of the plasma samples were sent to Maxxam Analytics Inc. (Mississauga, ON, Canada) for determination of maraviroc concentration. Blood and plasma samples were sent to Pfizer for ultralow level scintillation counting and plasma metabolite profiling. Urine and faeces were collected from predose until at least 120 h postdose. Further urine and faecal collections were made up to 196 h postdose or until 95% of the administered radioactivity had been recovered or daily excretion was <1%. Duplicate 1-ml urine samples were sent to BioDynamics (Cardiff, UK) for measurement of radioactivity, and duplicate 50-ml urine samples were sent to Pfizer for metabolite profiling. Each faecal collection was weighed and two 50-g samples were sent to BioDynamics to measure radioactivity. Up to 100 g of faeces were sent to Pfizer for metabolite profiling. Plasma and urine samples were also sent to Pfizer for analysis of UK-463,977 (an unlabelled metabolite).
In study 2, blood samples (5 ml) were collected predose and at intervals up to 48 h postdose for the i.v. and oral doses for the analysis of maraviroc. Samples were collected into beadless heparinized tubes; plasma was isolated by centrifugation and stored at −20°C within 1 h of collection. In cohort 1, urine was collected up to 24 h postdose at each treatment visit. Urine samples were stored at approximately −20°C until assay for maraviroc and UK-408,027 (secondary amine metabolite).
Assay details
For determination of radioactivity in blood, plasma, faeces and urine in study 1, the following procedures were used. Plasma samples (∼0.5 g) were mixed with 19 ml scintillation cocktail (Starscint; PerkinElmer, Waltham, MA, USA) prior to liquid scintillation counting (Guardian; PerkinElmer). Whole blood (∼0.25 g) was prepared for scintillation counting by the addition of bleach (2 ml) and mixing with 17 ml scintillation cocktail (Starscint; PerkinElmer) prior to liquid scintillation counting (Guardian; PerkinElmer). Replicate weighed aliquots of urine (∼1 g) were mixed with 2 ml scintillation cocktail (Ultima Gold XR; PerkinElmer), and the radioactivity was measured by liquid scintillation counting (Canberra Packard 2300 TR Scintillation Counter). Faecal samples were homogenized in water (sufficient to make a paste), using a Silverson blender. Replicate weighed aliquots (∼0.2–0.4 g) were combusted in a biological oxidizer (Model 307 MK2 Tri-Carb®; PerkinElmer, Beaconsfield, UK) and the evolved 14CO2 measured by liquid scintillation counting in 8 ml scintillation cocktail (Permafluor E+; PerkinElmer).
For pharmacokinetic determination of maraviroc in plasma (study 1), the overall method imprecision values for the analysis of plasma quality control (QC) samples were 5.4%, 1.1% and 2.2% for maraviroc at target concentrations of 1, 90 and 180 ng ml−1, respectively. The mean inaccuracy of the assay ranged from to −6.7% to 3.0% over the QC concentration range. The calibration range was 0.5–200 ng ml−1. For the metabolite UK-463,977 (a carboxylic acid metabolite resulting from N-dealkylation) assay in plasma, the mean imprecision values of the QC samples were 11.3%, 7.6% and 2.2% for UK-463,977 target concentrations of 0.1, 0.5 and 5.0 ng ml−1, respectively. The mean inaccuracy of the assay ranged from −1.5% to 5.0% over the QC concentration range. The calibration range in plasma was 0.1–5.0 ng ml−1. For UK-463,977 assay in urine, the mean imprecision values of the QC samples were 2.0%, 4.2% and 1.4% for UK-463 977 target concentrations of 10, 100 and 1000 ng ml−1, respectively. The mean inaccuracy of the assay ranged from 4.5% to 14% over the QC concentration range. The calibration range in urine was 10–1000 ng ml−1.
In study 2, plasma and urine samples were analysed at Maxxam Analytics, Inc., using validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) methods. The overall method imprecision values for the analysis of plasma QC samples were 5.8%, 3.1% and 3.9% for maraviroc at target concentrations of 1, 90 and 180 ng ml−1, respectively. Mean inaccuracy of the assay ranged from −5.6% to 8.0% over the QC concentration range. The calibration range was 0.5–200 ng ml−1. The overall method imprecision values for the analysis of urine QC samples were 2.6%, 5.2% and 4.0% for maraviroc at target concentrations of 15, 300 and 900 ng ml−1, respectively. Mean inaccuracy of the assay ranged from −7.3% to 10.7% over the QC concentration range, and the calibration range was 5–1000 ng ml−1. For analysis of UK-408, 027 in urine, the overall method imprecision values for the analysis of QC samples were 13.2%, 6.6%, 3.3%, 3.3% and 3.5% and inaccuracy was −0.5%, −1.8%, 2.6%, 5.0% and −2.5% at target concentrations of 5, 10, 50, 500 and 1000 ng ml−1, respectively. The calibration range was 5–1000 ng ml−1.
Safety assessments
In both studies, adverse events were monitored throughout the studies. Safety assessments included clinical laboratory testing (including haematology and clinical chemistry), physical examination, blood pressure, pulse rate, and electrocardiogram (ECG) data, and were conducted at appropriate intervals throughout the studies. In study 2, the subjects in cohort 1 were also monitored by ECG telemetry from 30 min predose until at least 4 h postdose.
Statistical analysis
No formal sample size calculations were performed in study 1; the sample size was limited to three, allowing determination of metabolite profile but limiting the number of subjects exposed to radioactivity. Descriptive statistics were used to summarize maraviroc pharmacokinetic parameters.
In study 2, cohort 1, the sample size of eight subjects was chosen as a compromise between the need to minimize first exposure of an i.v. dose of maraviroc in humans and the need to provide adequate safety and tolerability information at each dose level. For cohort 2, it was estimated that a sample size of 12 would provide a 95% confidence interval (CI) of ±0.303 on the natural log scale for area under the plasma concentration–time curve (AUC) with 80% coverage probability. Assuming 100% absolute bioavailability, it was estimated that the 95% CI would be (73.9%, 135%). The standard deviation of differences (natural log scale) of 0.414 for AUC was calculated using the within-subject variability (0.0855 for AUC) from the analysis of a previous Phase 1 study.
In cohort 1, dose proportionality for the 3-, 10- and 30-mg i.v. infusions was investigated using both the dose divide method and power law analysis. For the power law analysis the AUC from zero to the time of the last measurable concentration (AUCt) and maximum observed drug concentration (Cmax) were divided by dose and log transformed prior to analysis; they were subjected to an analysis of variance (anova) allowing for variation due to subject with log dose as a covariate (period was not included in the model as it was confounded with dose). An estimate of the dose proportionality constant β was obtained from the model (slope estimate +1) and presented together with its 95% CIs. The statistical significance of the dose proportionality constant from 1 and the size of the confidence limits were used to determine whether dose proportionality could be concluded. AUCt and Cmax were also presented using the dose divide method for further information.
In cohort 2, natural log-transformed AUC, dose-normalized to 1 mg, was subjected to anova appropriate for the two-period, two-treatment crossover design, allowing for variation due to sequence, subject within sequence, period, and treatment. The comparison of interest was 100 mg maraviroc (oral tablet) vs. 30 mg maraviroc i.v. For AUC, the differences between oral and i.v. formulation means, standard errors associated with the differences, and 95% CIs for the differences were presented. The difference in means was back-transformed to give a ratio between antilogged geometric treatment means, and the corresponding antilogged 95% CIs were presented. The ratio between geometric means of AUC for the oral and i.v. doses provided the estimate of absolute bioavailability. The model assumptions of constant variance and normality were assessed through examination of the plots of the residuals.
Mass-balance model
Data from both studies, as well as predictions of absorption from other Phase I studies, were used to construct a mass-balance model to describe the disposition of maraviroc. Mass-balance was used to apportion the relative amounts of parent maraviroc and (combined) metabolites to various pathways, including first-pass effects, renal clearances (CLR), and metabolic and other (secretion) nonrenal clearance (CLnR). The main assumption was that after absorption, all further disposition of maraviroc and all metabolites is strictly linear, including first-pass metabolism/clearance. It was further assumed that total systemic plasma clearance (CL) consisted of only a renal component (CLR) and a nonrenal component (CLnR) that had a metabolic component (CLm) and a nonmetabolic component (CLs). The full methodology for this analysis will be presented in a separate publication.
Results
Subjects
Three subjects entered and completed study 1 and all were included in the pharmacokinetic and safety analyses. All subjects were White men aged 45–53 years (mean 49 years) and ranged in weight from 82 to 85 kg (mean 83 kg). Twenty subjects entered and completed study 2 and all were included in the pharmacokinetic and safety analyses. All subjects were White men aged 22–42 years (cohort 1 mean 35 years; cohort 2 mean 32 years). Subjects in cohort 1 ranged in weight from 65 to 93 kg (mean 82 kg) and subjects in cohort 2 ranged in weight from 66 to 89 kg (mean 78 kg).
Pharmacokinetics
Plasma/whole blood radioactivity and maraviroc pharmacokinetics
In study 1, radiolabelled 14C maraviroc oral solution (300 mg) was rapidly absorbed, with time to Cmax (Tmax) for maraviroc and total radioactivity being reached by 2 h postdose for all subjects (Tables 1 and 2, Figure 1). The interindividual variation was low. The relative amount of radioactivity was higher in plasma than in whole blood, with blood/plasma ratios for AUCt and Cmax of 0.624 and 0.612, respectively (Table 1).
Table 1.
Mean pharmacokinetic parameters for radioactivity in plasma and whole blood for 300 mg oral solution dose (study 1)
| Parameter | Medium | Mean (% CV or SD) | Blood/plasma ratio (% CV) |
|---|---|---|---|
| AUCt (ng equiv g−1 h)* | Plasma | 4497 (4.6%) | nc |
| Blood | 2251 (45%) | 0.624 (24%)‡ | |
| Cmax (ng equiv g−1)* | Plasma | 800 (28%) | nc |
| Blood | 489 (34%) | 0.612 (6.6%) | |
| Tmax (h)† | Plasma | 1.33 (0.58) | nc |
| Blood | 1.33 (0.58) | nc |
Geometric means (% CV).
Arithmetic means (SD).
AUCt ratio determined using time-matched AUCt values. CV, coefficient of variation; nc, not calculated; SD, standard deviation.
Table 2.
Mean maraviroc plasma pharmacokinetics for 300-mg oral solution dose (study 1)
| Parameter | Mean (%CV or SD) |
|---|---|
| AUCt (ng ml−1 h)* | 2055 (8.6%) |
| AUC (ng ml−1 h)* | 2086 (8.0%) |
| Cmax (ng ml−1)* | 496 (24%) |
| Tmax (h)† | 0.83 (0.58) |
| t1/2 (h)† | 23 (3.6) |
Geometric means (%CV).
Arithmetic means (SD). CV, coefficient of variation; SD, standard deviation; t1/2, terminal elimination half-life.
Figure 1.
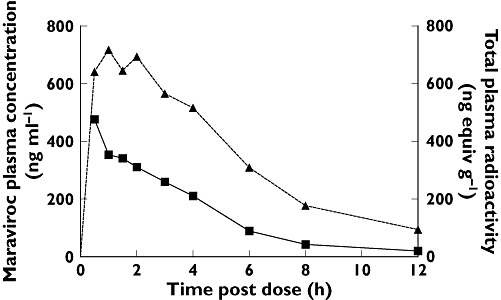
Mean plasma concentrations after dosing with 300 mg maraviroc (14C-labelled) oral solution (study 1). Plasma radioactivity (14C) (▴); Plasma Maraviroc (▪)
Maraviroc (as measured by specific assay) accounted for approximately 40% of total plasma radioactivity. The plasma maraviroc concentration/total plasma radioactivity ratio for AUCt was 0.402. The mean ratio for Cmax was 0.622.
Radioactivity in urine and faeces
The mean amount of total administered radioactivity excreted in the urine was 19.6% [coefficient of variation (CV) 12.9%], with the major proportion being excreted in the 36-h period after dosing. The mean amount of radioactivity excreted in the faeces was 76.4% (CV 3.4%), and the majority was excreted in the 96-h period after dosing. The mean total recovery of dosed radioactivity was 96% over the 168-h postdose period with low intersubject variation (CV 0.3%).
Maraviroc metabolite profiling
Metabolite profiling of precipitated plasma (time normalized pool [7]; 0–18 h postdose) by high-performance liquid chromatography and LC/MS/MS analysis revealed that unchanged maraviroc was the major circulating component, accounting for 42% of circulating radioactivity (cf., 40% as measured by specific assay of maraviroc). A secondary amine metabolite resulting from N-dealkylation adjacent to the tropane moiety (UK-408 027) and an analogue of the amine involving oxidation of the methyl group of the triazole moiety, were also identified in the plasma and accounted for 22% and 11% of the circulating radioactivity, respectively. Plasma concentration of the unlabelled carboxylic acid metabolite (UK-463 977) resulting from the N-dealkylation pathway, was determined by specific assay, and the Cmax and Tmax for this metabolite were 37.3 ng ml−1 and 1.5 h, respectively. The molecular weight normalized Cmax for UK-463 977 was approximately 10% compared with total radioactivity. Elimination of UK-463 977 from plasma followed a similar profile to the radioactivity.
Metabolite profiling of urine and faecal homogenates showed similar and extensive metabolism of maraviroc in all subjects. Unchanged maraviroc accounted for 33.3% of the total radioactive dose (Figure 2) excreted in both the urine (8.0%) and faeces (25.3%). Other major excreted components were four metabolites resulting from mono-oxidation in the difluorocyclohexane ring, each accounting for 5–9% of radioactivity; a metabolite resulting from oxidation in the triazole group (10%); and the secondary amine (UK-408 027) resulting from N-dealkylation adjacent to the tropane moiety (7%). Quantification of the unlabelled carboxylic acid metabolite (UK-463 977) in urine showed that this metabolite represented ∼3% of the total dose. A comprehensive schematic for observed maraviroc metabolites is presented in Figure 2, along with the relative abundance of each as a percentage of the total administered radioactive dose.
Figure 2.
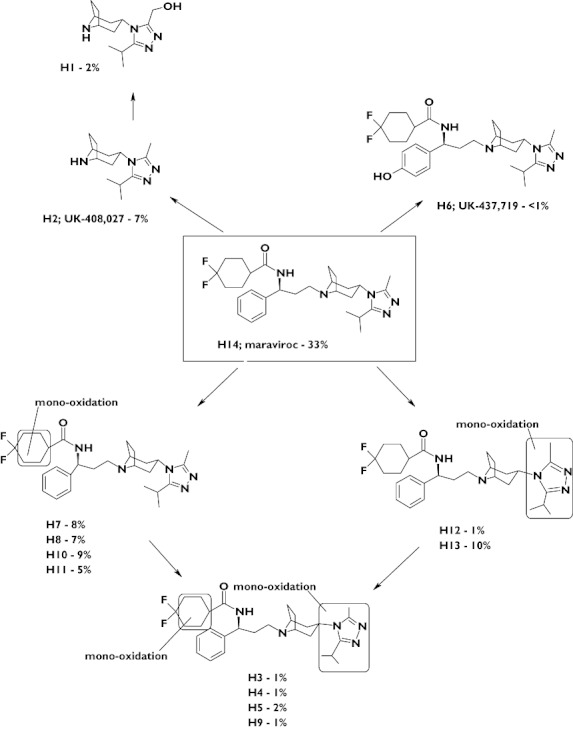
Proposed metabolic pathways for maraviroc following a single oral 300-mg dose and percentage of total excreted dose for each metabolite
Dose proportionality and absolute bioavailability
Mean plasma pharmacokinetic parameters for maraviroc following i.v. and oral dosing are presented in Tables 3 and 4 (study 2). For most subjects, Tmax occurred at the end of the 1 h i.v. infusion (Figure 3). Analysis of the relationship between Cmax and AUCt and dose following i.v. maraviroc confirmed that there was no evidence of a significant deviation from linearity. The dose proportionality constants calculated using Cmax and AUCt were 1.031 (95% CI 0.958, 1.105) and 1.065 (95% CI 1.012, 1.119), respectively. Following a 30-mg i.v. dose (cohort 1), the mean total clearance of maraviroc was calculated as 44 l h−1. Approximately 23% of total clearance was accounted for by CLR (10.2 l h−1) and 77% by CLnR (33.8 l h−1). Volume of distribution at steady state (Vss) was 194 l (cohort 1). CLR was similar for all doses.
Table 3.
Summary of maraviroc pharmacokinetic parameters (study 2, cohort 1)
| Mean (CV% or SD) | |||
|---|---|---|---|
| Parameter | 3 mg i.v. (n = 8) | 10 mg i.v. (n = 8) | 30 mg i.v. (n = 8) |
| AUCt (ng ml−1 h)* | 57.6 (15.3%) | 201 (12.0%) | 670 (12.5%) |
| AUC (ng ml−1 h)* | nc | nc | 687 (12.0) |
| Cmax (ng ml−1)* | 36.9 (18.6%) | 122 (14.6%) | 397 (13.8%) |
| Tmax (h)† | 0.94 (0.12) | 0.94 (0.18) | 0.91 (0.19) |
| t1/2 (h)† | nc | nc | 13.2 (2.80) |
| CL (l h−1)† | nc | nc | 44.0 (5.55) |
| CLR (l h−1)† | 11.2 (1.52) | 10.5 (1.63) | 10.2 (1.69) |
| CLnR (l h−1)† | nc | nc | 33.8 (4.67) |
| Vss (l)† | nc | nc | 194 (60.7) |
Unadjusted geometric means (%CV).
Unadjusted arithmetic means (SD). CV, coefficient of variation; nc, not calculated; SD, standard deviation; Vss, volume of distribution at steady state.
Table 4.
Summary of maraviroc pharmacokinetic parameters (study 2, cohort 2)
| Mean (CV or SD) | ||
|---|---|---|
| Parameter | 30 mg i.v. | 100 mg oral |
| AUCt (ng ml−1 h)* | 645 (15.0%) | 492 (34.3%) |
| AUC (ng ml−1 h)* | 656 (14.7%) | 506 (33.8%) |
| Cmax (ng ml−1)* | 374 (12.1%) | 122 (53.0%) |
| Tmax (h)† | 0.98 (0.07) | 3.08 (1.00) |
| t1/2 (h)† | 12.0 (1.66) | 12.5 (1.69) |
| CL (l h−1)† | 46.2 (6.87) | nc |
| Vss† | 182 (43.1) | nc |
Geometric means (%CV).
Arithmetic means (SD). CV, coefficient of variation; nc, not calculated; SD, standard deviation; Vss, volume of distribution at steady state.
Figure 3.
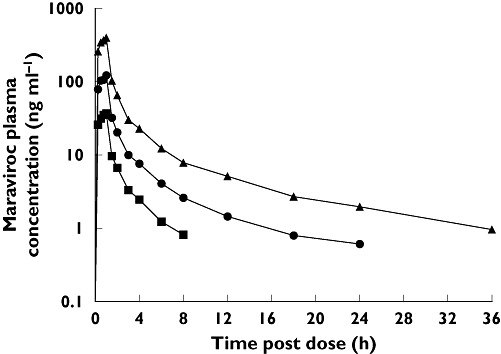
Semilog plot of mean plasma maraviroc concentrations after escalating i.v. doses (study 2, cohort 1). 3 mg IV maraviroc (▪); 10 mg IV maraviroc ( ); 30 mg IV maraviroc (▴)
); 30 mg IV maraviroc (▴)
In study 2, a comparison of maraviroc exposure between oral (100-mg tablet) and i.v. (30-mg solution) administration routes (cohort 2) estimated the absolute oral bioavailability of 100 mg oral maraviroc to be 23.1% (95% CI 19.2%, 27.8%). The mean plasma profiles for these doses are presented in Figure 4. A summary of plasma pharmacokinetic parameters is presented in Table 4.
Figure 4.
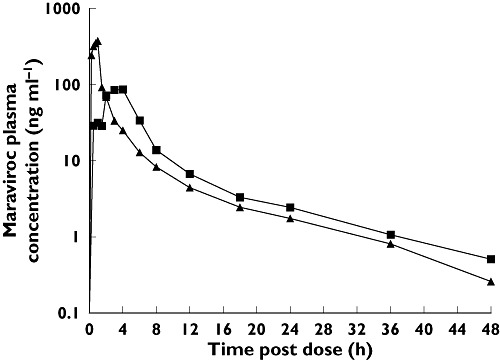
Semilog plot of mean plasma maraviroc concentrations after dosing with a 100-mg oral tablet or 30 mg i.v. solution (study 2, cohort 2). 100 mg maraviroc oral tablet (▪); 30 mg maraviroc IV (▴)
Mass balance model
A detailed diagram of the disposition of maraviroc and its metabolites after a 300-mg oral solution dose of maraviroc is presented in Figure 5. It shows that at this dose, approximately 84% of maraviroc is absorbed. First pass extraction (E) was determined as approximately 60% and implies that the maximum possible bioavailability of maraviroc, even with 100% absorption, would be no more than 40%. The absolute bioavailability of maraviroc at 300 mg is estimated to be approximately 33% (cf., 23% determined for 100 mg maraviroc).
Figure 5.
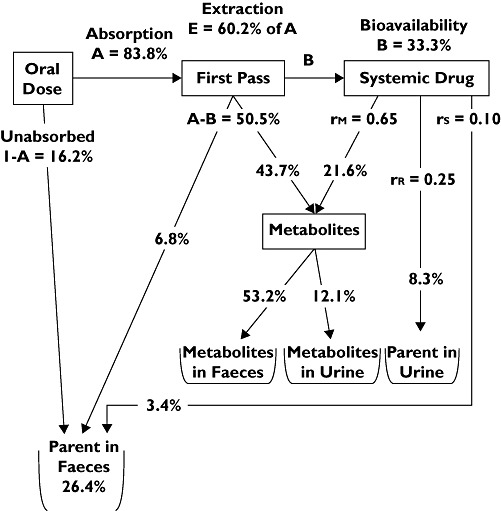
Model of mass balance for a 300-mg oral solution dose of maraviroc. rR, renal fraction; rM, metabolic fraction; rS, secreted, or nonmetabolic fraction.
Safety and toleration
All i.v. and oral doses in both studies were well tolerated, and there were no serious adverse events or discontinuations. Most adverse events were mild or moderate in severity. There were no clinically significant changes in laboratory test parameters or in blood pressure, pulse rate, electrocardiographic or physical examination findings.
Discussion
Maraviroc was rapidly absorbed following oral dosing. Tmax for maraviroc and total radioactivity both occurred within 2 h of dosing for all subjects. The blood/plasma partition coefficient for radioactivity was 0.6 for AUCt and Cmax, suggesting maraviroc is mainly confined to the plasma with little distribution into red blood cells. Approximately 96% of the administered 14C-labelled maraviroc dose was recovered, most of which was excreted in the faeces (76.4%), with 19.6% excreted in the urine. Unchanged maraviroc was the major circulating component and accounted for approximately 42% of the radioactivity in plasma. Full metabolite profiling of plasma, urine and faecal samples showed that maraviroc is extensively metabolized with the major pathways involving oxidation and N-dealkylation reactions.
The pharmacokinetics of i.v. maraviroc was shown to be essentially linear across the dose range studied. Approximately 23% of total clearance was accounted for by CLR and 77% by CLnR. CLR was not affected by dose.
The absolute bioavailability of 100-mg oral maraviroc was 23%. The mass-balance model predicts that the absolute bioavailability of maraviroc at 300 mg is approximately 33%.
Maraviroc doses of up to 300 mg orally and 30 mg by i.v. infusion were well tolerated.
Competing interests
S.A., D.R., L.A.W., C.E.R., A.N.R.N. and D.K.W. were employees of Pfizer Ltd at the time of this research.
These studies were sponsored by Pfizer. Editorial assistance was provided by Dylan Harris, PhD and Janet E. Matsuura, PhD at Complete Healthcare Communications, Inc., and was funded by Pfizer Inc., New York, NY, USA.
REFERENCES
- 1.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427 857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–32. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman AN, Smith DA. Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab Dispos. 2005;33:587–95. doi: 10.1124/dmd.104.002626. [DOI] [PubMed] [Google Scholar]
- 3.Hyland R, Jones B, Muirhead G. In vitro assessment of the CYP-based drug–drug interaction potential of UK-427 857. 5th International Workshop on Clinical Pharmacology of HIV Therapy; 2004 April 1–3; Rome, Italy. [Google Scholar]
- 4.Abel S, van der Ryst E, Rosario MC, Ridgway CE, Medhurst CG, Taylor-Worth RJ, Muirhead GJ. Assessment of the pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist, in healthy volunteers. Br J Clin Pharmacol. 2008;65(1):5–18. doi: 10.1111/j.1365-2125.2008.03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan PLS, Weatherley B, McFadyen L. A population pharmacokinetic meta-analysis of maraviroc in healthy volunteers and asymptomatic HIV-infect subjects. Br J Clin Pharmacol. 2008;65(1):76–85. doi: 10.1111/j.1365-2125.2008.03139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag M, Goebel F, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 7.Hop CE, Wang Z, Chen Q, Kwei G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J Pharm Sci. 1998;87:901–3. doi: 10.1021/js970486q. [DOI] [PubMed] [Google Scholar]