

Abstract
P. gingivalis, an opportunistic pathogen in periodontal disease, can reside within the epithelial cells that line the gingival crevice. A proteomic analysis revealed that infection of gingival epithelial cells with P. gingivalis induces broadly based changes in the level and phosphorylation status of proteins that exert multi-level control on the eukaryotic cell cycle. Pathways that were impacted by P. gingivalis included those involving cyclins, p53 and PI3K. The predicted infection-dependent phenotype was confirmed by cytofluorimetry that showed an enhanced proliferation rate of gingival epithelial cells infected with P. gingivalis associated with accelerated progression through the S-phase. Elevated cell proliferation was dependent on the presence of the long fimbriae of P. gingivalis. The ability of P. gingivalis, a common inhabitant of the subgingival crevice, to accelerate cell cycling could have biological consequences for barrier and signaling functions, and for physiological status, of the gingival epithelium.
Keywords: microbe-cell interactions, periodontal disease, cell proliferation
1. Introduction
The cell cycle of mammalian cells is a fundamental aspect of their biological status and uncontrolled proliferation leads to tumorigenesis [1]. In addition, immune responses, barrier function and cellular differentiation can all vary with cell cycle progression. A complex network of interconnected regulatory pathways controls the cell cycle and ensures that each step is performed in the proper sequence. These regulatory pathways involve highly choreographed protein kinase/phosphatase and proteolysis events [2–4]. Microbial pathogens that initiate infections on mucosal membranes have devised means to manipulate host cells in order to facilitate survival and persistence [5]. In many instances bacteria can direct their own uptake by the epithelial cells, and survive intracellularly. This process can involve cytoskeletal remodeling, and reprogramming of signal transduction pathways [6, 7]. More recently it has become apparent that bacterial interference with host cell physiology can also impact the host cell cycle. Bacterial effectors, termed cyclomodulins, have been identified that can either activate or inhibit cell cycle progression [8]. Examples of these include cytolethal distending toxin, produced by a number of organisms, that causes arrest at the G2/M transition; and the CagA protein of H. pylori that interacts with multiple signaling proteins to induce cell proliferation [9].
Porphyromonas gingivalis is a gram-negative anaerobe that is associated with severe and chronic manifestations of periodontal disease, one of the most common bacterial diseases of humans [10]. The organism is intensely invasive and can enter primary cultures of gingival epithelial cells (GECs) rapidly, and persist in high numbers in the perinuclear area [11]. Invasion requires epithelial cell microfilament and microtubule rearrangements along with modulation of MAP kinase family member activity [12, 13]. GECs that are infected with P. gingivalis do not undergo apoptotic or necrotic death, and indeed are rendered resistant to chemically induced apoptosis [14, 15]. The anti-apoptotic phenotype induced by P. gingivalis in GEC is associated with activation of the PI3K/Akt and JAK/Stat pathways [15, 16]. Moreover, cytochrome c release from the mitochondria is inhibited and caspase 3 activity is downregulated following P. gingivalis infection, and there is upregulation of anti-apoptosis genes encoding Bcl-2 and survivin [14–16].
The transcriptional responses of gingival and other oral epithelial cells to P. gingivalis have been studied on a global scale [17, 18], and pathways relating to apoptosis, cell cycle control and proliferation are consistently impacted. However, cell cycle activity is controlled on numerous levels, including protein degradation and phosphorylation/dephosphorylation, that can not be deduced from transcriptional array data. Hence, we have adopted proteomic and phenotypic approaches to identify the levels and activation status of cell cycle effectors, and their impact on GEC cell cycle in response to P. gingivalis.
2. Methods
2.1. Bacterial strains and media
P. gingivalis 33277 and YPF1 (FimA−) were cultured to mid-log phase in Trypticase soy broth (TSB) supplemented with yeast extract (1 μg ml−1), menadione (1 μg ml−1) and hemin (5 μg ml−1), at 37°C under anaerobic conditions of 85% N2, 10% H2 and 5% CO2.
2.2. Eukaryotic cells
Primary cultures of GEC were generated as described previously [19]. Briefly, healthy gingival tissue was collected from patients undergoing surgery for removal of impacted third molars and following Institutional Review Board Guidelines. Basal epithelial cells were separated and cultured in keratinocyte growth medium (KGM; Cambrix, East Rutherford, NJ) in the absence of antibiotics at 37 °C in 5% CO2. GECs were used at passage four.
2.3. Proteomic analysis
GECs (60% confluence) were co-cultured with P. gingivalis at a Multiplicity of Infection (MOI) of 100 for 6 h. An MOI of 100 has been shown to activate anti-apoptotic pathways in GECs [16]; and differential protein expression in GEC following P. gingivalis infection can take up to 6 h [20]. P. gingivalis-infected and control GECs were pelleted and suspended in lysis buffer (20 mM MOPS, 60 mM β-glycerophosphate, 5 mM EDTA, 2 mM EGTA, 1 mM Na3VO4, 30 mM NaF, 0.5% Nonidet P-40, and 1 mM DTT, supplemented with 1 mM PMSF, 10 μM leupeptin, 4 μg/ml aprotinin and 5 μM pepstatin A). Fifty micrograms of protein were fluorescently labeled by Kinexus (Vconcouver, BC). Purified labeled proteins from infected and control samples were incubated simultaneously on a Kinex KAM-1.1 antibody microarray side by side. Each Kinex antibody microarray has 2 identical fields of antibody grids containing 603 antibodies each that target cell signaling proteins. These include 346 pan-specific antibodies for measurement of the expression of 240 protein kinases and 106 other signaling proteins, as well as 257 phosphosite specific antibodies. After probing, arrays were scanned using a ScanArray scanner (Perkin Elmer, Wellesley, MA) with a resolution of 10 μm, and the resulting images were quantified using ImaGene (BioDiscovery, El Segundo, CA). Detailed information on the arrays can be found on Kinexus Bioinformatics Corporation’s website at http://www.kinexus.ca
Data was filtered by the application of the following parameters: % range </= 15, Flag 0, S/N ratio > 1.5, and Ave >/= 1000. % range shows how tightly the normalized net signal median for adjacent duplicate spots of the same protein cluster around the average. A flag of 0 indicates that the spot is acceptable based on morphology and background. S/N ratio is the ratio of signal median intensity to background median intensity. Ave is the average of normalized net signal median for adjacent duplicate spots of the same protein. Proteins passing these screens were selected on the basis of %CFC >/= 20. %CFC is the percentage change from control and is a measure of the change in normalized signal intensity averages between P. gingivalis-infected and control samples over two independent experiments.
2.4. GEC proliferation
GECs were cultured to approximately 50% confluence and labeled with 5μM flourescein diacetate derivative carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) for 45 min at 37°C in a 5% CO2 incubator in KBM growth medium. The cells were washed with PBS, KBM medium replaced and infected with P. gingivalis strains for 24 h at the MOI indicated. Uninfected samples were also included in order to determine basal level of cell proliferation. At the end of the incubation, cells were dissociated with 0.05% trypsin-0.53 mM EDTA, collected into flow-cytometry tubes, washed with PBS and fixed in 10% neutral buffered formalin for 20 min. Flow-cytometry was with excitation at 488 nm and a 515-nm band pass filter for CFSE dye and data were analyzed using Summit V3.1 software. CFSE unlabeled cells served as control for appropriate electronic compensation of the instrument. Dual parameter histograms of forward light scatter (cell size) and fluorescence were performed to quantitate the variation in fluorescence intensity between samples. Relative growth rate was expressed as reduction in mean fluorescence per cell relative to uninfected controls. All experiments were repeated 3 times with GEC from different donors.
2.5. Cell cycle analysis
40% confluent GECs were incubated in KGM without supplements for 48 h to reach 50% confluence. Following starvation, the synchronized GECs (2×106) were co-cultured with P. gingivalis at 37 °C in 5% CO2 for 2 h at a MOI of 1:100. After infection, the cells were washed three times with PBS and further cultured for 4 h, 18 h and 42 h in complete KGM. The cells were dissociated, washed with PBS and fixed in ice-cold 70% ethanol overnight. The cell pellets were washed with PBS three times and incubated in 1 ml 0.6 % RNase A solution at RT for 30 min. Propidium iodide (50 μg/ml; Invitrogen) was added to stain double-strand nucleic acids. Cell cycle analysis of 30,000 cells per sample was with excitation at 488 nm. Data were analyzed with CellQuest software (BD biosciences) and ModFit LT V 3.1 (Verity software). The reduced chi-square value was less than 5.0, and the CV was < 8% in all analyses. All experiments were repeated three times with GEC from different donors.
3. Results and Discussion
3.1. P. gingivalis modulates the expression and activation status of proteins involved in cell cycle control
In the current study we have applied a proteomics approach to determine the impact of P. gingivalis on pathways relating to control of the cell cycle. These studies are facilitated by the availability of primary low passage cultures of gingival epithelial cells. Unlike transformed cells, the GECs have normal growth and differentiation characteristics, and will eventually reach senescence [19]. P. gingivalis-infected GEC were examined by antibody microarrays (Kinexus) and compared to uninfected controls. Of the 346 proteins represented on the array, 299 were detected, with 69 increasing in concentration and 89 decreasing in concentration following P. gingivalis infection. 186 phosphorylated epitopes were detected, 48 of which increased in intensity and 36 decreased in intensity after P. gingivalis infection. Regulated proteins were interrogated and assembled into pathways based on the literature and databases including GeneCards (http://www.genecards.org), LINNEA™ Pathways (http://escience.invitrogen.com/ipath), PhosphoSite (http://www.phosphosite.org) and Kegg (http://www.genome.ad.jp/kegg/pathway.html). This analysis revealed several pathways that exert control over cell cycle were impacted by P. gingivalis infection (summarized in Fig 1A), including those involving cyclins, p53 and PI3K. Cyclins are nuclear proteins that are transiently expressed in order to activate cognate cyclin dependent kinases (Cdks) [21]. Cyclins and Cdks form complexes which facilitate cell cycle progression through various cell cycle stages. Expression of Cyclin A, that is essential for control of the cell cycle at the G1/S (start) and the G2/M (mitosis) transitions, was increased by P. gingivalis (Fig 1B). In addition, Cdk2, which shows maximal activity during S and G2 phases [2], although reduced in amount, was activated by phosphorylation at Thr 160. Levels of Cdk4 and Cdk6 that regulate the G1/S transition [2] were elevated in infected cells, whereas levels of INK4, that inhibits Cdk4/6 and can induce cell cycle arrest in G1 and G2 phases [2] were diminished. Cyclin D, which regulates progression through G1 [2], was downregulated following P. gingivalis infection. Collectively these data indicate that at the collection point (6h) infected GEC had progressed through the G1 phase and were beginning to enter S and G2, while non-infected cells were still on the transition of the G0/G1 phase.
Fig 1.
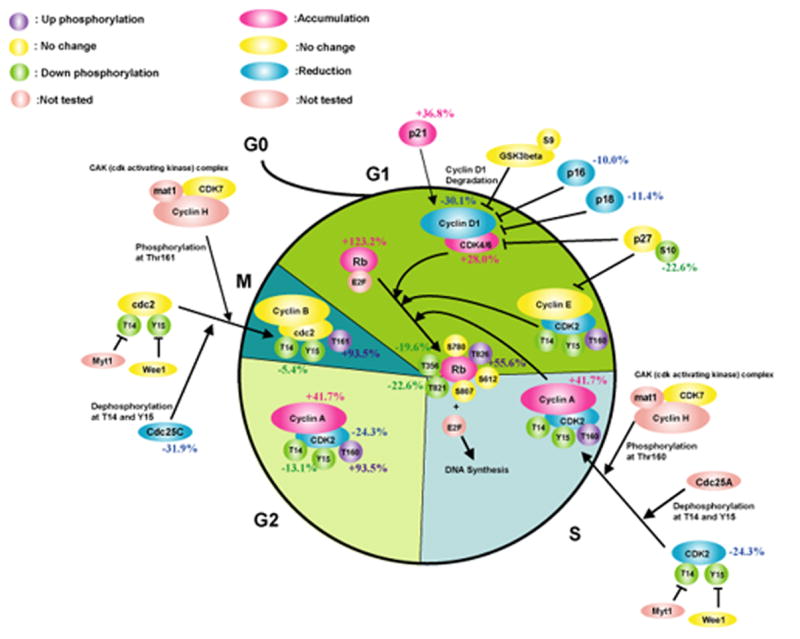
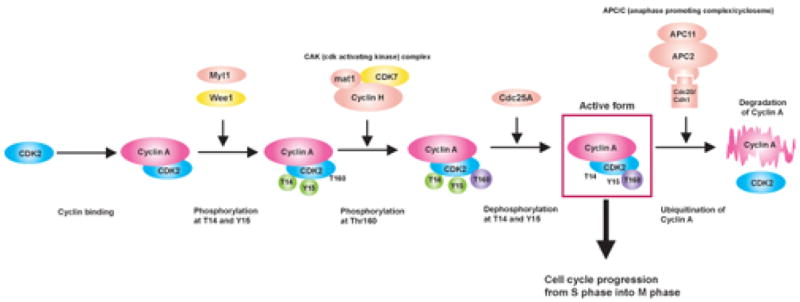
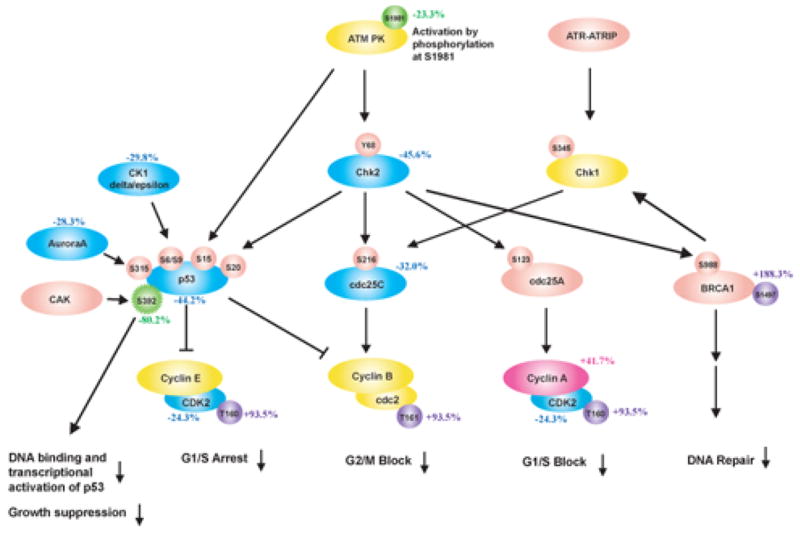
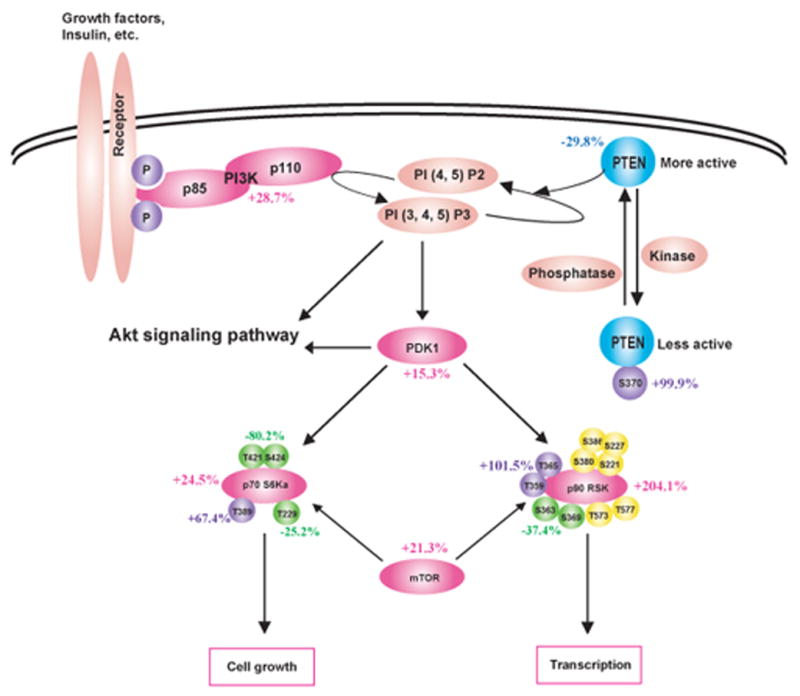
P. gingivalis impacts cell cycle control in GECs. Schematic representation of Kinexus antibody array results showing cell cycle pathways modulated by P. gingivalis infection (MOI 100, 6h). Pointed arrow indicates molecular interaction resulting in activation, flat arrow indicates molecular interaction resulting in inhibition. Magenta ellipsoids represent proteins that accumulate in infected cells as compared to non-infected cells, blue ellipsoids represent proteins reduced in amount, yellow ellipsoids indicate proteins whose expression was not affected by P. gingivalis infection, and coral pink ellipsoids represent proteins that were not present on the array. Violet and green circles respectively indicate more phosphorylated sites and less phosphorylated sites in infected cells, yellow circles show the sites that were equally phosphorylated in both cells, and coral pink circles represent phosphorylation sites that were not tested. % change in accumulation of phosphorylation of individual proteins is given. (A) Cell cycle checkpoint overview (B) Phosphorylation of CDK2 at Thr160 and accumulation of Cyclin A suggested an increase in cell cycle progression into S and G2 phases. (C) Both the level and the activity of p53 were diminished by P. gingivalis. (D) Activation of the PI3K/Akt pathway by P. gingivalis.
Infection of GEC with P. gingivalis resulted in a reduction of p53 levels. p53 is a tumor suppressor that plays a major role in cellular response to DNA damage and other genomic aberrations, and its activation can lead to either cell cycle arrest and DNA repair, or to apoptosis [22]. p53 is stabilized and activated by phosphorylation at multiple sites through the action of kinases such as Chk2, Aurora A, CK1delta and CK1epsilon [23]. All of these kinases were downregulated by P. gingivalis, and the remaining p53 was under-phosphorylated at Ser392 in infected cells as compared to uninfected controls (Fig 1C). Thus both the level and the activity of p53 are diminished by P. gingivalis, consistent with an increased proliferation rate of infected GEC.
The PI3K pathway is another important component of the regulation of cell cycle progression [24]. Infection with P. gingivalis increased levels of PI3K and of phosphoinositide-dependent protein-serine kinase 1 (PDK1), a key molecule that couples PI3K to cell proliferation and survival signals by activating Akt, PKC isoenzymes, p70S6K and p90RSK [24] (Fig 1D). Consistent with this, both the level and phosphorylation state of p70S6K and of p90RSK were upregulated by P. gingivalis. The PI3K pathway is negatively regulated by PTEN a lipid phosphatase that removes the 3-phosphate from the PI3K lipid product PtdIns (3,4,5)P3 to produce PtdIns (4,5)P2 which prevents Akt activation [25]. PTEN was downregulated and inactivated by phosphorylation at Ser370 after P. gingivalis infection. These data concur with previous work [15] showing activation of the PI3K/Akt pathway in GEC by P. gingivalis, which was associated with resistance to apoptotic cell death.
3.2. P. gingivalis enhances the proliferation rate on GEC in a fimbriae dependent manner
The results of the proteomic analysis corroborate our earlier investigation of the transcriptional responses of GEC, showing overpopulation of the Cell Cycle pathway (Kegg hsa04110; http://www.genome.ad.jp/kegg/pathway.html), in response to infection with P. gingivalis [17]. Furthermore, transcriptional profiling with a fimbriae deficient mutant of P. gingivalis indicated that the long (FimA) fimbriae of the organism are responsible for activation of over 200 genes associated with cell cycle and cell proliferation [17]. Hence we sought to verify both the proteomic data indicating an increase in cell cycle progression, and previous transcriptomic data indicating the involvement of fimbriae in this process, by examining the effects of P. gingivalis parental and fimbrial deficient mutant strains on GEC proliferation at the phenotypic level. Accordingly, we determined the proliferation rates of GECs infected with either P. gingivalis wild type (33277) or its FimA deficient derivative, YPF1. GECs were labeled with CFSE (carboxyfluorescein diacetate succinimidyl ester) that stains viable cells without compromising their functional properties and does not interfere with spontaneous apoptosis [26]. Moreover CFSE is retained by both apoptotic and necrotic cells. Each successive doubling of proliferating GECs will thus be marked by a halving of cellular fluorescence intensity, which was traced by flow cytometry. As shown in Fig 2, 24h after infection with wild type P. gingivalis, at MOI 10 or 100, there was an almost twofold increase in the proliferation rate of GECs. In contrast there was no significant difference in the growth rate of GEC infected with the fimbriae deficient mutant strain YPF1.
Fig 2.
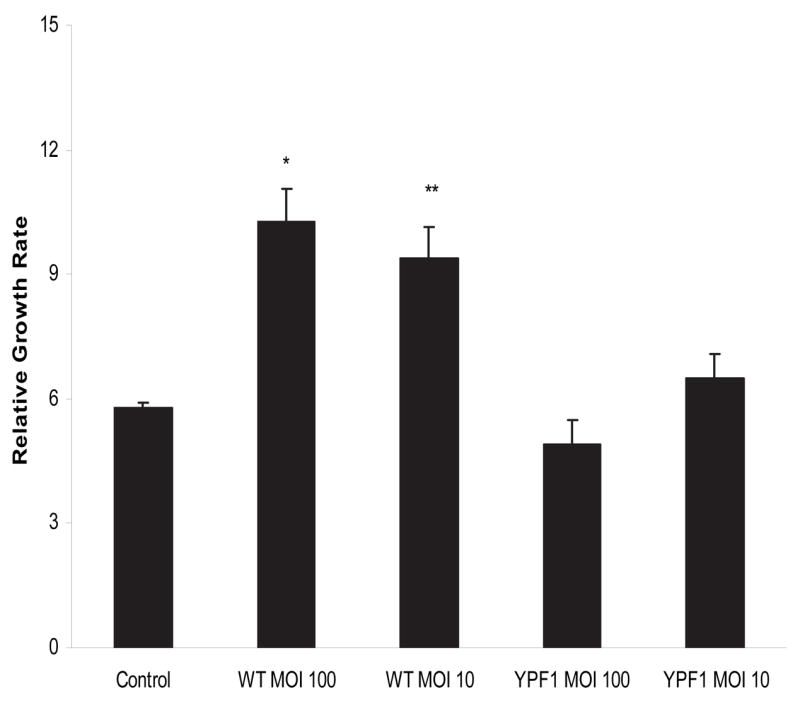
P. gingivalis increases GEC proliferation rate. Flourescein-labeled GEC were infected with P. gingivalis 33277 (WT) or a FimA deficient mutant (YPF1) for 24h at a MOI of 10 or 100. Relative growth rate was calculated based on a reduction in mean fluorescence by cytofluorimetry per cell normalized to day 0 population (not shown). A t-test was used for comparison of infected and uninfected cells: ** p < 0.05; * p < 0.01.
3.3. P. gingivalis accelerates progression through S-phase
Having established that P. gingivalis can stimulate cell proliferation of GECs, we next sought to quantify progression through the phases of the cell cycle. Starvation synchronized cells were infected with P. gingivalis strains and double stranded DNA was stained with propidium iodide. Flow cytometry showed that 6 hours after infection, 33% of the cells exposed to P. gingivalis wild type were at the S phase, as compared to 19% of control cells and 20% of cells exposed to YPF1 (Fig 3). Twenty hours after infection, over 64% of the cells exposed to parental P. gingivalis had progressed to S phase, actively replicating DNA, while control cells (34%) and the cells exposed to YPF1 (38%) showed less progression. After 44 hours, the majority of the cells exposed to P. gingivalis wild type had moved to G2 or G0/G1 phase, while the control cells (47%) and the cells exposed to YPF1 (40%) were still predominantly at S phase.
Fig. 3.
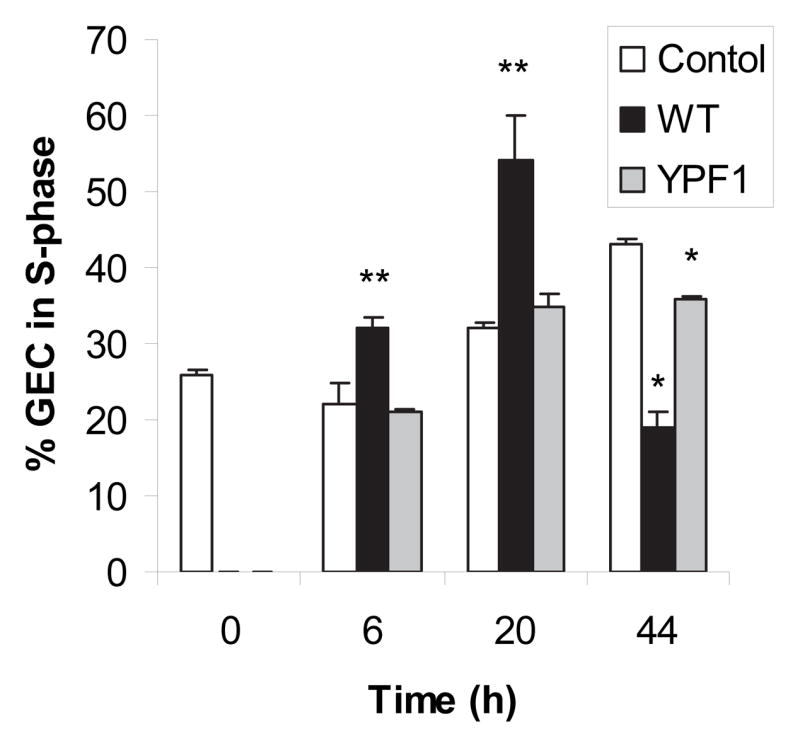
Infection with P. gingivalis accelerates GEC cell cycle progression. GEC were infected with P. gingivalis 33277 (WT) or a FimA deficient mutant (YPF1) at a MOI of 100 for the times indicated. Control cells were uninfected. % of cells in the S-phase was calculated from intensity of propidium iodide staining of double stranded DNA by cytofluorimetry. A t-test was used for comparison of infected and uninfected cells: ** p < 0.05; * p < 0.01.
The FimA fimbriae are the major adhesion and invasion effectors of the organism and act through engagement of integrin receptors on the GEC surface [27]. FimA deficient mutants are delayed in entry of GECs and do not impact the GEC cytoskeleton to the same degree as the parent [28]. The inability of the FimA mutant to enhance cell division may result from failure to activate integrin signaling and/or a delay in establishing an intracellular location. Given the broadly based nature of the proliferative phenotype induced by P. gingivalis it is likely that timing, context and physical location are parameters that all are operational in determining the overall GEC response to P. gingivalis challenge.
3.4. Conclusions
Increased GEC proliferation caused by P. gingivalis could have significant effects on the integrity and turnover of the gingiva in vivo. Loss of cell cycle control could impact wound healing in the periodontal pocket which in turn could facilitate bacterial penetration of the periodontal tissues. In addition to their barrier function, epithelial cells also participate in a signaling network that alerts the immune system to the presence of colonizing bacteria. GECs that are rapidly proliferating may be less efficient in these signaling functions and so immune surveillance in the periodontal area may be compromised. Furthermore, while traditionally bacteria have been considered bystanders in proliferative diseases such as cancer, the ability of a range of organisms to cause DNA damage and induce cellular proliferation has led to a reappraisal of this role [29, 30]. Although carcinogenesis is a protracted multistage, multifactorial process, the possibility of a contribution by a proliferation-enhancing organism such as P. gingivalis can not be excluded.
Acknowledgments
Supported by DE11111 and DE16593
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 4.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 5.Galan JE, Cossart P. Host-pathogen interactions: a diversity of themes, a variety of molecular machines. Curr Opin Microbiol. 2005;8:1–3. doi: 10.1016/j.mib.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 6.Gruenheid S, Finlay BB. Microbial pathogenesis and cytoskeletal function. Nature. 2003;422:775–781. doi: 10.1038/nature01603. [DOI] [PubMed] [Google Scholar]
- 7.Schlumberger MC, Hardt WD. Salmonella type III secretion effectors: pulling the host cell’s strings. Curr Opin Microbiol. 2006;9:46–54. doi: 10.1016/j.mib.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Nougayrede JP, Taieb F, De Rycke J, Oswald E. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005;13:103–110. doi: 10.1016/j.tim.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Oswald E, Nougayrede JP, Taieb F, Sugai M. Bacterial toxins that modulate host cell-cycle progression. Curr Opin Microbiol. 2005;8:83–91. doi: 10.1016/j.mib.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belton CM, Izutsu KT, Goodwin PC, Park Y, Lamont RJ. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223. doi: 10.1046/j.1462-5822.1999.00022.x. [DOI] [PubMed] [Google Scholar]
- 12.Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe K, Yilmaz O, Nakhjiri SF, Belton CM, Lamont RJ. Association of mitogen-activated protein kinase pathways with gingival epithelial cell responses to Porphyromonas gingivalis infection. Infect Immun. 2001;69:6731–6737. doi: 10.1128/IAI.69.11.6731-6737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakhjiri SF, Park Y, Yilmaz O, Chung WO, Watanabe K, El-Sabaeny A, Park K, Lamont RJ. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol Lett. 2001;200:145–149. doi: 10.1111/j.1574-6968.2001.tb10706.x. [DOI] [PubMed] [Google Scholar]
- 15.Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, Stavropoulos MF, Yilmaz O, Lamont RJ. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997–2007. doi: 10.1111/j.1462-5822.2007.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Handfield M, Mans JJ, Zheng G, Lopez MC, Mao S, Progulske-Fox A, Narasimhan G, Baker HV, Lamont RJ. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell Microbiol. 2005;7:811–823. doi: 10.1111/j.1462-5822.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 18.Milward MR, Chapple IL, Wright HJ, Millard JL, Matthews JB, Cooper PR. Differential activation of NF-kappaB and gene expression in oral epithelial cells by periodontal pathogens. Clin Exp Immunol. 2007;148:307–324. doi: 10.1111/j.1365-2249.2007.03342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oda D, Watson E. Human oral epithelial cell culture I. Improved conditions for reproducible culture in serum-free medium. In Vitro Cell Dev Biol. 1990;26:589–595. doi: 10.1007/BF02624208. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa Y, Mans JJ, Mao S, Lopez MC, Baker HV, Handfield M, Lamont RJ. Gingival epithelial cell transcriptional responses to commensal and opportunistic oral microbial species. Infect Immun. 2007;75:2540–2547. doi: 10.1128/IAI.01957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 22.Vousden KH. Outcomes of p53 activation--spoilt for choice. J Cell Sci. 2006;119:5015–5020. doi: 10.1242/jcs.03293. [DOI] [PubMed] [Google Scholar]
- 23.Knippschild U, Milne DM, Campbell LE, DeMaggio AJ, Christenson E, Hoekstra MF, Meek DW. p53 is phosphorylated in vitro and in vivo by the delta and epsilon isoforms of casein kinase 1 and enhances the level of casein kinase 1 delta in response to topoisomerase-directed drugs. Oncogene. 1997;15:1727–1736. doi: 10.1038/sj.onc.1201541. [DOI] [PubMed] [Google Scholar]
- 24.Garcia Z, Kumar A, Marques M, Cortes I, Carrera AC. Phosphoinositide 3-kinase controls early and late events in mammalian cell division. Embo J. 2006;25:655–661. doi: 10.1038/sj.emboj.7600967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko S, Ning J, He L, Yang H, Sun M, Nicosia SV, Cheng JQ. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005;10:975–987. doi: 10.2741/1592. [DOI] [PubMed] [Google Scholar]
- 26.Dumitriu IE, Mohr W, Kolowos W, Kern P, Kalden JR, Herrmann M. 5,6-carboxyfluorescein diacetate succinimidyl ester-labeled apoptotic and necrotic as well as detergent-treated cells can be traced in composite cell samples. Anal Biochem. 2001;299:247–252. doi: 10.1006/abio.2001.5415. [DOI] [PubMed] [Google Scholar]
- 27.Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- 28.Yilmaz O, Young PA, Lamont RJ, Kenny GE. Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology. 2003;149:2417–2426. doi: 10.1099/mic.0.26483-0. [DOI] [PubMed] [Google Scholar]
- 29.Lax AJ, Thomas W. How bacteria could cause cancer: one step at a time. Trends Microbiol. 2002;10:293–299. doi: 10.1016/s0966-842x(02)02360-0. [DOI] [PubMed] [Google Scholar]
- 30.Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]