

Abstract
Aims
To evaluate the pharmacokinetics, safety and tolerability of single and multiple oral doses of maraviroc in healthy volunteers.
Methods
Three double-blind, placebo-controlled, dose-escalation studies with either single or multiple doses of maraviroc were conducted in healthy volunteers. Plasma and urine samples were collected to investigate the pharmacokinetics of maraviroc and evaluate any changes with respect to dose and duration/frequency of dosing. Safety and toleration of maraviroc were also assessed.
Results
Maraviroc is rapidly absorbed following oral administration, and plasma Tmax is achieved within 0.5–4.0 h postdose. Steady-state plasma concentrations are achieved after 7 consecutive days of dosing. Although the pharmacokinetics of maraviroc is nonproportional over the dose range studied (3–1200 mg), the degree of nonproportionality is small at clinically relevant doses. Renal clearance is approximately 10–12 l h−1 and appears unaffected by increasing maraviroc doses. Maraviroc does not significantly modulate the activity of CYP2D6 or CYP3A4 at clinically relevant doses. There were no serious adverse events in any of these studies, and doses up to 900 mg were generally well tolerated, with postural hypotension being the dose-limiting event. There was no pattern or dose relationship observed with maraviroc with regard to laboratory abnormalities, including hepatic transaminases. No clinically significant increases in QTc were noted at clinically relevant doses.
Conclusions
Maraviroc is absorbed into the systemic circulation and reaches steady state by day 7 of multiple dosing. It does not significantly influence the activity of major drug-metabolizing enzymes and is well tolerated at clinically relevant doses, with most adverse events being mild or moderate.
Keywords: bioavailability, CCR5 antagonist, exposure, maraviroc, pharmacokinetics, safety
Introduction
At the end of 2006, approximately 40 million people were living with human immunodeficiency virus (HIV) infection [1]. The development of different classes of antiretroviral agents for combating HIV infection has increased survival in patients to whom these drugs are available. Nucleoside inhibitors of the HIV reverse transcriptase enzyme (NRTIs) were the first class of drugs used to treat HIV patients on a grand scale. The second and third classes of antiretroviral drugs (ARVs), protease (PIs) and non-NRTI inhibitors, followed in the 1990s and, when used in combination with NRTIs, dramatically increased the survival rate for HIV/AIDS patients.
A hallmark of HIV is the high rate of genetic mutability seen during its replication [2], which allows the virus to evade host immune response and also to develop resistance to drugs that inhibit replication [3]. Resistance has been demonstrated against all currently used HIV treatments, and hence there is still a high medical need for new agents, with different resistance profiles, that are well tolerated and conveniently administered.
Enfuvirtide (Fuzeon®; Roche, Basel, Switzerland), an injectable peptide inhibitor of HIV attachment to host cells, represents the first agent in a fourth major class of drugs called entry inhibitors that prevent the entry of the HIV-1 virus into cells, effectively precluding infection and viral replication. Whereas enfuvirtide disrupts the interaction between the viral gp41 protein and the CD4 cell surface receptor, it does not affect the interaction between gp120 and CCR5. Inhibition of viral entry through the CCR5 coreceptor is predicted to be clinically effective, as individuals who are homozygous for a mutant allele encoding a 32-base pair deletion in CCR5 (rendering them effectively CCR5 negative) are highly (although not completely) resistant to HIV-1 infection [4]. Furthermore, individuals with polymorphisms potentially resulting in reduced CCR5 expression tend to have slower disease progression and lower viral loads [5]. CCR5-tropic viruses predominate in primary infection and early disease stages, but even in antiretroviral-treated patients with detectable viraemia, >50% still carry CCR5-tropic virus exclusively [6].
Maraviroc is a potent, orally bioavailable CCR5 antagonist recently approved for use along with other antiretroviral agents for treatment-experienced patients infected with CCR5-tropic HIV-1. Maraviroc has been shown to be effective and well tolerated in preclinical [7, 8] and clinical studies [9–11]. Here, we present data from three Phase I clinical studies describing the pharmacokinetics, safety and tolerability of escalating oral doses of maraviroc in healthy subjects.
Methods
Subjects
Subjects were healthy male and female volunteers aged 18–45 years, with body weight between 60 and 100 kg for men and between 50 and 100 kg for women, and a body mass index of 18–28 kg m−2. Female participants had to be postmenopausal, surgically sterilized or practising accepted methods of contraception for 3 months before entering the study and had to have negative pregnancy tests before and during the study.
Subjects were excluded if they had any evidence of clinically significant disease or allergies, especially drug hypersensitivity, or clinically significant abnormalities identified by laboratory testing or physical examination; were taking or had taken any prescribed or over-the-counter medication (with the exception of paracetamol, up to 3 g day−1) in the 3 weeks before the first dose of study medication; had received any investigational drug within the previous 4 months prior to the first dosing day of the study; had evidence of drug abuse or excessive alcohol or cigarette use; tested positive for HIV-1, hepatitis C virus, or hepatitis B virus; or had a history of cardiac dysrhythmias (studies 1 and 2 only). Women who were lactating were also excluded. All subjects provided written informed consent before participating in these studies.
Study designs
All three studies were double-blind (third party open), placebo-controlled studies designed to assess the safety, toleration and pharmacokinetics of maraviroc (Table 1). In all studies, subjects were randomized to various treatment arms using a computer-generated pseudorandom code. Predefined rules for stopping dose escalation were in place for all studies, and each dose was administered to subjects only if the previous dose level had been well tolerated. All subjects had a screening visit within 4 weeks of the first dose and a follow-up visit 7–10 days after the last dose.
Table 1.
Description of study design for three maraviroc dose-escalation studies
| Study | Design | Treatment, daily dose |
|---|---|---|
| Study 1 | Double-blind (third-party open), placebo-controlled, 4-way crossover, escalating oral single-dose study | Cohort A: 1, 10, 100, 900 mg, placebo (oral solution) |
| Cohort B: 3, 30, 300, 1200 mg, placebo (oral solution) | ||
| Study 2 | Double-blind (third-party open), placebo-controlled, parallel group, single- and multiple-escalating dose study | Cohort 1: 100 mg b.i.d. or placebo (ratio 3 : 1) (oral solution) |
| Cohort 2: 300 mg b.i.d. or placebo (ratio 3 : 1) (oral solution) | ||
| Cohort 3: 600 mg q.d. or placebo (ratio 3 : 1) (oral tablet) | ||
| Cohort 4: 25 mg b.i.d. or placebo (ratio 3 : 1) (oral solution) | ||
| Cohort 5: 600 mg q.d. or placebo (ratio 3 : 1) (oral tablet) | ||
| Cohort 6: 3 mg b.i.d., 10 mg b.i.d., or placebo (ratio 5 : 5 : 2) (oral solution) | ||
| Study 3* | Double-blind (third-party open), placebo-controlled, parallel group, multiple-escalating dose study | Cohort 1: 300 mg b.i.d. or placebo (days 1–7); 600 mg b.i.d. or placebo (days 8–14) (ratio 3 : 1) (oral tablets) |
| Cohort 2: 600 mg b.i.d. or placebo (days 1–7); 900 mg b.i.d. (days 8–14) (ratio 3 : 1) (oral tablets) | ||
| Cohort 3: 900 mg q.d. (days 1–7); 1200 mg q.d. (days 8–14) (ratio 3 : 1) (oral tablets) |
Subjects allocated to placebo in study 3 remained on placebo from days 1–14.
Study 1 was a dose-escalating, four-way crossover study in two parallel cohorts of 12 subjects each. Subjects received single oral solution doses in each dosing period, and doses were separated by at least 7 days. Subjects received escalating maraviroc doses, with one dosing period substituted by placebo. For dosing and schedules, see Table 1. Subjects fasted from 22.00 h the day before each dose until at least 4 h postdose. Maraviroc was administered under supervision with 250 ml of water, except for the 1200-mg dose, which was administered with up to 450 ml of water. Other fluids were restricted 1 h pre- and postdose. Subjects ate a standard lunch and dinner after providing the 4- and 10-h blood samples, respectively.
Study 2 was a parallel-group study consisting of six separate cohorts of 12 subjects each (Table 1). Subjects were administered single oral doses on study day 1, followed by q.d. or b.i.d. dosing on days 3–11. Subjects in cohorts 1–5 were randomized to receive study drug or double-blind placebo in a 3 : 1 ratio, whereas subjects in cohort 6 were randomized to receive either 3 mg or 10 mg or double-blind placebo in a 5 : 5 : 2 ratio. A final dose was administered to all cohorts on the morning of day 12. Doses of 3–300 mg were administered as solution, and the 600-mg dose was given as four 150-mg tablets. Subjects were fasted from midnight until at least 2 h postdose, except on days −1, 1, 7 and 12, when they were fasted until 4 h postdose. Maraviroc was administered under supervision with 250 ml of water; other fluids were restricted 1 h pre- and postdose. A standard breakfast, lunch, a light snack (if desired) and dinner were served daily. Breakfast was not served on days −1, 1, 7 and 12.
Study 3 was a parallel-group study consisting of three separate cohorts of 12 subjects each (Table 1). Subjects in all cohorts were randomized to receive study drug or double-blind placebo in a 3 : 1 ratio. Subjects allocated to study drug received a lower dose level or placebo on days 1 through 7, followed by an escalation to a higher dose level on days 8 through 14. Maraviroc was administered as 100-mg tablets. Subjects allocated to placebo stayed on it from days 1 through 14. For dosing and schedules, see Table 1. On days 1–14, treatment was administered between 08.00 h and 10.00 h, and between 20.00 h and 22.00 h. Subjects on a q.d. regimen received their treatment in the morning only. Subjects ate a standard breakfast 1 h postdose and had lunch, dinner and evening snack 4, 9 and 13 h post morning dose, respectively. Subjects fasted from 22.00 h the evening before dosing on days 1, 7 and 14, and fluids were restricted 1 h pre- and postdose.
Pharmacokinetic assessments
Blood and urine samples
All blood samples were collected into beadless, heparinized tubes. Plasma samples were isolated by centrifugation and stored at −20°C within 1 h of collection. All urine samples were collected and stored at −20°C until assayed. In study 1, blood samples were collected predose and at intervals up to 24 h postdose. Additional samples were taken at 36 and 48 h postdose for subjects who received 900-mg and 1200-mg doses. Urine samples were also collected from subjects predose and up to 24 h postdose. In study 2, blood samples were collected on day 1 predose and at intervals up to 12 h postdose for cohort 6 and up to 48 h postdose for cohorts 1–5. During the multiple-dose phase, blood samples were taken predose in the morning on days 3–12 and at intervals up to 12 h postdose on day 7 and up to 72 h postdose on day 12. Cohort 3 stopped dosing on day 7 due to poor toleration. Consequently, the additional samples (18–72 h) planned for day 12 were taken following the morning dose on day 7 for this cohort. There was no 72-h postdose blood sample collected for cohort 6 on day 12. Urine samples were collected predose in the morning on days 1, 7 and 12 and postdose on days 1 and 12 (up to 24 h postdose), and day 7 (up to 12 h postdose); no day 12 urine was collected for cohort 3. In study 3, blood samples were collected before the morning dose on days 1 through 14 and at intervals up to 12 h after the morning dose on days 1, 7 and 14. Urine samples were collected predose on day 1 and up to 12 h post morning dose on days 1, 7 and 14.
Assay details
Plasma samples were analysed by liquid chromatography/tandem mass spectrometry (LC/MS/MS). In study 1, the overall assay quality control (QC) imprecision values for the MS-MS analysis of maraviroc plasma concentration were 8.7%, 8.2% and 8.0% and the respective percentage inaccuracies were 2.9%, −0.1% and −3.4% at the target concentrations of 0.5, 10 and 40 ng ml−1. The 200 ng ml−1 diluted QC sample gave an overall imprecision of 10.9% and an overall inaccuracy of −7.0% based on dilution factors of between one in five and one in 150. The calibration range was 0.25–50 ng ml−1. In study 2, the overall method imprecision values for the analysis of plasma QC samples for cohorts 1–5 were 8.57%, 3.28%, 2.64%, 1.85%, 2.12% and 3.82%, and the percentage inaccuracy was 0.144%, −1.31%, −1.03%, 1.27%, 1.71% and −1.30% at target concentrations of 1.5, 60, 160, 1500 (×10 dilution), 1500 (×20 dilution) and 160 (×5 dilution) ng ml−1, respectively. For cohort 6, the imprecision values were 12.6%, 2.7% and 2.7% and the percentage inaccuracy was −0.8%, −1.3% and −1.0% at target concentrations of 0.3, 5 and 8 ng ml−1. The calibration range was 0.5–200 ng ml−1 and 0.1–10 ng ml−1 for cohorts 1–5 and cohort 6, respectively. In study 3, the overall method imprecision values for the analysis of plasma QC samples were 7.8%, 5.3% and 5.1% at target maraviroc concentrations of 1, 90 and 180 ng ml−1, respectively. The mean inaccuracy of the assay ranged from −1.6% to 4.5%. The calibration range was 0.5–200 ng ml−1.
Maraviroc concentrations in urine were analysed using LC/MS/MS. The overall QC imprecision values for the high-performance liquid chromatography (HPLC) MS-MS analysis of maraviroc urine concentration were 4.0%, 6.1% and 4.2% at concentrations of 15, 500 and 800 ng ml−1, respectively. The overall inaccuracy of the assay as determined by the same QC samples was 13.4%, 8.6% and 6.5%, respectively. The calibration range was 5–1000 ng ml−1. In study 2, the overall method imprecision values for the analysis of urine QC samples were 12.0%, 4.7% and 5.3% at concentrations of 15, 500 and 800 ng ml−1, respectively. The overall inaccuracy of the assay was 3.8%, 3.5% and 1.7%, respectively. The calibration range was 5–1000 ng ml−1. In study 3, the overall method imprecision values for the analysis of urine QC samples were 7.8%, 5.7% and 4.1% for maraviroc at target concentrations of 15, 300 and 900 ng ml−1, respectively. The mean inaccuracy of the assay ranged from −1.3 to 11.3%. The calibration range was 5–1000 ng ml−1.
Debrisoquine metabolic ratio
In study 2, the debrisoquine metabolic ratio (MR) was determined as an indicator of cytochrome P450 enzyme 2D6 (CYP2D6) inhibition in cohorts 1–5. Subjects received a single oral debrisoquine tablet (10 mg) alone on day −1 and another on day 10, administered immediately after maraviroc or placebo. Urine was collected up to 8 h following the debrisoquine dose, for determination of debrisoquine and its major metabolite, 4-OH-debrisoquine. Debrisoquine and 4-OH-debrisoquine concentrations were determined by use of a HPLC method at Biodynamics Research, Ltd (Walton, UK). The QC samples met the predefined acceptance criteria that at least two-thirds of them were within 15% of their respective nominal concentrations and there was at least one acceptable QC at each concentration.
6β-OH cortisol/cortisol ratio
Urine samples were also taken in study 2 from −12 to 0 h on day −1 and from 12 to 24 h after the morning dose on day 9 to assess the endogenous 6β-OH-cortisol/cortisol ratio as an indicator of CYP3A4 activity. Cohort 3 subjects had urine collected on day 7 instead of day 9. The 6β-OH cortisol/cortisol ratio in urine was determined using a previously validated LC/MS/MS method at BAS Analytics, Ltd (Kenilworth, UK). Quantification was acceptable if the intrabatch precision [coefficient of variation (CV)] was <15% (<20% at the lower limit of quantification) and the mean inaccuracy (bias) was within 15% (20% at the lower limit of quantification). The calibration range was 10–2000 ng ml−1 and 5–1000 ng ml−1 for 6β-OH-cortisol and cortisol, respectively.
Pharmacodynamic assessments
Preclinical pharmacology studies showed maraviroc to have weak affinity for seven transmembrane receptors other than CCR5 (including μ opioid and muscarinic receptors – data on file). Although the affinity was weak, selected pharmacodynamic assessments were carried out to assess any clinical relevance of these activities.
Salivary flow was measured predose and at 2 and 4 h postdose in studies 1 (cohorts 1–5) and 2 and also at 24 h postdose in study 1. Subjects swallowed all saliva and then sucked on a sweet, dispensed from a preweighed (with the sweet) container for 1 min without chewing, crunching or swallowing. They then expelled the sweet along with saliva into the container, which was then reweighed. The difference between the two weights was the salivary flow rate (ml min−1).
Visual near point and pupillometry were measured in study 1 and study 2 (cohorts 1–5). For study 1, these parameters were measured predose and at 2, 4 and 24 h postdose. In study 2, visual near point and pupillometry were evaluated at screening (visual near point only); day −1; predose; and then at 2 and 4 h postdose on days 1, 7 and 12. The measurements were only taken after the morning dose. Cohort 3 had measurements taken on days −1, 1 and 7. Visual near point (cm) was the distance, as measured with the Royal Air Force (RAF accommodation and vergence measurement) rule, in which a standard piece of text had to be moved away from the face before the first line came into focus. Pupil diameter was measured using a commercially available pupillometer.
CCR5 receptor occupancy was also determined in studies 1 and 2. These data are presented separately [12]. Immune effector monitoring was performed in several early studies, the results of which will be discussed in a separate publication [13].
Safety assessments
Subjects were monitored for adverse events (AEs) throughout the studies, had blood and urine samples taken for laboratory safety tests, and underwent comprehensive physical examinations at appropriate intervals in all three studies.
In study 1, supine blood pressure and pulse rate were measured at screening, predose, and at specified times up to 24 h postdose and at follow-up. Subjects were monitored with continuous telemetry from at least 30 min predose to 8 h postdose and had 12-lead electrocardiograms (ECGs) predose and at specified times up to 24 h postdose.
In study 2, safety parameters included extensive supine and standing blood pressure and pulse rate, 12-lead ECG measurements, and lipid profile tests. Cohorts 2, 3 and 5 (300 mg b.i.d. and 600 mg q.d. doses) also had cardiac monitoring using a Recollect™ (Quantum Research UK Ltd, Welwyn Garden City, UK) recording device from at least 30 min predose on day −1 until at least 24 h after the last dose of study medication.
In study 3, subjects had supine and standing blood pressure and pulse rate and 12-lead ECG measurements taken at specified times.
Ethics Approval was obtained for all studies, and all subjects gave written informed consent prior to participation. All studies were conducted in compliance with the ethical principles set forth in the Declaration of Helsinki (1989) and with local laws and regulations relevant to the use of new therapeutic agents in Belgium and the UK.
Pharmacokinetic analysis
The following pharmacokinetic parameters were calculated for maraviroc in at least one of the three studies:
Aet Total urinary recovery of unchanged drug up to time t
Aet(%) Total urinary recovery of unchanged drug expressed as a percentage of the dose excreted
AUC Area under the plasma concentration–time curve from zero to infinity calculated as AUC = AUCt + Ct/kel, where Ct is the last measurable concentration
AUC12 Area under the plasma concentration–time curve from zero to 12 h postdose; calculated using the linear trapezoidal method
AUCt Area under the plasma concentration–time curve from time zero to the time of the last measurable concentration, calculated using the linear trapezoidal method
AUCτ Area under the plasma concentration–time curve over the interdosing interval τ; calculated using the linear trapezoidal method
CLR Renal clearance; calculated as Aet/AUCt
Cmax Maximum observed plasma concentration; taken directly from the plasma concentration–time data
Tmax Time to first occurrence of Cmax; taken directly from the plasma concentration–time data
kel The apparent terminal elimination phase rate constant, calculated by linear regression of the log-linear plasma concentration–time curve. Only those data points judged to describe the terminal log-linear decline were used in the regression
t1/2 The apparent terminal elimination phase half-life, calculated as ln(2)/kel
Pharmacodynamic analysis
The following pharmacodynamic parameters were calculated for maraviroc in at least one of studies 1, 2 and 3:
AUEC0–4/4 Mean change from baseline in average blood pressure and pulse rate over 4 h postdose
AUEC(0–x) The area under the effect curve of blood pressure change from baseline vs. time from 0 to x, where x = 4 or 12 h
Maximum decrease in blood pressure (supine, standing, systolic and diastolic)
Maximum increase in supine and standing pulse rate
Maximum postural change in blood pressure and pulse rate (= standing − supine; systolic and diastolic)
Maximum decrease in salivary flow
Maximum increase in visual near point
Maximum increase in initial, minimum and maximum pupil diameter
Maximum decrease in pupil constriction velocity
% receptor (CCR5) occupancy
Statistical analysis
Sample sizes in studies 1, 2 and 3 were chosen as a compromise between combinations of the following: (i) the need to minimize the exposure of human subjects to a new chemical entity; (ii) the need to provide sufficient safety and tolerability data; and (iii) the need to maintain sufficient residual degrees of freedom to enable useful conclusions to be drawn.
Summary statistics were calculated for all maraviroc pharmacokinetic parameters in all three studies. For study 1, pharmacokinetic parameters AUC, Cmax, Tmax and t1/2, and pharmacodynamic parameters salivary flow, visual near point, and pupillometry end-points were analysed using analysis of variance (anova).
For study 2, pharmacokinetic parameters AUCτ, Cmax, Tmax and t1/2 were analysed using anova. Debrisoquine/4-OH debrisoquine metabolic ratio at baseline vs. on-treatment, and 6β-OH-cortisol/cortisol ratio at baseline vs. on treatment were compared using anova.
For study 3, pharmacokinetic parameters AUC12, Cmax and Tmax were analysed using anova. Day 7 vs. day 1 comparisons for AUC12 estimated the accumulation ratios. Day 14 vs. day 7 comparisons for AUC12 estimated the increase in exposure given the increase in dose.
The standard errors and 95% confidence intervals (CIs) for the comparisons were presented. Log-transformed data were back-transformed to provide the ratio between doses and associated 95% CIs.
QTcF (QT interval corrected for heart rate using Fridericia's formula; QTcF = QT/RR1/3) and QTcP (a study-specific population QTc value derived using the predose and placebo data for all subjects to correct for variations in heart rate; QTcP = QT/RRslope(av), where average slope was 0.267 and 0.28 for studies 1 and 2, respectively) were calculated for at least two of the three studies.
Results
Subjects
There were 24 male subjects in study 1, who ranged in age from 21 to 45 years (mean 28.6 years) and in weight from 61 to 93 kg (mean 76.3 kg). All subjects were White except for one Black subject. There were 72 male subjects in study 2, ranging in age from 18 to 41 years (mean 29.3 years) and in weight from 59 to 93 kg (mean 75.4 kg); 66 (92%) were White, three (4%) Black, and three (4%) were of another race. There were 36 subjects in study 3 (19 male and 17 female) ranging in age from 18 to 44 years (mean 31.3 years). All subjects were White. The weights of the men ranged from 61 to 91 kg and of the women from 50 to 86 kg (total mean 74.6 kg).
Pharmacokinetics
Plasma pharmacokinetics
Maraviroc was absorbed, with Tmax generally occurring between 0.5 and 4 h in all studies (Figures 1 and 2). Maraviroc Tmax did not appear to change with dose or duration of dosing. The pharmacokinetics of maraviroc (Cmax and AUC) after oral dosing were nonproportional across the dose range (1–1200 mg) in study 1 (Table 2), although the degree of nonproportionality seemed to decrease with increasing dose (Figure 3). Following single oral doses, mean t1/2 was approximately 9–14 h in study 1. Mean t1/2 values appeared to increase from single to multiple dosing (study 2); however, estimates are highly dependent on the sampling duration postdose, and hence the apparent difference in t1/2 may be artefactual. Coefficients of variation for Cmax and AUC parameters at doses ≥100 mg were typically within the range of 20–40%, suggesting moderate intersubject variability.
Figure 1.
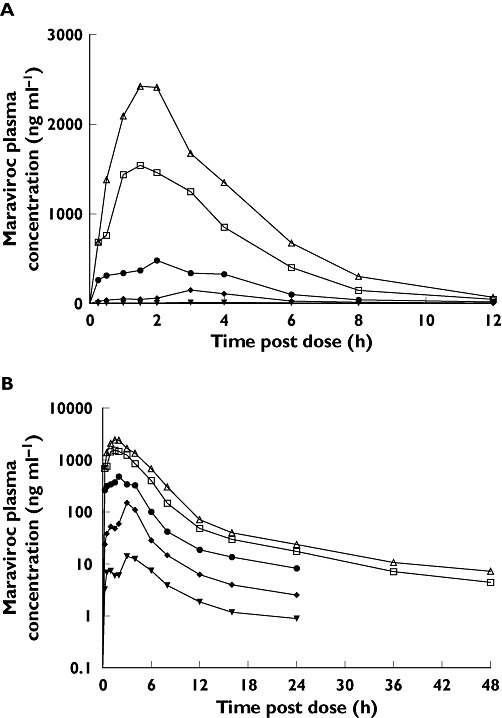
(A) Linear and (B) semi-log plots of mean plasma maraviroc concentrations following single oral doses (study 1). 30 mg (▾); 100 mg (♦); 300 mg ( ); 900 mg (□); 1200 mg (▵)
); 900 mg (□); 1200 mg (▵)
Figure 2.
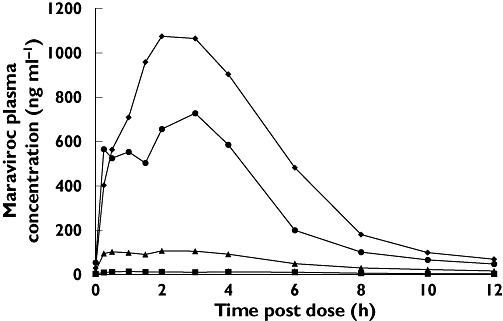
Linear plot of mean plasma maraviroc concentrations measured at steady state on day 12 (study 2, cohorts 1, 2, 4 and 5). 25 mg b.i.d. (▪); 100 mg b.i.d. (▴); 300 mg b.i.d. ( ); 600 mg q.d. (♦)
); 600 mg q.d. (♦)
Table 2.
Mean maraviroc pharmacokinetics for single oral doses ranging from 1 mg to 1200 mg (study 1)
| Maraviroc dose (mg) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | 1 | 3 | 10 | 30 | 100 | 300 | 900 | 1200 |
| AUC (ng ml−1 h)* | † | 2.25‡ | 17.2‡ | 117 | 619 | 2313 | 7358 | 11 432 |
| Mean (CV%) | (31) | (43) | (42) | (16) | (24) | (22) | (26) | |
| Cmax (ng ml−1)* | † | 0.58 | 2.92 | 15.3 | 172 | 621 | 1632 | 2 807 |
| Mean (CV%) | (47) | (74) | (71) | (18) | (33) | (37) | (23) | |
| Tmax (h)§ | † | 2.1 | 2.5 | 2.9 | 3.1 | 1.6 | 1.9 | 1.8 |
| Mean (SD) | (2.4) | (2.2) | (1.7) | (0.6) | (1.1) | (0.8) | (0.4) | |
| t1/2 (h)§ | nc | nc | nc | 8.9 | 9.9 | 10.6 | 11.3 | 12.5 |
| Mean (SD) | (0.9) | (2.0) | (2.7) | (2.5) | (2.5) | |||
| Aet (%)§ | 1.5 | 1.6 | 2.3 | 3.1 | 6.1 | 9.5 | 9.7 | 12 |
| CLr (ml min−1)§ | nc | nc | nc | 10.5 | 10.3 | 12.9 | 11.5 | 12.7 |
Geometric means.
Only two subjects had quantifiable data.
AUCt values presented because AUC could not be calculated.
Arithmetic means. nc, not calculated. 1-, 10-, 100- and 900-mg doses were from cohort A; 3-, 30-, 300- and 1200-mg doses were from cohort B.
Figure 3.
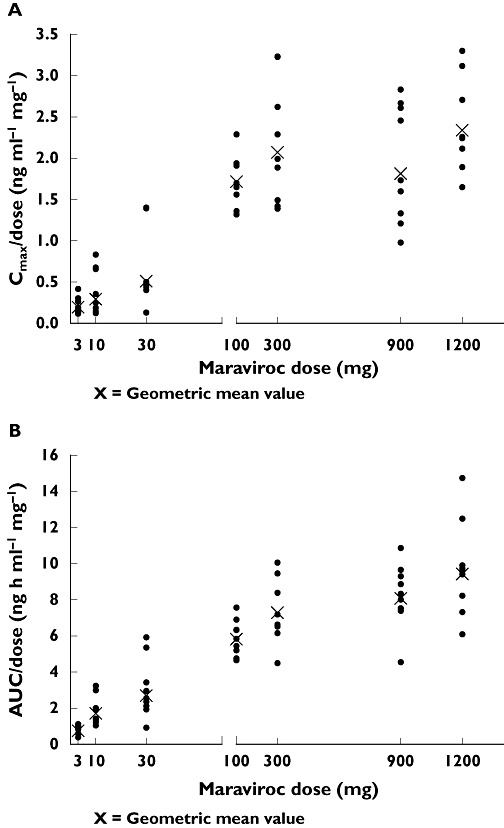
Dose-normalized (A) Cmax and (B) AUC vs. maraviroc dose (study 1)
Dosing in the range of 3–600 mg was examined during the 12-day treatment period in study 2. Pharmacokinetic parameters for all cohorts determined on days 1, 7 and 12 are summarized in Table 3. Visual inspection of trough concentrations in studies 2 and 3 suggests that steady state was achieved for all doses by 7 days of dosing (Figure 4).
Table 3.
Mean maraviroc pharmacokinetics for 12-day multiple-dose study (study 2)
| Maraviroc dose | Day (n) | AUCτ*,† (ng ml−1 h) Mean (CV%) | AUC* (ng ml−1 h) Mean (CV%) | Cmax* (ng ml−1) Mean (CV%) | Tmax‡ (h) Mean (SD) | t1/2‡ (h) Mean (SD) |
|---|---|---|---|---|---|---|
| 3 mg b.i.d. | 1 (5) | nc | nc | 0.67 (52) | 0.9 (0.4) | nc |
| 7 (5) | 6.6 (32) | nc | 1.32 (31) | 1.1 (0.2) | nc | |
| 12 (4) | 4.2 (69) | nc | 0.83 (35) | 0.6 (0.3) | nc | |
| 10 mg b.i.d. | 1 (5) | 11.8 (33) | nc | 2.26 (37) | 1.8 (1.4) | nc |
| 7 (5) | 19.0 (25) | nc | 2.71 (35) | 1.9 (2.3) | nc | |
| 12 (5) | 22.2 (24) | 44.8 (8.6) | 3.33 (41) | 1.3 (1.5) | 15.2 (3.1) | |
| 25 mg b.i.d. | 1 (9)§ | 46.1 (51) | 74.6 (50) | 8.72 (62) | 3.3 (2.3) | 10.8 (2.7) |
| 7 (8) | 92.0 (47) | nc | 18.6 (51) | 3.1 (1.7) | nc | |
| 12 (8)§ | 98.6 (49) | 236 (35) | 16.2 (48) | 3.3 (2.1) | 13.9 (2.2) | |
| 100 mg b.i.d. | 1 (9) | 512 (21) | 579 (21) | 187 (43) | 2.2 (0.9) | 7.76 (0.57) |
| 7 (9) | 636 (26) | nc | 159 (42) | 2.5 (1.4) | nc | |
| 12 (9) | 686 (24) | 1018 (23) | 181 (30) | 2.5 (1.9) | 18.5 (2.5) | |
| 300 mg b.i.d. | 1 (9) | 2157 (47) | 2422 (46) | 538 (46) | 1.6 (1.4) | 8.63 (2.2) |
| 7 (9) | 2641 (43) | nc | 674 (47) | 1.5 (1.3) | nc | |
| 12 (9) | 3609 (32) | 4561 (31) | 854 (32) | 2.6 (1.5) | 16.4 (2.3) | |
| 600 mg q,d, (cohort 3) | 1 (9) | 5877 (20) | 6074 (20) | 1317 (38) | 3.3 (0.9) | 7.74 (0.99) |
| 7 (9) | 6982 (27) | 7650 (26) | 1351 (38) | 2.6 (1.3) | 15.3 (2.2) | |
| 12 (0) | – | – | – | – | – | |
| 600 mg q.d. (cohort 5) | 1 (9) | 5545 (23) | 5717(22) | 1322 (41) | 2.1 (1.4) | 7.84 (1.4) |
| 7 (9) | nc | nc | 1204 (45) | 2.8 (1.8) | nc | |
| 12 (9) | 6440 (12) | 7177 (13) | 1361 (20) | 2.3 (1.1) | 17.2 (3.9) |
Geometric mean.
τ = 12 h for b.i.d. dosing and 24 h for q.d. dosing.
Arithmetic mean.
AUC and t1/2 were only calculated for five subjects. nc, not calculated.
Figure 4.
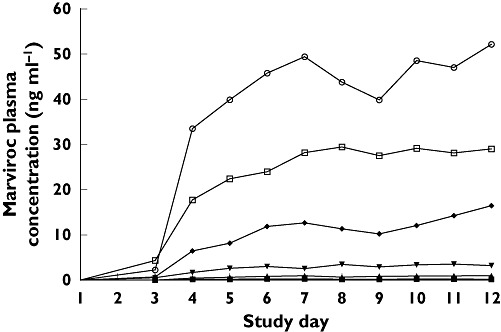
Mean trough maraviroc plasma concentrations by treatment group (study 2). 3 mg b.i.d. (▪); 10 mg b.i.d. (▴); 25 mg b.i.d. (▾); 100 mg b.i.d. (♦); 300 mg b.i.d. (○); 600 mg q.d. (□)
Maraviroc accumulated in plasma upon multiple dosing in study 2, with mean accumulation (AUCτ) ratios (day 12/day 1) on day 12 ranging from 134% to 206% following 10–300-mg b.i.d. dosing and 123% following 600-mg q.d. dosing (Table 4). The pharmacokinetic parameters for study 3 are summarized in Table 5. The mean accumulation ratios were 1.21 and 1.20 after 300 and 600 mg b.i.d. for 7 days, respectively, whereas the ratios for Cmax were 1.14 and 1.07, respectively. The mean accumulation ratio was 1.09 after 900 mg q.d. for 7 days with no change in Cmax. A doubling of the dose from 300 to 600 mg b.i.d. increased AUC12 and Cmax by 2.4-fold and 2.2-fold, respectively (Table 6).
Table 4.
Statistical analysis of AUC12 and Cmax (study 2)
| Ratio (%) of means and 95% Cis | |||
|---|---|---|---|
| Maraviroc dose | Comparison* | AUCt | Cmax |
| 3 mg b.i.d. | Day 7/day 1 | nd | 197 (125, 311) |
| Day 12/day 1 | nd | 129 (79, 212) | |
| 10 mg b.i.d. | Day 7/day 1 | 162 (119, 219) | 120 (76, 189) |
| Day 12/day 1 | 189 (139, 255) | 148 (94, 232) | |
| 25 mg b.i.d. | Day 7/day 1 | 193 (152, 245) | 208 (146, 299) |
| Day 12/day 1 | 206 (162, 262) | 181 (126, 259) | |
| 100 mg b.i.d. | Day 7/day 1 | 124 (99, 155) | 85 (61, 120) |
| Day 12/day 1 | 134 (107, 168) | 97 (69, 136) | |
| 300 mg b.i.d. | Day 7/day 1 | 123 (98, 154) | 125 (89, 176) |
| Day 12/day 1 | 167 (134, 210) | 159 (113, 223) | |
| 600 mg q.d. (cohort 3) | Day 7/day 1 | 119 (95, 149) | 103 (73, 144) |
| Day 12/day 1 | nd | nd | |
| 600 mg q.d. (cohort 5) | Day 7/day 1 | nd | 91 (65, 128) |
| Day 12/day 1 | 123 (98, 154) | 103 (73, 145) | |
Day 7 = 5 days of multiple dosing. nd, no data.
Table 5.
Mean maraviroc pharmacokinetics (study 3)
| Maraviroc dose | Day | AUC12* (ng ml−1 h) Mean (CV %) | Cmax* (ng ml−1) Mean (CV %) | Tmax† (h) Mean (SD) |
|---|---|---|---|---|
| 300 mg b.i.d. | 1 | 1 896 (41) | 770 (40) | 1.8 (0.5) |
| 300 mg b.i.d. | 7 | 2 102 (27) | 807 (38) | 2.0 (0.0) |
| 600 mg b.i.d. | 14‡ | 4 980 (26) | 1808 (34) | 2.0 (0.0) |
| 600 mg b.i.d. | 1 | 3 232 (16) | 1096 (19) | 2.0 (0.0) |
| 600 mg b.i.d. | 7 | 3 872 (20) | 1177 (24) | 2.3 (0.71) |
| 900 mg b.i.d. | 14‡ | 5 884 (21) | 1629 (21) | 2.3 (0.71) |
| 900 mg q.d. | 1 | 5 993 (24) | 1993 (25) | 1.7 (0.56) |
| 900 mg q.d. | 7 | 6 549 (23) | 1960 (31) | 2.0 (0.0) |
| 1200 mg q.d. | 14‡ | 10 394 (21) | 2987 (31) | 1.8 (0.5) |
Unadjusted geometric mean.
Unadjusted arithmetic mean.
Parameters on day 1 and day 7 were measured after the lower dosing level and on day 14 after 7 days of the higher dosing level.
Table 6.
Statistical analysis of AUC12, Cmax and Tmax (study 3)
| Maraviroc dose | Comparison | AUC12 ratio % (95% CI) | Cmax ratio % (95% CI) | Tmax difference (95% CI) |
|---|---|---|---|---|
| 300 mg b.i.d. | Day 7/day 1* | 121 (106, 140) | 114 (93.2, 140) | 0.19 (−0.26, 0.63) |
| 600 mg b.i.d. | Day 14/day 7† | 237 (210, 267) | 224 (181, 277) | 0.00 (−0.22, 0.22) |
| 600 mg b.i.d. | Day 7/day 1* | 120 (104, 138) | 107 (87.5, 132) | 0.25 (−0.19, 0.69) |
| 900 mg b.i.d. | Day 14/day 7† | 152 (135, 171) | 138 (112, 171) | 0.00 (−0.22, 0.22) |
| 900 mg q.d. | Day 7/day 1* | 109 (95.7, 125) | 98.3 (81.1, 119) | 0.28 (−0.14, 0.70) |
| 1200 mg q.d. | Day 14/day 7† | 159 (142, 178) | 152 (125, 186) | −0.17 (−0.38, 0.04) |
The day 7/day 1 ratio shows the accumulation for the first dose with time.
The day 14/day 7 ratio shows the increase in exposure following the increase in dose, at steady state. CI, confidence interval.
Urine pharmacokinetics
Study 1 demonstrated that although the percentage of unchanged drug recovered in urine increased with dose (1.5% following 1-mg single dose to 12% following 1200-mg single dose), renal clearance (CLR) remained constant across the dosing range (Table 2) and appeared similar following single and multiple dosing (study 2, data not shown), indicating no change in renal clearance with dose or time.
Debrisoquine and cortisol MR
There was no apparent difference in debrisoquine MR with or without maraviroc at doses up to and including 300 mg b.i.d. (Table 7). At 600 mg q.d., there was an increase in MR on treatment compared with baseline, suggesting some potential of maraviroc to inhibit weakly CYP2D6 metabolism at this dose; however, no subjects were converted to CYP2D6 poor metabolizers (i.e. no debrisoquine MR >12.6). The 6β-OH-cortisol/cortisol ratios were similar in the presence and absence of maraviroc at all doses, indicating that maraviroc (up to 600 mg q.d.) does not inhibit or induce CYP3A4.
Table 7.
Maraviroc effects on debrisoquine and cortisol metabolism (study 2)
| Maraviroc dose | Assessment | Debrisoquine/4-OH-debrisoquine ratio | 6β-OH-cortisol/cortisol ratio |
|---|---|---|---|
| Placebo | Baseline | 0.96 | 4.90 |
| On treatment | 0.85 | 4.55 | |
| 25 mg b.i.d. | Baseline | 0.86 | 7.01 |
| On treatment | 0.72 | 6.03 | |
| 100 mg b.i.d. | Baseline | 0.63 | 4.09 |
| On treatment | 0.65 | 3.75 | |
| 300 mg b.i.d. | Baseline | 1.52 | 3.80 |
| On treatment | 1.13 | 3.90 | |
| 600 mg q.d. (cohort 3)* | Baseline | nd | 4.31 |
| On treatment | nd | 5.48 | |
| 600 mg q.d. (cohort 5) | Baseline | 1.23 | 3.99 |
| On treatment | 4.11 | 4.78 |
Results for cohort 3 are from day 7 instead of day 9. nd, no data.
Pharmacodynamics
There were no clinically relevant changes in pharmacodynamic parameters, including salivary flow, visual near point, and pupillometry, assessed at intervals up to 24 h postdose in studies 1 and 2 (data not shown).
Safety and tolerability
There were no serious AEs reported in these three studies. Most AEs were mild or moderate in severity and resolved without intervention.
In study 1, there were no discontinuations or dose reductions due to AEs and no treatment-related AEs reported at doses <100 mg. The only treatment-related AEs were abdominal pain (1/9) with 100 mg and asthenia (1/9) and headache (1/9) with 300 mg. At the higher doses, the most common treatment-related AEs were asthenia (five subjects each in the 900- and 1200-mg groups), postural hypotension reported by four subjects in the 1200-mg group, and headache reported by three subjects in the 1200-mg group (Table 8). Postural hypotension was considered the dose-limiting AE in this study. There were no clinically significant laboratory value abnormalities or discontinuations due to abnormal laboratory results. There were no subjects with a QTc ≥450 ms or an increase in QTc from baseline of ≥60 ms. There was no evidence of changes in supine blood pressure, pulse rate, or QTc with doses of maraviroc up to 900 mg. Mean supine pulse rate did increase by 9.7 bpm at 1 h after administration of the 1200-mg dose. In addition, there was evidence of a potential QTc effect at 1200 mg, with a mean QTcF increase from baseline of 10.7 ms at 2 h postdose. When the population-specific QTc (QTcP) was analysed, the mean increase from baseline was 7.8 ms. The difference in these parameters is indicative of the sensitivity of QTc correction factors to changes in pulse rate. The effect of maraviroc on QTc was further investigated in a thorough QT study [14].
Table 8.
Treatment-related adverse events in study 1
| Cohort A | Cohort B | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo n = 12 | 1 mg n = 9 | 10 mg n = 9 | 100 mg n = 9 | 900 mg n = 9 | Placebo n = 12 | 3 mg n = 9 | 30 mg n = 9 | 300 mg n = 9 | 1200 mg n = 9 | |
| Number of subjects with >1 Adverse event* | 2 | 0 | 0 | 1 | 6 | 2 | 0 | 0 | 1 | 8 |
| Abdominal pain | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 |
| Asthenia | 2 | 0 | 0 | 0 | 5 | 1 | 0 | 0 | 1 | 5 |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 3 |
| Hot flushes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Postural hypotension | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 |
| Dry mouth | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Dizziness | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 |
| Abnormal vision | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 |
Table shows adverse events that were reported by more than one subject in any treatment group.
In study 2, there were four discontinuations due to treatment-related AEs: three in the 600-mg dose group (cohort 3; two after maraviroc 600 mg q.d., one after placebo) due to severe postural hypotension, and one in the 3-mg dose group (cohort 6) due to elevated hepatic transaminases. The entire cohort 3 (600 mg q.d./placebo) was discontinued on day 7 owing to severe postural hypotension experienced by the three subjects cited above (2/9 on 600 mg q.d. maraviroc, 1/3 on placebo). This dose was well tolerated when given to a second group of subjects (cohort 5), with only one subject experiencing a mild episode of postural hypotension. Table 9 summarizes treatment-related AEs for study 2.
Table 9.
Treatment-related adverse events in study 2
| Maraviroc dose group | ||||||||
|---|---|---|---|---|---|---|---|---|
| Placebo n = 17 | 3 mg b.i.d. n = 5 | 10 mg b.i.d. n = 5 | 25 mg b.i.d. n = 9 | 100 mg b.i.d. n = 9 | 300 mg b.i.d. n = 9 | 600 mg q.d.*n = 9 | 600 mg q.d.†n = 9 | |
| Number of subjects with >1 Adverse event‡ | 12 | 4 | 0 | 2 | 3 | 5 | 8 | 4 |
| Asthenia | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Headache | 5 | 1 | 0 | 1 | 2 | 1 | 3 | 1 |
| Postural hypotension | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 1 |
| Diarrhoea | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Flatulence | 4 | 0 | 0 | 0 | 0 | 3 | 5 | 0 |
| Nausea | 2 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| Dizziness | 3 | 2 | 0 | 0 | 0 | 0 | 4 | 2 |
| Somnolence | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| Abnormal vision | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 |
Cohort 3.
Cohort 5.
Table shows adverse events that were reported by more than one subject in any treatment group.
As mentioned, there was a single discontinuation in the 3-mg dose group due to increased hepatic transaminases. This subject also showed mild elevation of alanine aminotransferase at screening (66 IU l−1), and the observed increase to more than three times the upper limit of normal (ULN) occurred concurrent with an episode of food poisoning. There were eight subjects out of the total of 72 who exhibited transaminase levels above the ULN during the study. These elevations were mild and showed no dose relationship. With the exception of one subject in the 100-mg b.i.d. group, whose gamma glutamyl transpeptidase levels rose to more than three times ULN during the study, no other subjects who had normal serum liver enzyme levels at baseline experienced increases three or more times times ULN, and there was no associated increase in bilirubin or alkaline phosphatase in any subject with increased serum liver enzyme markers.
There were apparent dose-related increases in total cholesterol and low-density lipoprotein (LDL)-cholesterol compared with baseline, although it should also be noted that the median baseline values appear to be higher in the placebo subjects than in those receiving maraviroc. Table 10 shows the median baseline and median change from baseline to last observation in total cholesterol, high-density lipoprotein-cholesterol, LDL-cholesterol and triglycerides for each maraviroc dose and placebo.
Table 10.
Median baseline and changes from baseline in lipid parameters (study 2)
| Total cholesterol (median; mg dl−1) | HDL-cholesterol (median; mg dl−1) | LDL-cholesterol (median; mg dl−1) | Triglycerides (median; mg dl−1) | |||||
|---|---|---|---|---|---|---|---|---|
| Dose | Baseline | Change from baseline | Baseline | Change from baseline | Baseline | Change from baseline | Baseline | Change from baseline |
| 25 mg b.i.d. | 160 | 4 | 44 | 0 | 99 | 7 | 84 | −11 |
| 100 mg b.i.d. | 169 | 1 | 55 | −7 | 96 | 7 | 77 | −10 |
| 300 mg b.i.d. | 172 | 10 | 53 | −4 | 91 | 15 | 95 | −1 |
| 600 mg q.d. (cohort 3) | 159 | 19 | 42 | 4 | 98 | 17 | 86 | −8 |
| 600 mg q.d. (cohort 5) | 149 | 14 | 49 | 2 | 80 | 16 | 89 | −8 |
| Placebo | 200 | −3 | 45 | −1 | 131 | −6 | 104 | 9 |
Compared with placebo, there were dose-related increases in the mean maximum decrease from baseline (over 0–4 h) for standing systolic blood pressure for 300 mg b.i.d. and 600 mg q.d. and for standing diastolic blood pressure for 600 mg q.d. (Table 11). These trends were not observed for supine blood pressure. There was an increase in the maximum change from baseline for standing pulse rate (bpm) at 600 mg q.d. compared with placebo. There was no obvious clinically relevant treatment-related effect on the QTc interval over the dose range and duration studied and there was no clear relationship between maraviroc plasma concentration and QTc.
Table 11.
Mean maximum decreases from baseline for standing blood pressure and mean maximum increases from baseline for standing pulse rate over 0–4 h postdose (study 2)
| Dose group | Day | SBP (mmHg) | DBP (mmHg) | PR (bpm) |
|---|---|---|---|---|
| 3 mg b.i.d. | 1 | −4.14 | −8.90 | 1.90 |
| 7 | −9.94 | −5.70 | 2.10 | |
| 12 | −6.34 | −2.90 | 18.90 | |
| 10 mg b.i.d. | 1 | −6.23 | −6.37 | 5.57 |
| 7 | −5.23 | −4.37 | 6.17 | |
| 12 | −10.23 | −8.17 | 12.97 | |
| 25 mg b.i.d. | 1 | −9.85 | −6.35 | −2.47 |
| 7 | −6.65 | −4.02 | 1.75 | |
| 12 | −7.02 | −3.77 | 6.00 | |
| 100 mg b.i.d. | 1 | −8.72 | −5.09 | 7.63 |
| 7 | −8.39 | −0.54 | 7.74 | |
| 12 | −5.28 | −3.09 | 9.96 | |
| 300 mg b.i.d. | 1 | −12.75 | −7.84 | 5.98 |
| 7 | −17.42 | −7.84 | 10.20 | |
| 12 | −16.08 | −7.40 | 7.65 | |
| 600 mg q.d. (cohort 3) | 1 | −12.97 | −8.92 | 12.56 |
| 7 | −25.64 | −17.36 | 21.23 | |
| 12 | nd | nd | nd | |
| 600 mg q.d. (cohort 5) | 1 | −13.12 | −11.10 | 11.35 |
| 7 | −18.90 | −12.88 | 19.91 | |
| 12 | −15.34 | −14.10 | 13.91 | |
| Placebo | 1 | −11.27 | −5.59 | 5.20 |
| 7 | −11.86 | −6.36 | 9.38 | |
| 12 | −9.76 | −5.83 | 11.71 |
DBP, diastolic blood pressure; nd, not determined; PR, pulse rate; SBP, systolic blood pressure.
In study 3, two subjects discontinued treatment due to AEs, one considered unrelated to treatment and one due to a rash after taking placebo for 3 days, which was considered treatment-related. The most common treatment-related AE was dizziness (Table 12). One subject experienced severe dizziness after a 600-mg b.i.d. dose (cohort 2), and three subjects in the 900–1200-mg q.d. cohort experienced severe postural hypotension. There were no discontinuations due to abnormal laboratory test results, and no clear pattern in changes in laboratory tests at any dose. There was no significant effect on mean supine systolic or diastolic blood pressure. There were decreases in mean standing blood pressure and increases in mean pulse rate at all doses, and the magnitude of the effect increased with dose (Table 13). There were no clinically significant changes in ECG parameters.
Table 12.
Treatment-related adverse events in study 3
| Cohort 1 | Cohort 2 | Cohort 3 | |||||
|---|---|---|---|---|---|---|---|
| Placebo n = 9 | 300 mg b.i.d. n = 9 | 600 mg b.i.d. n = 8 | 600 mg b.i.d. n = 9 | 900 mg b.i.d. n = 8 | 900 mg b.i.d. n = 9 | 1200 mg q.d. n = 9 | |
| Number of subjects with >1 Adverse event* | 9 | 9 | 6 | 8 | 8 | 8 | 8 |
| Abdominal pain | 2 | 1 | 1 | 2 | 1 | 0 | 2 |
| Asthenia | 1 | 0 | 0 | 4 | 1 | 6 | 4 |
| Headache | 3 | 2 | 1 | 3 | 5 | 3 | 2 |
| Postural hypotension | 0 | 0 | 0 | 0 | 0 | 5 | 6 |
| Dry mouth | 0 | 0 | 2 | 0 | 1 | 2 | 3 |
| Flatulence | 1 | 2 | 0 | 1 | 0 | 1 | 1 |
| Nausea | 1 | 2 | 2 | 4 | 3 | 4 | 2 |
| Hyperlipidaemia | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
| Dizziness | 2 | 2 | 2 | 6 | 7 | 3 | 3 |
| Somnolence | 0 | 0 | 0 | 4 | 6 | 0 | 0 |
| Cough increased | 1 | 1 | 0 | 1 | 2 | 0 | 0 |
| Epistaxis | 0 | 0 | 0 | 2 | 4 | 0 | 1 |
| Pharyngitis | 1 | 3 | 2 | 0 | 0 | 0 | 0 |
| Rhinitis | 0 | 1 | 3 | 2 | 4 | 2 | 3 |
| Rash | 2 | 0 | 1 | 0 | 0 | 0 | 1 |
| Abnormal vision | 0 | 0 | 1 | 2 | 3 | 3 | 0 |
| Conjunctivitis† | 0 | 0 | 0 | 0 | 5 | 4 | 5 |
| Eye pain | 1 | 0 | 0 | 1 | 2 | 1 | 1 |
Table shows adverse events that were reported by more than one subject in any treatment group.
The investigator term for all the cases of conjunctivitis was bloodshot eyes.
Table 13.
Mean maximum decreases from baseline for blood pressure and mean maximum increases from baseline for pulse rate (study 3)
| Standing | Supine | ||||||
|---|---|---|---|---|---|---|---|
| Dose | Days | SBP (mmHg) | DBP (mmHg) | PR (bpm) | SBP (mmHg) | DBP (mmHg) | PR (bpm) |
| Placebo | 1–14 | −21.0 | −14.8 | 21.9 | −16.6 | −14.2 | 21.5 |
| 300 mg b.i.d. | 1–7 | −22.1 | −19.8 | 33.2 | −13.3 | −14.8 | 24.5 |
| 600 mg b.i.d. | 8–14 | −21.3 | −17.0 | 32.3 | −12.6 | −14.3 | 25.0 |
| 600 mg b.i.d. | 1–7 | −20.1 | −13.5 | 27.9 | −17.4 | −13.0 | 14.6 |
| 900 mg b.i.d. | 8–14 | −23.6 | −17.0 | 25.3 | −17.3 | −15.5 | 9.1 |
| 900 mg q.d. | 1–7 | −32.0 | −22.1 | 47.3 | −12.7 | −9.8 | 17.0 |
| 1200 mg q.d. | 8–14 | −34.6 | −21.4 | 49.0 | −15.8 | −13.3 | 22.8 |
DBP, diastolic blood pressure; PR, pulse rate; SBP, systolic blood pressure.
Discussion
Because of the frequent emergence of HIV genetic variants that are resistant to existing therapies, there is a compelling need to develop new drugs that target different phases of the virus' life cycle. Orally bioavailable inhibitors of HIV entry represent a potential major advance in the treatment of HIV-infected patients. Blocking the CCR5 receptor prevents entry of HIV into host cells and therefore should reduce viral load. Pharmacological studies have shown that maraviroc is highly specific for CCR5 [11]. Although maraviroc exhibited some weak affinity for the muscarinic and opioid receptors in the rat brain, appreciable binding to these receptors occurred only at concentrations near 10 μM (data on file), roughly 2000-fold higher than the mean antiviral IC90[7] and approximately 25-fold higher than the mean free Cmax at 300 mg b.i.d. observed in studies 2 and 3, predicting negligible biological significance for these interactions. The results of pharmacodynamic assessments, designed to reveal any interaction with these cell surface receptors, showed no physiological changes following maraviroc dosing in these clinical studies.
Assessment of maraviroc pharmacokinetics and tolerability in human subjects is critical for determining safety, appropriate dosing, and feasibility of further efficacy studies. Pharmacokinetics and safety of single and multiple oral doses up to 900 mg b.i.d. and 1200 mg q.d. are described here, and results indicate that maraviroc is absorbed with a Tmax occurring within 4 h of dosing. Steady-state plasma concentrations were achieved by 7 days of dosing at all doses. Up to 12% of the maraviroc dose was excreted unchanged in the urine, and renal clearance was unaffected by dose or time.
The pharmacokinetics of maraviroc appeared to be nonproportional across the dose range studied. The degree of nonproportionality appeared greatest at doses ≤100 mg. This nonproportionality has been further studied in a population pharmacokinetic meta-analysis of maraviroc in healthy subjects and HIV patients [15]. Maraviroc is a substrate for the transporter P-glycoprotein (Pgp) which is thought to limit the absorption of many compounds [8]. It is hypothesized that at low intestinal concentrations of maraviroc, Pgp mediates efflux of maraviroc back into the lumen of the small intestine, thereby limiting the amount of maraviroc that is absorbed, resulting in reduced bioavailability and exposure. As the dose of maraviroc increases and concentrations in the gut lumen increase, Pgp efflux may become saturated and reduce the ability of Pgp to limit bioavailability. A similar mechanism has been suggested for other compounds exhibiting nonproportional pharmacokinetics [16].
Maraviroc's effects on the activity of CYP2D6 and CYP3A4 were evaluated to assess the potential for adverse drug–drug interactions. Maraviroc did not appear to affect debrisoquine MR (an indicator of CYP2D6 activity) at doses up to 300 mg b.i.d. At 600 mg QD, the mean debrisoquine MR was higher in the presence of maraviroc, suggesting a potential for weak CYP2D6 inhibition at this dose. However, it should be noted that none of the subjects' MRs rose above 12.6, a value above which subjects are considered to be CYP2D6 poor metabolizers. The 6β-OH-cortisol/cortisol ratio was unaffected by the presence of maraviroc at all doses studied (up to 600 mg q.d.), suggesting no induction or inhibition of CYP3A4. Another study examining the influence of maraviroc on other drugs has revealed no clinically relevant effect on exposure of midazolam [17], a probe CYP3A4 substrate, consistent with a lack of influence on the activity of this major drug-metabolizing enzyme.
Maraviroc was well tolerated at single oral doses up to 900 mg, with asthenia and headache being the most common AEs and the majority of AEs being described as mild to moderate in nature. Supine blood pressure and pulse rate were not affected at doses up to 900 mg. Multiple doses of maraviroc were well tolerated up to 300 mg b.i.d., with few AEs. The dose-limiting adverse event in study 1 was postural hypotension (4/9 subjects at 1200 mg). In study 2, cohort 3, postural hypotension was reported on day 7 by three subjects (2/9 on 600 mg q.d. maraviroc and 1/3 on placebo). When a different group of subjects in study 2 (cohort 5) were administered the same 600-mg q.d. dosing regimen, only one subject reported mild postural hypotension. There were no reports of postural hypotension in the 600–900-mg b.i.d. cohort in study 3, although dizziness was reported. The reporting of postural hypotension in study 3 was confined to the 900–1200-mg q.d. cohort. It should be noted that in all of the studies, these events generally occurred within the first 4 h after dosing. Whilst this is also the time period associated with peak maraviroc concentrations, it is also the period over which subjects are generally fasted and relatively inactive, except for standing for postural blood pressure assessments. The relevance of the incidence of these events under these circumstances to a real-life situation is currently unclear.
The incidence of AEs (including postural hypotension) at doses up to and including 300 mg b.i.d. maraviroc was similar to placebo. The results of study 3 suggest there was no obvious toleration benefit of titrating to a higher dose of maraviroc.
No clinically significant changes in QTc were observed for single oral maraviroc doses up to 900 mg. There was evidence of a potential QTc effect after dosing with 1200 mg single-dose maraviroc; however, the magnitude of the effect was highly dependent on the correction factor used. No clinically significant changes in QTc were observed in study 3, which investigated doses up to 1200 mg q.d. for 7 days. No subject in any study had a QTc value of ≥450 ms or an increase from baseline of ≥60 ms. The importance of appropriately correcting QTc for changes in heart rate is well recognized [18], The QTcP values calculated in studies 1 and 2 employed population-specific correction factors to increase the accuracy of the QTc measurement. Another Phase I maraviroc study specifically addressing potential effects on QT interval [14] has shown that maraviroc does not significantly impact QTc at clinically relevant doses.
Hepatic safety and monitoring of liver enzymes is a critical consideration in the development of any new drug. Although several subjects showed increases in hepatic transaminases during study 2, there was no clear relationship with dose, and most cases were not significantly above the ULN. None of the increases in transaminases was associated with increases in bilirubin. Small apparently dose-related increases in total cholesterol and LDL-cholesterol were seen in study 2. However the clinical relevance of these changes in an inpatient setting where subjects have a change in diet and lifestyle is unclear. Subsequent to this study, a 28-day study examining the safety and toleration of maraviroc 100-mg and 300-mg b.i.d. doses in healthy volunteers confirmed that these doses were well tolerated, with no trend changes in liver enzymes, lipid profile, blood pressure, pulse rate or ECG parameters [19].
In summary, the pharmacokinetic and safety profile of maraviroc ascertained in these three studies supported its further clinical development.
Competing interests
Samantha Abel, Elna van der Ryst, Maria C. Rosario, Caroline E. Ridgway, Christine G. Medhurst, Richard J. Taylor-Worth, and Gary J. Muirhead were employed by Pfizer Ltd at the time of this research.
These studies were sponsored by Pfizer. Editorial assistance was provided by Dylan Harris, PhD and Janet E. Matsuura, PhD at Complete Healthcare Communications, Inc., and was funded by Pfizer Inc., New York, NY, USA.
REFERENCES
- 1.Joint United Nations Programme on HIV/AIDS. AIDS Epidemic Update: Special Report on HIV/AIDS. Geneva, Switzerland: UNAIDS/WHO; 2006. [Google Scholar]
- 2.Preston BD, Poiesz BJ, Loeb LA. Fidelity of HIV-1 reverse transcriptase. Science. 1988;242:1168–71. doi: 10.1126/science.2460924. [DOI] [PubMed] [Google Scholar]
- 3.Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–8. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, Kunstman K, Erickson D, Dragon E, Landau NR, Phair J, Ho DD, Koup RA. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–3. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 5.Walli R, Reinhart B, Luckow B, Lederer E, Loch O, Malo A, Wank R, Schlondorff D, Goebel FD. HIV-1-infected long-term slow progressors heterozygous for delta32-CCR5 show significantly lower plasma viral load than wild-type slow progressors. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:229–33. doi: 10.1097/00042560-199807010-00005. [DOI] [PubMed] [Google Scholar]
- 6.Hunt PW, Harrigan PR, Huang W, Bates M, Williamson DW, McCune JM, Price RW, Spundich SS, Lampiris H, Hoh R, Leigler T, Martin JN, Deeks SG. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis. 2006;194:926–30. doi: 10.1086/507312. [DOI] [PubMed] [Google Scholar]
- 7.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427 857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–32. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman AN, Smith DA. Species differences in the disposition of the CCR5 antagonist, UK-427 857, a new potential treatment for HIV. Drug Metab Dispos. 2005;33:587–95. doi: 10.1124/dmd.104.002626. [DOI] [PubMed] [Google Scholar]
- 9.Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TH, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 10.Lalezari J, Goodrich J, DeJesus E, Lampiris H, Gulick R, Saag M, Ridgway C, McHale M, van der Ryst E, Mayer H. Efficacy and safety of maraviroc (MVC) plus optimized background therapy (OBT) in viremic antiretroviral treatment experienced patients infected with CCR5-tropic HIV-1: 24-week results of a phase 2b/3 study in the USA and Canada. 14th Conference on Retroviruses and Opportunistic Infections; 2007 February 25–28; Los Angeles, CA. [Google Scholar]
- 11.Nelson M, Fätkenheuer G, Konourina I, Lazzarin A, Clumeck N, Horban A, Tawadrous M, Sullivan J, Mayer H, van der Ryst E. Efficacy and safety of maraviroc (MVC) plus optimized background therapy (OBT) in viremic, antiretroviral treatment experienced patients infected with CCR5-tropic (R5) HIV-1 in Europe, Australia and North America: 24-week results. 14th Conference on Retroviruses and Opportunistic Infections; 2007 February 25–28; Los Angeles, CA. [Google Scholar]
- 12.Rosario MC, Jacqmin P, Dorr P, James I, Jenkins TM, Abel S, van der Ryst E. Population pharmacokinetic-pharmacodynamic analysis of CCR5 receptor occupancy by maraviroc in healthy subjects and HIV-positive patients. Br J Clin Pharmacol. 2008;65(1):86–94. doi: 10.1111/j.1365-2125.2008.03140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayoub A, Van der Ryst E, Turner K, McHale M. A review of the markers of immune function during the maraviroc phase I and IIa studies (abstract 509). 14th Conference on Retroviruses and Opportunistic Infections; 2007 February 25–28; Los Angeles, CA. [Google Scholar]
- 14.Davis J, Hackman F, Layton G, Higgins T, Sudworth D, Weissgerber G. The effect of single doses of maraviroc on the QT/QTc interval in healthy subjects. Br J Clin Pharmacol. 2008;65(1):68–75. doi: 10.1111/j.1365-2125.2008.03138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan PLS, Weatherley B, McFadyen L. A population pharmacokinetic meta-analysis of maraviroc in healthy volunteers and asymptomatic HIV-infected subjects. Br J Clin Pharmacol. 2008;65(1):76–85. doi: 10.1111/j.1365-2125.2008.03139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abel S, Beaumont KC, Crespi CL, Eve MD, Fox L, Hyland R, Jones BC, Muirhead GJ, Smith DA, Venn RF, Walker DK. Potential role for P-glycoprotein in the non-proportional pharmacokinetics of UK-343 664 in man. Xenobiotica. 2001;31:665–76. doi: 10.1080/00498250110052779. [DOI] [PubMed] [Google Scholar]
- 17.Abel S, Russell D, Whitlock LA, Ridgway CE, Muirhead GJ. Effect of maraviroc on the pharmacokinetics of midazolam, lamivudine/zidovudine, and ethinyloestradiol/levonorgestrel in healthy volunteers. Br J Clin Pharmacol. 2008;65(1):19–26. doi: 10.1111/j.1365-2125.2008.03132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malik M, Farbom P, Batchvarov V, Hnatkova K, Camm AJ. Relation between QT and RR intervals is highly individual among healthy subjects: implications for heart rate correction of the QT interval. Heart. 2002;87:220–8. doi: 10.1136/heart.87.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell D, Bakhtyari A, Jazrawi RP, Whitlock L, Ridgway C, McHale M, et al. Multiple dose study to investigate the safety of UK-427 857 (100 mg or 300 mg) BID for 28 days in healthy males and females. 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2003 September 14–17; Chicago, IL. [Google Scholar]