

Abstract
Fatty acyl-CoAs participate in numerous cellular processes. This article describes a method for the quantitation of subpicomole amounts of long-chain and very-long-chain fatty acyl-CoAs by reverse-phase LC combined with electrospray ionization tandem mass spectrometry in positive ion mode with odd-chain-length fatty acyl-CoAs as internal standards. This method is applicable to a wide range of species [at least myristoyl- (C14:0-) to cerotoyl- (C26:0-) CoA] in modest numbers of cells in culture (∼106–107), with analyses of RAW264.7 cells and MCF7 cells given as examples. Analysis of these cells revealed large differences in fatty acyl-CoA amounts (12 ± 1.0 pmol/106 RAW264.7 cells vs. 80.4 ± 6.1 pmol/106 MCF7 cells) and subspecies distribution. Very-long-chain fatty acyl-CoAs with alkyl chain lengths > C20 constitute <10% of the total fatty acyl-CoAs of RAW264.7 cells versus >50% for MCF7 cells, which somewhat astonishingly contain approximately as much C24:0- and C26:0-CoAs as C16:0- and C18:0-CoAs and essentially equal amounts of C26:1- and C18:1-CoAs. This simple and robust method should facilitate the inclusion of this family of compounds in “lipidomics” and “metabolomics” studies.
Keywords: lipidomics, metabolomics, RAW264.7, MCF7, lignoceroyl-CoA, nervonoyl-CoA, cerotoyl-CoA
Fatty acyl-CoAs participate in energy metabolism, the biosynthesis and recycling of complex lipids, the posttranslational modification of proteins, the regulation of gene expression, and other cellular processes (1). They are derived from de novo fatty acid biosynthesis and the recycling of preexisting fatty acids (2) and serve as intermediates of most fatty acid modification reactions, such as oxidation (3), elongation, and desaturation (4). Hence, for both metabolic and regulatory studies, it would be useful to have an analytical method to quantitate a wide variety of these compounds.
Mass spectrometry and tandem mass spectrometry have been shown to be particularly useful for the analysis of fatty acyl-CoAs because they provide high levels of structural specificity and sensitivity (5–11), especially when combined with LC and ESI (5–9). Previously published methods have used negative ion mode MS (5) and MS/MS (7, 8) as well as positive ion mode MS/MS using a precursor ion scan (6), but they have not been applied to a wide spectrum of fatty acyl-CoA molecular species, especially the very-long-chain fatty acyl-CoAs with alkyl chain lengths of >20 carbons.
This report describes an LC-ESI MS/MS protocol that builds on some of the features of these methods for the analysis of a wider profile of fatty acyl-CoAs than has heretofore been reported, with alkyl chain lengths varying from C14 (myristoyl-CoA) to C26 (cerotoyl-CoA), and is potentially applicable to species as short as C2 (acetyl-CoA) and free coenzyme A (CoASH). Analysis of extracts from RAW264.7 cells (a mouse macrophage line) and MCF7 cells (a human breast carcinoma line) demonstrated the method's utility for profiling and quantifying multiple molecular species of fatty acyl-CoAs in relatively small samples and revealed interesting differences in the fatty acyl-CoA profiles for these cell lines. The availability of this fairly simple and robust method should facilitate the inclusion of this family of compounds in a wide range of studies.
EXPERIMENTAL PROCEDURES
Reagents
Fatty acyl-CoAs (C14 to C26 and the odd-chain-length internal standards C15:0-, C17:0-, C23:0-, and C25:0-CoA) were obtained as triammonium salts from Avanti Polar Lipids (Alabaster, AL), and according to the manufacturer, analysis of the fatty acyl-CoAs by TLC, HPLC, MS, NMR, and other tests such as the Bartlett assay for phosphorus were consistent with the purity being >99%. CoASH (lithium salt monohydrate), acetyl-CoA (C2:0) (sodium salt), octanoyl-CoA (C8:0) (sodium salt), and lauroyl-CoA (C12:0) (sodium salt) were from Sigma-Aldrich (St. Louis, MO) and were 95–99% pure. HPLC-grade CH3OH (catalog number MX0475-1), CH3CN (AX0145-1), and CHCl3 (CX1058-1) were from EMD (Darmstadt, Germany). Triethylammonium acetate (TEAA) was from Fluka (Buchs, Switzerland), and triethylamine (TEA) was from Sigma-Aldrich. The water (>18 MΩ/cm) was purified with the Barnstead Diamond Nanopure system (Boston, MA).
Cell culture
MCF7 cells (HTB-22) and RAW264.7 cells (TIB-71) were obtained from the American Type Culture Collection (Manassas, VA). RAW264.7 cells were grown in DMEM (Cellgro catalog number 10-013; it contains 4 mM l-glutamine and 4.5 g/l glucose; Mediatech, Manassas, VA) and 10% heat-inactivated fetal calf serum (HyClone catalog number SH30071.03; Logan, UT). MCF7 cells were grown in MEM supplemented with 10% FBS (HyClone catalog number SV30071.03), 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 0.01 mg/ml bovine insulin. Media for both cell types included tissue culture-grade sodium bicarbonate (1.5 g/l) and 100 U/ml penicillin plus 0.1 mg/ml streptomycin (Gibco catalog number 15140-122; Invitrogen, Carlsbad, CA). Both cell types were cultured at 37°C, 95% relative humidity, and 5% CO2. Dishes (60 mm; catalog number 430166; Corning, Inc., Corning, NY) of RAW264.7 cells were seeded at 2 × 106 cells per dish and grown for ∼30 h before harvesting for lipid analysis. Dishes (100 mm; catalog number 430293; Corning, Inc.) of MCF7 cells were seeded at 2 × 106 cells/ml and grown for ∼48 h. For the RAW264.7 cells, the cell culture conditions followed the standard protocol adopted by the Lipid MAPS Consortium (www.lipidmaps.org).
Cell extraction
Dishes of cells cultured as described above (typically 0.5–2 × 107 cells) were placed on ice, the medium was aspirated, and adherent cells were gently washed twice with 5 ml of chilled PBS then once with 5 ml of chilled distilled water (which did not cause cell lysis as assayed by trypan blue exclusion, if added and removed rapidly). Using a rubber policeman, the cells were rapidly scraped into a corner of the tilted dish and mixed to a uniform suspension in the residual water (∼0.5 ml) with a pipette, from which 50 μl was removed for cell quantitation by counting (or analysis of protein or DNA) and 200 μl for analysis of the fatty acyl-CoAs. This procedure avoids centrifugation of the cells after they have been removed from the Petri dish, because a substantial percentage of RAW264.7 cells were broken during scraping; analysis of more robust cells can use centrifugation for the washes.
The aliquot for fatty acyl-CoA analysis was placed in a screw-cap tube (13 × 100 mm; catalog number 73750-13100; with a Teflon-lined cap; Kimble Chase, Vineland, NJ) on ice. To this was added 500 μl of CH3OH containing 1 mM EDTA (added to the CH3OH from a 0.5 M aqueous stock) and 10 μl of an internal standard mixture that contained 100 pmol each of C15:0-, C17:0-, C23:0-, and C25:0-CoA, which were prepared in CH3OH/CHCl3 (2:1, v/v) containing 30 mM TEAA. After a brief sonication (∼0.5 min), 250 μl of CHCl3 was added, followed by another brief sonication, and the single-phase extraction mixture was incubated for 30 min in a 50°C heating block. After cooling to room temperature, CHCl3 and water (250 μl each) were added with mixing by vortexing after each addition. After brief centrifugation using a table-top centrifuge, the fatty acyl-CoAs were in the upper phase and interface and most of the other lipids (which would interfere with the subsequent reverse-phase LC) were in the lower phase. The upper layer was removed with a Pasteur pipette and transferred into a screw-cap tube (as above), and the remainder (interface and lower layer) was reextracted twice (each time with brief centrifugation to separate the two phases cleanly) with 0.5 ml each of synthetic upper phase (H2O/CH3OH/CHCl3, 45:50:5, v/v/v), from which the upper phases were added to the same screw-cap tube as the first extract. To the pooled upper phases was added 180 μl of CH3OH/CH3(CH2)2CH2OH/CHCl3 (50:25:25, v/v/v), which was found to help keep the very-long-chain fatty acyl-CoAs in solution for at least 24 h at room temperature and/or at least 3 days at −20°C.
Chromatographic conditions
The HPLC separations used a Shimadzu SCL-10A VP system controller, two LC-10AD VP pumps, a DGU-14A degassing unit, a Perkin-Elmer series 200 autosampler, a Phenomenex (Torrance, CA) Gemini C18 column (2 mm inner diameter × 150 mm with 5 μm particles), and a 2 × 4 mm guard column with the same packing material. The column was maintained at 40°C using a MetaTherm (Torrance, CA) column oven.
The flow rate was 200 μl/min in binary gradient mode with the following elution program: the column was equilibrated with mobile phase A [H2O/CH3CN (85:15, v/v), containing 0.05% TEA], the sample was injected, and mobile phase A was continued for 5 min, followed by a 14 min gradient to 50% mobile phase A and 50% mobile phase B [H2O/CH3CN (10:90, v/v), containing 0.05% TEA], during which the long-chain and very-long-chain fatty acyl-CoAs eluted. Afterward, the column was washed by a 1 min gradient to 100% B and a 5 min hold at 100% B, followed by reequilibration of the column by a 1 min gradient to 100% A and a 5 min hold at 100% A before injection of the next sample.
Mass spectrometry
The analyses were conducted using an Applied Biosystems (Foster City, CA) 4000 QTrap triple quadrupole/linear ion trap mass spectrometer. Dry N2 gas was used to nebulize the column effluent (gas 1) and for the desolvation gas (gas 2), curtain, and collision gases, with instrument settings of 35, 25, 15 (arbitrary units), and medium, respectively. The interface heater was on (100°C), and the Turbo V ESI source temperature was 350°C. The ESI needle voltage was 5.5 kV in positive ion mode and −4.5 kV in negative ion mode.
To establish the optimal parameters for MS and MS/MS, stocks of the fatty acyl-CoA standards (∼0.5 mM each) were prepared in CH3OH/CHCl3 (2:1, v/v) and then diluted before use to 1 μM with H2O/CH3CN (50:50, v/v) containing 30 mM TEAA. The compounds were infused at 5 μl/min to optimize desolvation potential (DP), collision energy (CE), and collisionally activated dissociation (CAD) gas to yield maximum sensitivity for the singly protonated (M + H)+ fatty acyl-CoA precursor ions in positive ion mode and for the singly deprotonated (M − H)− species in negative ion mode. A narrow (6 Da) width was used to determine the optimum DP for each standard.
The MS/MS parameters were determined by identifying the acyl chain-retaining product ions, which in positive ion mode resulted from the loss of 507.0 Da from the (M + H)+ precursor, as has been observed previously (6, 11), and optimizing CE, collision cell exit potential (CXP), and CAD gas values for each molecular species (Table 1).
TABLE 1.
Mass spectrometer settings and chromatographic properties of selected fatty acyl-CoAs
| Acyl-CoAa | Q1 | Q3 | Desolvation Potential, Collision Energy, Collision Cell Exit Potential | LC Retentionb | Peak Area per pmolc |
|---|---|---|---|---|---|
| m/z | m/z | eV | min | cps × 105 | |
| 14:0 | 978.3 | 471.3 | 180, 50, 12.7 | 11.4 | 2.2 |
| 15:0 | 992.4 | 485.4 | 190, 50, 13.0 | 12.2 | 2.3 |
| 16:0 | 1,006.4 | 499.4 | 180, 50, 13.3 | 12.7 | 2.5 |
| 17:0 | 1,020.4 | 513.4 | 180, 53, 13.5 | 13.3 | 2.5 |
| 18:0 | 1,034.4 | 527.4 | 190, 52, 14.3 | 14.1 | 2.9 |
| αOH-18:0 | 1,050.3 | 543.4 | 195, 55, 14.5 | 13.1 | 2.1 |
| 18:1 | 1,032.4 | 525.4 | 185, 52, 13.9 | 13.1 | 2.1 |
| 18:2 | 1,030.4 | 523.4 | 180, 52, 14.0 | 12.7 | ND |
| 18:3 | 1,028.4 | 521.4 | 190, 50, 14.5 | 12.3 | ND |
| 20:0 | 1,062.4 | 555.4 | 190, 52, 15.3 | 14.5 | 2.4 |
| 20:4 | 1,054.4 | 547.4 | 180, 52, 15.0 | 12.3 | 1.8 |
| 22:0 | 1,090.4 | 583.4 | 190, 53, 16.3 | 15.1 | 2.0 |
| 22:6 | 1,078.4 | 571.4 | 190, 51, 16.0 | 12.3 | 1.1 |
| 23:0 | 1,104.5 | 597.5 | 200, 55, 16.5 | 15.5 | 1.9 |
| 24:0 | 1,118.5 | 611.5 | 210, 57, 17.0 | 15.9 | 1.3 |
| 24:1 | 1,116.5 | 609.5 | 208, 57, 17.0 | 15.2 | 2.0 |
| 25:0 | 1,132.5 | 625.5 | 218, 59, 17.5 | 16.2 | 1.1 |
| 26:0 | 1,146.4 | 639.5 | 220, 58, 17.5 | 16.6 | 0.84 |
Number of linear chain carbon atoms:number of double bonds; αOH denotes an α-hydroxy fatty acyl chain.
During LC, the ion source conditions were as follows: temperature, 350°C; gas 1, 35 arbitrary units; gas 2, 25 arbitrary units; electrospray needle voltage, 5.5 kV.
Slope of the linear regression of a calibration curve for each analyte [abcissa = pmol, ordinate = peak area (cps), y intercept = 0]; the slope has units of cps/pmol. All R2 values were ⩾0.999, except for C26:0 (0.991).
After the elution time for each fatty acyl-CoA of interest was determined using the LC conditions described in the preceding section, the compounds were dissolved in the corresponding proportions of mobile phase A and B for flow injection analysis to reoptimize source-dependent parameters to values appropriate for the higher flow rate (200 μl/min) of the HPLC used in this study (Table 1).
Quantitation of fatty acyl-CoAs in cultured cells
For each new biological sample, the fatty acyl-CoA species of a cell extract that was not spiked with internal standards was first profiled to determine the minimum number of precursor-product pairs that were needed for the multiple reaction monitoring (MRM) protocol. This was conducted using neutral loss (507.0 Da) scans during the HPLC elution with three periods having DP, CE, and CXP settings that were appropriate for analytes that eluted during those times: period 1 (0–11 min) scanned m/z values 750–980, which encompassed CoASH to C14:0-CoA; period 2 (11–15.1 min) scanned m/z values 980–1,070, which encompassed C14:0-CoA to C20:0-CoA; and period 3 (15.1–18 min) scanned m/z values 1,050–1,150, which encompassed C20:0-CoA to C26:0-CoA. After identification of the analytes that were detectable (three times signal-to-noise), the MRM program was built to include these, the internal standards, and sometimes additional analytes of potential interest.
The fatty acyl-CoAs were quantified by LC-ESI MS/MS in positive ion mode using the MRM pairs shown in Table 1, which correspond to (M + H)+ precursor ions (selected with Q1) and the structure-specific product ion resulting from a neutral loss of 507.0 Da (selected with Q3). The amount of each analyte of interest was calculated as follows: 1) LC chromatogram peaks for internal standards and endogenous fatty acyl-CoAs were integrated using Analyst 1.4.2 software (Applied Biosystems) and peak areas were copied to spreadsheets; 2) for each endogenous analyte, a cognate internal standard was selected based upon either similarity of acyl chain length or similarity of LC elution time (whichever is most appropriate); and 3) the following formula was used to calculate pmol of analyte (pmola):
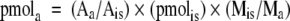 |
where Aa = analyte peak area, Ais = internal standard peak area, pmolis = spiked pmol of internal standard, Mis = slope of the linear regression of the internal standard's calibration curve, and Ma = slope of the linear regression of the analyte's calibration curve. If desired, the observed peak areas (representing monoisotopic ions) may be isotopically corrected; however, this is not usually necessary if the analyte and internal standard are similar (i.e., the correction is <1% per methylene difference in the fatty acyl chain length).
The calibration curves for the fatty acyl-CoAs were prepared by serially diluting each compound and analysis by LC-ESI MS/MS as described above. In addition, calibration curves were generated for select fatty acyl-CoAs by spiking six different quantities from 0.1 to 5 pmol of the internal standards (C15:0-, C17:0-, C23:0-, and C25:0-CoA) into RAW264.7 cell extracts and analyzing by LC-ESI MS/MS. The limit of detection and the limit of quantitation (LOQ) were defined as 3 times signal-to-noise and 10 times signal-to-noise, respectively. Plotting of the quantities analyzed (abcissa) versus observed peak areas (ordinate) was followed by linear regression (with y intercept = 0) to calculate the slope of this regression line (Table 1).
Validation of the extraction and LC-ESI MS/MS conditions
To determine the efficiency with which fatty acyl-CoAs were recovered, the three upper phases from the cell extraction procedure (see above) were collected individually and the lower layer was extracted an additional fourth time, then each was analyzed separately by LC-ESI MS/MS and compared with the peak areas for fatty acyl-CoAs not carried through these extraction procedures.
Statistical analysis
All data shown are the results of at least three analytical replicates (LC-ESI MS/MS analyses) of at least three biological replicates (culture dishes) and are representative of at least two independent experiments (conducted on different days). Data are shown as means ± SD; all statements of significant differences are based upon Student's t-test with P ⩽ 0.05.
RESULTS
The goal of these experiments was to develop a quantitative LC-ESI MS/MS analysis method for a wide range of fatty acyl-CoAs suitable for small samples, such as cultured mammalian cells. The strategy that was followed was to examine the ionization and fragmentation of purified fatty acyl-CoAs, to select candidate precursor ion-product ion pairs for MRM analysis, to determine LC conditions that separated a substantial number of species to allow unambiguous quantitation, and to identify internal standards and extraction conditions that are effective with biological samples, using two mammalian cell lines (RAW264.7 and MCF7) as prototypes.
Characterization of fatty acyl-CoAs by MS and MS/MS
The MS spectra of standard C16:0-CoA contain both (M + H)+ ions and (M + Na)+ adducts in positive ion mode (Fig. 1A) and (M − H)− ions and (M − 2H + Na)− adducts in negative ion mode (Fig. 1B) at the electrospray needle voltage yielding the most intense signal for singly (de)protonated precursor ions. The (M − H)− signal was ∼7-fold more intense than the (M + H)+ signal for identical sample, infusion, solvent, and ion source conditions, except for the polarity of the electrospray needle, which was at the maximum voltage setting in both ionization modes.
Fig. 1.

Mass spectra of C16:0-CoA in the high mass region using positive (A) and negative (B) ionization modes. Q1 scans using a triple quadrupole tandem mass spectrometer are shown. Labels above the peaks denote m/z values of the monoisotopic peaks and describe the (de)protonated or sodium adduct ions. Both ionization mode spectra were collected using the desolvation potential (DP) yielding the best peak height for the singly (de)protonated molecular ion and the same sample, solvent, and ion source conditions (except for the polarity of the needle voltage, which used the maximum voltage setting in both polarities). Spectra (summation of 1 min scans) were collected during the infusion (5 μl/min) of C16:0-CoA at a concentration of 1 pmol/μl.
The MS/MS spectra for collision-induced fragmentation of the (M + H)+ molecular ions of C16:0-CoA (m/z 1,006.4) yielded highly abundant product ions of m/z 499.4 (Fig. 2, upper panel), which correspond to cleavage of the C-O bond of the 5′-β-phosphate with charge retention on the acyl portion of the molecule. Less abundant product ions of m/z 428.4, 397.4, 261.1, 159.0, and 136.0 were also detected. Proposed fragmentations are shown (Fig. 2, upper panel) that correspond to the m/z 428.4, 261.1, and 136.0 product ions. The m/z 397.4 product ion also retains the acyl chain (see below).
Fig. 2.

Tandem mass spectra of C16:0-CoA using positive (A) and negative (B) ionization modes. Both ionization mode spectra were collected using the same sample and ion source conditions, the DP to yield the best peak height for the singly (de)protonated molecular ion, and the collision energy (CE), collisionally activated dissociation (CAD) gas setting, and collision cell exit potential (CXP) to yield the best peak height for the most abundant product ion retaining the acyl chain moiety (499.4 m/z in positive mode and 657.4 m/z in negative mode). Uppercase letter labels by the peaks denote the systematic nomenclature proposed in Fig. 3. The C16:0-CoA structures shown with each ionization mode propose fragmentations consistent with the tandem mass spectra. The horizontal arrow from the precursor ion to the product ion indicates a neutral loss of 507.0 Da. Spectra (summation of 1 min scans) were collected during the infusion (5 μl/min) of C16:0-CoA at a concentration of 1 pmol/μl.
Collision-induced fragmentation of the (M − H)− molecular ions of C16:0-CoA (m/z 1,004.4) yielded product ions of m/z 924.5, 657.4, 408.0, 328.1, and 159.0. The ion of m/z 657.4 corresponds to cleavage of the C-O bond of the 5′-α-phosphate, subsequent loss of water, and charge retention on the acyl portion of the molecule; the other product ions correspond to the proposed fragmentations shown (Fig. 2, lower panel).
The MS/MS fragmentations of all of the fatty acyl-CoAs analyzed shared similarities in positive and negative ion modes, and a systematic nomenclature for these product ions is proposed in Fig. 3. Positive ion mode MS/MS resulted in abundant product ions retaining the acyl moiety (Z ions; Fig. 3A, B) as well as product ions derived from the CoA moiety (A, B, and C ions; Fig. 3A, B). At least one additional product ion resulting from a neutral loss of 609 Da retained the acyl moiety (m/z 383 and 523; Fig. 3A, B, respectively). Similarly, negative ion mode MS/MS resulted in product ions retaining the acyl moiety (X ions; Fig. 3D, E) as well as product ions derived from the CoA moiety (A, B, and C ions; Fig. 3D, E).
Fig. 3.

MS/MS spectra of C15:0-CoA and C25:0-CoA and nomenclature for fatty acyl-CoA collision-induced dissociation. Positive ion mode MS/MS (A, B) and negative ion mode MS/MS (D, E) are shown for the representative compounds C15:0-CoA (A, D) and C25:0-CoA (B, E). A generic fatty acyl-CoA structure with fragmentations and proposed nomenclature is shown in C, with R representing the fatty acyl chain. Product ions derived from the CoA moiety were observed in both ionization modes (B and C ions) as well as product ions retaining the acyl chain moiety (Z ions in positive mode; X ions in negative mode). The horizontal arrows from the precursor ion to the product ion indicate a neutral loss of 507.0 Da. In all spectra shown, the CE and CXP were optimized for nearly complete fragmentation of the singly (de)protonated precursor ion. Spectra (summation of 1 min scans) were collected during the infusion (5 μl/min) of standards at a concentration of 1 pmol/μl.
Many of these product ions have been noted in earlier publications. For example, positive ion mode ESI MS/MS has been shown to yield Z, B, and C ions and product ions resulting from a neutral loss of 609 Da (6, 9) as well as A ions and m/z 261 and 159 products (6). Negative ion mode ESI MS/MS has been shown to yield X, X-H2O, X-HPO3, C, and C-H2O ions (7, 9) as well as B ions (9).
The results from the MS and MS/MS analyses were used to choose precursor (selected by Q1) and product (selected by Q3) ion pairs for MRM. For maximum sensitivity and specificity, the product ion should be both abundant and structure-specific for the acyl moiety, and these criteria are met by the positive mode Z product ion (e.g., m/z 499.4 for C16:0-CoA; Fig. 2A), which is easily optimized as the base peak of the tandem mass spectrum. In the negative ion mode, the X-H2O product ion was more intense than the positive mode Z product ion under virtually identical infusion conditions (Fig. 2A, B), although neither of the product ions that retain the acyl chain (e.g., m/z 657.4 and 577.4 for C16:0-CoA; Fig. 2B) could be optimized as the base peak of the tandem mass spectrum.
Analysis of fatty acyl-CoAs by LC-ESI MS/MS
LC-ESI MS/MS analysis of fatty acyl-CoAs used MRM pairs corresponding to singly (de)protonated precursor ions (selected by Q1) and the most abundant structure-specific product ion retaining the acyl chain (selected with Q3). For example, positive ion mode MRM analysis of C16:0-CoA used the MRM pair 1,006.4→499.4, whereas negative ion mode MRM analysis used the pair 1,004.4→657.4. The DPs, CEs, and CXPs were optimized to yield maximum peak heights for the selected precursor and product ions in both polarities. Comparison of LC-ESI MS/MS peak areas of C16:0-, C18:1-, and C24:1-CoA using both negative and positive mode MRM revealed that the positive ion mode was ∼3-fold more sensitive (Fig. 4A); therefore, ionization, dissociation, and separation conditions were optimized for maximum sensitivity and specificity for each individual acyl-CoA species in positive ion mode (Table 1, Fig. 4B).
Fig. 4.

LC-ESI MS/MS of palmitoyl-, oleoyl-, and nervonoyl-CoA in positive and negative ion modes (A) and a wider range of fatty acyl-CoAs in positive ion mode (B). A: Representative chromatograms of 5 pmol of each fatty acyl-CoA determined using multiple reaction monitoring (MRM) pairs that had been optimized for that species as described in Experimental Procedures (the ion source conditions were identical except that the electrospray needle voltage was 5.5 kV in positive mode and −4.5 kV in negative mode, the maximum settings). Positive ion mode MRM used the settings shown in Table 1, whereas negative ion mode MRM used the following Q1 m/z, Q3 m/z, DP (eV), CE (eV), and CXP (eV): 1,004.4, 657.4, −185, −60, and −7.5 (C16:0-CoA); 1,030.4, 683.4, −190, −60, and −8.0 (C18:1-CoA); and 1,114.5, 767.5, −200, −65, and −10.0 (C24:1-CoA). + and − denote peaks from positive and negative ionization modes. B: A cocktail of fatty acyl-CoA standards (10 pmol of each in 5 μl) was analyzed in positive ion mode as described in Experimental Procedures using the MRM pair for each analyte (Table 1). The individual ion chromatograms have been superimposed with labels above each peak to designate the analyte. The small peaks coeluting with C18:1-CoA (MRM for C18:0) and C24:1-CoA (MRM for C24:0) are from the ion chromatograms for the saturated species but reflect the naturally occurring (M + 2) isotopomers of C18:1- and C24:1-CoA.
The chromatographic conditions for Fig. 4B (described in Experimental Procedures) were optimized for the separation of saturated and monounsaturated fatty acyl-CoAs with chain lengths of C14 to C26, because of the interest of our laboratory in the precursors of sphingolipids. There is baseline resolution of acyl-CoAs with acyl chain differences of only one methylene unit and/or a single double bond (Fig. 4B). The latter separation is advantageous because the (M + 2) isotopomer of monounsaturated species matches the MRM pair for the cognate saturated species, as seen in the small peak with the MRM pair for C18:0-CoA coeluting with C18:1-CoA in Fig. 4B as well as for the MRM pair for C24:0-CoA that coelutes with C24:1-CoA. These chromatographic conditions also resolve many other species, including α-hydroxy-fatty acyl-CoAs (as shown for α-hydroxy-C18:0-CoA with a retention time 13.1 vs. 14.1 min for the nonhydroxylated species C18:0-CoA; Table 1). Even when there is only partial chromatographic resolution for some of the fatty acyl-CoAs [such as C22:0- and C24:1-CoA (Fig. 4B) and C20:4- and C22:6-CoA (data not shown)], the analytes are usually distinguishable by their characteristic MRM pairs. Shorter fatty acyl-CoAs elute earlier (e.g., octanoyl-CoA, C8:0-CoA, elutes ∼1 min after the void volume), and free CoASH elutes in the 5 min hold at 100% A after sample injection (data not shown). Therefore, this procedure might be usable for shorter chain species, including other categories of CoA thioesters that can be distinguished by the MRM pairs and LC mobility.
For all of the fatty acyl-CoAs that we have studied in depth by LC-ESI MS/MS (i.e., species from C14 to C26), the LOQ was on the order of 5 fmol and the linear range of quantitation was ∼3 orders of magnitude (from the LOQ to at least 5 pmol), with R2 > 0.99 from linear regression analysis (Fig. 5A, Table 1). Figure 5A shows the log-log relationship between the signal intensities for several representative fatty acyl-CoAs, from which the slopes and other results presented in Table 1 were derived by linear regression analysis. In general, the covariance (SD divided by the mean) for 10 replicate injections of each standard was found to be <10% for 0.1–1 pmol of the analyte, ∼15% for 10–50 fmol, and ∼20% at the LOQ (5 fmol) (data not shown).
Fig. 5.

Relationships between peak area and fatty acyl-CoA amounts for the analysis by LC-ESI MS/MS. A mixture of 17 fatty acyl-CoA standards (each at 0.17 μM) was prepared in methanol-water-chloroform (50:45:5, v/v/v) and serially diluted as needed to obtain the amounts shown (in a 30 μl injection volume) for analysis by LC-ESI MS/MS using the conditions optimized for each analyte as described in Experimental Procedures and Table 1. A: Log-log plot of the means of the peak areas ± SD (n = 4) for four representative analytes (curved arrows). B: Relationship between the slopes of the linear regression plots from Table 1 (cps/pmol) and the chain lengths of the fatty acyl-CoAs, with the saturated species represented by the closed circles, the unsaturated species represented by the open circles, and α-hydroxy-C18:0-CoA represented by the open square.
It is evident from Figs. 4B and 5A that the relationship between peak area and pmol varies somewhat for different fatty acyl-CoAs, with a range of ∼3-fold for the highest (C18:1-CoA) versus the lowest (C26:0-CoA) studied here. This is not attributable solely to the increase in 13C isotopomers with additional carbon atoms or, as far as we have been able to ascertain, to differences in the concentrations of the fatty acyl-CoA standards. To ensure that the differences in ion yield were not attributable to hydrolysis of the fatty acyl-CoA standards, representative stocks (C18:0-, C18:1-, C24:0-, and C24:1-CoA) were analyzed for the amounts of free CoASH using this LC-ESI MS/MS method, and the mol% of free CoASH versus the fatty acyl-CoA was only 2–3%. Thus, it appears that the signal intensities for the saturated species (Fig. 5B, closed circles) are fairly constant for alkyl chain lengths of 14–20 carbons (with, perhaps, a slight peak at C18), beyond which they decline. The simplicity of this relationship allows a reasonably accurate estimation of the amounts of the saturated fatty acyl-CoAs using a limited number of internal standards (such as the C15:0-, C17:0-, C23:0-, and C25:0-CoAs), as described in Experimental Procedures. Based on the similarity of the peak areas for C18:1- and C24:1-CoAs, it appears that alkyl chain length has less of an influence on monounsaturated fatty acyl-CoAs; however, it does appear that the number of double bonds affects the signal intensity. Only one α-hydroxy-fatty acyl-CoA was available for comparison (α-hydroxy-C18:0-CoA), and its behavior was similar to that of oleoyl-CoA. Therefore, each analyte of interest for a particular investigation should be examined to ascertain how it behaves under the LC-ESI MS/MS conditions.
Analysis of fatty acyl-CoAs in cell extracts
Cell extracts were first examined by a neutral loss scan (507.0 Da) in positive ion mode, because this fragmentation was common to all fatty acyl-CoA species, including CoASH (Fig. 3A, B, horizontal arrows) (6). Although this could be conducted by infusion of the extract, more analytes were detected when LC was used to remove contaminants that suppress ionization and to distinguish compounds that have overlapping MRM pairs, as discussed above. Accordingly, the chromatographic elution time was divided into three periods with distinct m/z scan, DP, CE, and CXP ranges, as described in Experimental Procedures. The results for an MCF7 cell extract analyzed by this procedure are shown in Fig. 6. The ions eluting at or near the void volume (MS/MS of region B in Fig. 6) were consistent with CoA (m/z 768.4), propionyl-CoA (m/z 824.4), and the isomers succinyl-CoA and methylmalonyl-CoA (m/z 868.4). The abundant peaks eluting from 11 to 15 min (MS/MS of region C in Fig. 6) were consistent with C14:0-CoA (m/z 978.4), C16:1-CoA (m/z 1,004.4), C16:0-CoA (m/z 1,006.4), C17:0-CoA (m/z 1,020.4; internal standard), C18:1-CoA (m/z 1,032.4), and C20:4-CoA (m/z 1,054.4). A cluster of low-abundance peaks eluting between 15 and 18 min (MS/MS of region D in Fig. 6) were consistent with C24:1-CoA (m/z 1,116.5), C24:0-CoA (m/z 1,118.5), C25:0-CoA (m/z 1,132.5; internal standard), C26:1-CoA (m/z 1,144.5), and C26:0-CoA (m/z 1,146.5). When the extracts were analyzed without the internal standard spikes, the odd-chain-length fatty acyl-CoAs were ⩽10 fmol/107 cells, which allowed the use of these compounds as internal standards. Additionally, the removal of sodium ions from samples by reverse-phase HPLC was confirmed by the absence of a sodium adduct ion (predicted m/z 1,042.4) of the abundant internal standard C17:0-CoA (Fig. 6, region C).
Fig. 6.

LC-ESI MS/MS with neutral loss scanning (507.0 Da) of MCF7 cell extract. An extract of 2 × 107 MCF7 cells was analyzed by LC-ESI MS/MS as described in Experimental Procedures. A shows the total ion chromatogram for a neutral loss scan (507.0 Da); the chromatographic time is divided into three periods with distinct m/z scan, DP, CE, and CXP ranges. Dashed boxes on the total ion chromatogram labeled B–D indicate the time window from which the neutral loss scans in panels B–D are derived. The abcissae of B–D indicate the m/z range of the cognate neutral loss scan, and settings for DP, CE, and CXP ranged from those appropriate for the analyte of the lowest m/z value to those appropriate for the analyte of the highest m/z value within that scan range. The ordinates of B–D are normalized to their respective base peaks, not to the total ion chromatogram intensity of A.
Subsequent LC-ESI MS/MS analyses of cell extracts used the LC conditions described in Experimental Procedures and MRM pairs for the internal standards, the analytes identified for the biological sample of interest by neutral loss scan profiling (see above), and any additional fatty acyl-CoA analytes of interest because they might be encountered during the course of the particular experiment. For the RAW264.7 and MCF7 cells, there were 19 MRM pairs, which could be accommodated using dwell and settling times of 40 and 5 ms, respectively. When the number of analytes exceeds the ability of the MRM program to scan them in the elution time frame, it is possible to divide the chromatographic elution time into additional periods.
Analysis of multiple samples can be conducted using an autoinjector, because the analytes are stable and remain in solution for at least 24 h at room temperature and for at least 3 days at −20°C, which was verified by repeated injections of samples stored under these conditions and analysis by LC-ESI MS/MS. However, this was only the case if the empirically derived conditions described in Experimental Procedures were followed, specifically, the addition of an adequate amount of CH3OH/CH3(CH2)2CH2OH/CHCl3 (50:25:25, v/v/v) to the final extract to keep the very-long-chain fatty acyl-CoAs in solution during storage and analysis. Samples stored at −20°C sometimes become cloudy, but a clear single phase can be restored by warming to room temperature and brief vortexing.
Depending on the nature of the biological sample, it may be necessary to replace the guard column somewhat frequently (after ∼50 samples) if the back-pressure increases from ∼700 p.s.i. to 900 or 1,000 p.s.i., which results in shifting retention times, peak splitting, et cetera, during the gradient. Clogging of needles and lines by the samples was uncommon. There was essentially no (i.e., ⩽1%) carryover of fatty acyl-CoAs from either standards or samples, which was tested by inserting autoinjector vials that contain only the solvents at various intervals between vials containing standards or samples.
Quantitation of fatty acyl-CoAs in cell extracts
For quantitation of the fatty acyl-CoAs in extracts, the peak areas of the analytes of interest were compared with the peak areas of the internal standards, as described in Experimental Procedures. To be reliable, this requires that the internal standard have the same recovery as the analyte under the extraction conditions used, and Fig. 7A shows that this was the case. It is evident from the results that the odd-chain-length spikes had recovery parameters similar to those of endogenous fatty acyl-CoAs of similar length and unsaturation. The necessity of the multiple extractions is also illustrated in Fig. 7A, because the recovery from each individual extraction differed among the species, whereas the sum of the first three extractions (which are pooled when the method is followed as described in Experimental Procedures) was > 90% for all internal standards and endogenous analytes. No significant difference between RAW264.7 and MCF7 cells was observed with respect to the percentage of total fatty acyl-CoAs recovered with each reextraction. Similarly, the percentage of the total fatty acyl-CoAs recovered with each reextraction did not change significantly if the number of cultured cells being extracted was between 5 × 106 and 2 × 107 (data not shown).
Fig. 7.

Recovery of fatty acyl-CoAs at each step of the extraction of MCF7 cells (A) and comparison of peak areas for analytes in solvent versus cell extracts (B). A: Approximately 106 MCF7 cells were spiked with odd-chain-length fatty acyl-CoAs and extracted as described in Experimental Procedures, with the exception that each of the reextractions was collected and individually analyzed by LC-ESI MS/MS as described (white, dark gray, and light gray bars). The lower layer of the extraction was reextracted a fourth time (black bars). After integrating peak areas for each analyte, the areas in all three extractions were summed, and the percentage of the total recovered in each extraction is shown. Results are means ± SD of analyses of three replicate Petri dishes. B: Two internal standards (C15:0- and C25:0-CoAs, in the amounts shown) were analyzed alone versus as spiked into a RAW264.7 extract then analyzed by LC-ESI MS/MS. The circles are the C15:0-CoA and the squares are the C25:0-CoA; closed symbols are the analytes in solvent alone, the open symbols are the analytes spiked into the cell extract.
The linearity of the method was not compromised by the cell extract, as shown in Fig. 7B, in which standard curves for two of the internal standards (C15:0- and C25:0-CoA) are compared for the standards alone versus standards spiked into a RAW264.7 cell extract (R2 ⩾ 0.98). However, the signal intensities were reduced by ∼50% in the biological extracts, which increased the LOQ to ∼10 fmol.
Comparison of fatty acyl-CoAs in RAW264.7 and MCF7 cells
Quantitation of fatty acyl-CoAs in RAW264.7 and MCF7 cell extracts was conducted using the LC positive ion mode ESI MS/MS method described in Experimental Procedures and the MRM pairs that were selected on the basis of the results of the neutral loss scan. As can been seen from Fig. 8, there are significant differences between the two cell lines with respect to the types and amounts of fatty acyl-CoAs. Very-long-chain fatty acyl-CoAs constitute a greater proportion of the total fatty acyl-CoAs in MCF7 cells than in RAW264.7 cells (i.e., >50% vs. <10%, respectively). Indeed, MCF7 cells contain approximately as much C24:0- and C26:0-CoAs as C16:0- and C18:0-CoAs and essentially equal amounts of C26:1- and C18:1-CoAs. Oleoyl-CoA (C18:1-CoA) is one of the major fatty acyl-CoA species in both cell lines, as has been noted for many types of biological samples (5–7, 12, 13), but we have been unable to find any previous reports of such high amounts of very-long-chain fatty acyl-CoAs in cells or tissues. Myristoyl-CoA (C14:0-CoA) was ∼20% of the total fatty acyl-CoAs in RAW264.7 cells versus ∼7% in MCF7 cells. Overall, MCF7 cells had much higher amounts of total fatty acyl-CoAs (80.4 ± 6.1 pmol/106 cells) than RAW264.7 cells (12 ± 1.0 pmol/106 cells) (n = 3; P < 0.01).
Fig. 8.

Quantitation of the fatty acyl-CoAs of RAW264.7 and MCF7 cells. The cells were extracted and analyzed as described in Experimental Procedures. Results are means ± SD of analyses of three replicate Petri dishes.
For comparison, the quantities of very-long-chain fatty acids that have been reported for RAW264.7 cells (www.lipidmaps.org) and MCF7 cells (14), as well as the alkyl chain length distribution of the ceramides from these cells, which were analyzed by published methods (15), are presented in Fig. 9.
Fig. 9.

Fatty acid and ceramide compositions of RAW264.7 and MCF7 cells. A: The mol% age of very-long-chain fatty acids relative to other fatty acids was calculated from data for RAW264.7 cells from Lipid MAPS (www.lipidmaps.org) and for MCF7 cells from Welsh et al. (12) (SD values are not shown because they were not given by the sources). The very-long-chain fatty acids are shown in a single category (⩾C20) because that is how the data were provided in Ref. 12. B: The ceramide subspecies were analyzed for this study using LC-ESI MS/MS (13); the results shown are means ± SD of analyses of three replicate Petri dishes.
DISCUSSION
This article describes an LC-ESI MS/MS method for the quantitation of a broad variety of fatty acyl-CoA molecular species and its application to RAW264.7 and MCF7 cells. In many respects, it is similar to previous methods that have combined LC with ESI MS and MS/MS for the analysis of fatty acyl-CoAs (5–9). However, the extraction and LC conditions were optimized to allow the quantitation of fatty acyl-CoAs from at least C14 to C26 using positive ion mode ESI MS/MS and MRM parameters for structure-specific precursor-product ion pairs for the analytes of interest and commercially available odd-chain-length fatty acyl-CoAs as internal standards. To the best of our knowledge, this is the first report of a method that is able to quantify intact fatty acyl-CoAs with saturated and monounsaturated alkyl chain lengths of >20 carbons, which include the very-long-chain fatty acids found in the ceramide backbones of sphingolipids (e.g., C24:0, C24:1, C26:0, and C26:1) (16).
Although it was somewhat surprising that LC-ESI MS/MS (MRM) was more sensitive in the positive than the negative mode despite the high pH of the LC eluent (Fig. 4), this may have been attributable to the removal of sodium ions by the LC separation, which increased the proportion of the (M + H)+ versus sodium adduct ions (Fig. 1). Because the LC is conducted at high pH, it is possible to conduct the LC-ESI MS/MS analysis in negative ion mode, if necessitated by particular samples or instrumentation limitations; however, the m/z 79.0 product (phosphate) ion that is used in the negative ion mode has a greater potential for artifacts.
A major advantage of coupling HPLC to MS/MS is the resolution of isobaric analytes with different molecular structures, such as the naturally occurring (M + 2) isotopomers of monounsaturated fatty acyl-CoAs, which can have the same precursor and product ion m/z values as the corresponding saturated species (e.g., C18:1-CoA vs. C18:0-CoA). This resolution is a feature of some (5, 6) but not all (7) previously published LC-ESI MS/MS analyses of fatty acyl-CoAs.
The chain length-determined differences in the recovery of fatty acyl-CoAs during repeated extraction (Fig. 7) demonstrate the importance of using internal standards appropriate for the analytes of interest. This method could potentially be modified to include other molecular species of fatty acyl-CoAs after the validation of their recoveries with respect to these, or more appropriate, internal standards.
The fatty acyl-CoA composition of RAW264.7 and MCF7 cells displayed a diverse set of fatty acyl-CoA species, including those that may participate in the modification of proteins, such as C14:0-CoA (17), and those that connect lipid biosynthesis with lipid signal transduction pathways, such as C20:4-CoA (18). The most obvious difference in the fatty acyl-CoA compositions of RAW264.7 versus MCF7 cells is the proportion of the total fatty acyl-CoAs that have C20 and longer chain lengths, which amounts to more than half of the total in MCF7 cells but is near the LOQ in RAW264.7 cells. It is interesting that the quantities of very-long-chain fatty acyl-CoAs in these cells do not exactly match the amounts of free fatty acids that have been reported for them (Fig. 9A) or the relative amounts of very-long-chain fatty acids that are found in a more complex sphingolipid, such as ceramide (Fig. 9B). Indeed, the proportions of ceramides with very-long-chain fatty acids are similar for RAW264.7 cells and MCF7 cells, despite the former having barely detectable very-long-chain fatty acyl-CoAs. Another difference was the 8-fold higher amounts of total fatty acyl-CoAs for MCF7 cells compared with RAW264.7 cells, which may reflect the often higher activity of fatty acid synthase in cancer cells, including this breast carcinoma cell line (19, 20).
Fatty acyl-CoA amounts have typically been estimated to be on the order of nanomoles per wet gram of tissue in previous studies of human muscle and adipose tissue (12), rabbit muscle (5), and rat tissues including liver (6–8, 21), brain (7, 13, 21), muscle (21), and kidney (21). Because 1 g of tissue typically contains 108–109 cells (22), the amounts of fatty acyl-CoAs found in those studies are on the same order of magnitude (∼10 pmol/106 cells) as the fatty acyl-CoAs obtained with RAW264.7 and MCF7 cells in this study.
Because of the sensitivity, breadth, and selectivity of this LC-ESI MS/MS analysis, fatty acyl-CoAs can now be included in the list of compounds that can be analyzed in “lipidomics” studies of the roles of fatty acyl-CoAs in normal cell regulation and disease (1, 23–26) as well as in studies of the effects of pharmacologic agents that affect the biosynthesis of fatty acyl-CoAs, such as triacsin C (27), sterculic acid (28), orlistat (29, 30), C75 (31), and cerulenin (32), or that disrupt the utilization of fatty acyl-CoAs, such as inhibitors of mitochondrial oxidation (etomoxir) (33), glycerolipid biosynthesis (hypoglycin) (34), and sphingolipid biosynthesis (myriocin) (35). In addition to being adaptable for the analysis of fatty acyl-CoAs with acyl chains longer or shorter than those described here (and stable isotope-labeled fatty acids for kinetic studies) (5), the neutral loss (507.0 Da) scan should be able to detect and analyze CoA thioesters with other compounds, such as bile acids (36) and certain xenobiotics (8).
Abbreviations
CAD, collisionally activated dissociation
CE, collision energy
CoASH, coenzyme A
CXP, collision cell exit potential
DP, desolvation potential
LOQ, limit of quantitation
MRM, multiple reaction monitoring
TEA, triethylamine
TEAA, triethylammonium acetate
Published, JLR Papers in Press, February 20, 2008.
Footnotes
These studies were funded by National Institutes of Health Grant GM-069338 to the Lipid MAPS Consortium. The authors thank Dr. Walter Shaw and Avanti Polar Lipids for the provision of fatty acyl-CoA standards.
References
- 1.Faergeman N. J., and J. Knudsen. 1997. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem. J. 323 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sul, H. S., and S. Smith. 2008. Fatty acid synthesis in eukaryotes. In Biochemistry of Lipids, Lipoproteins and Membranes. 5th edition. D. E. Vance and J. E. Vance, editors. Elsevier, Amsterdam. 155–190.
- 3.Schulz, H. 2008. Oxidation of fatty acids in eukaryotes. In Biochemistry of Lipids, Lipoproteins and Membranes. 5th edition. D. E. Vance and J. E. Vance, editors. Elsevier, Amsterdam. 131–154.
- 4.Miyazaki, M., and J. M. Ntambi. 2008. Fatty acid desaturation and chain elongation in mammals. In Biochemistry of Lipids, Lipoproteins and Membranes. 5th edition. D. E. Vance and J. E. Vance, editors. Elsevier, Amsterdam. 191–212.
- 5.Sun D., M. G. Cree, and R. R. Wolfe. 2006. Quantification of the concentration and 13C tracer enrichment of long-chain fatty acyl-coenzyme A in muscle by liquid chromatography/mass spectrometry. Anal. Biochem. 349 87–95. [DOI] [PubMed] [Google Scholar]
- 6.Magnes C., F. M. Sinner, W. Regittnig, and T. R. Pieber. 2005. LC/MS/MS method for quantitative determination of long-chain fatty acyl-CoAs. Anal. Chem. 77 2889–2894. [DOI] [PubMed] [Google Scholar]
- 7.Mauriala T., K. H. Herzig, M. Heinonen, J. Idziak, and S. Auriola. 2004. Determination of long-chain fatty acid acyl-coenzyme A compounds using liquid chromatography-electrospray ionization tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 808 263–268. [DOI] [PubMed] [Google Scholar]
- 8.Kalderon B., V. Sheena, S. Shachrur, R. Hertz, and J. Bar-Tana. 2002. Modulation by nutrients and drugs of liver acyl-CoAs analyzed by mass spectrometry. J. Lipid Res. 43 1125–1132. [DOI] [PubMed] [Google Scholar]
- 9.Hankin J. A., and R. C. Murphy. 1997. MALDI-TOF and electrospray tandem mass spectrometric analysis of fatty acyl-CoA esters. Int. J. Mass Spectrom. Ion Process. 165/166 467–474. [Google Scholar]
- 10.Millington D. S., D. L. Norwood, N. Kodo, R. Moore, M. D. Green, and J. Berman. 1991. Biomedical applications of high-performance liquid chromatography-mass spectrometry with continuous-flow fast atom bombardment. J. Chromatogr. 562 47–58. [DOI] [PubMed] [Google Scholar]
- 11.Norwood D. L., C. A. Bus, and D. S. Millington. 1990. Combined high-performance liquid chromatographic-continuous-flow fast atom bombardment mass spectrometric analysis of acylcoenzyme A compounds. J. Chromatogr. 527 289–301. [DOI] [PubMed] [Google Scholar]
- 12.Ellis B. A., A. Poynten, A. J. Lowy, S. M. Furler, D. J. Chisholm, E. W. Kraegen, and G. J. Cooney. 2000. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am. J. Physiol. Endocrinol. Metab. 279 E554–E560. [DOI] [PubMed] [Google Scholar]
- 13.Deutsch J., S. I. Rapoport, and A. D. Purdon. 1997. Relation between free fatty acid and acyl-CoA concentrations in rat brain following decapitation. Neurochem. Res. 22 759–765. [DOI] [PubMed] [Google Scholar]
- 14.Welsh C. J., M. Robinson, T. R. Warne, J. H. Pierce, G. C. Yeh, and J. M. Phang. 1994. Accumulation of fatty alcohol in MCF-7 breast cancer cells. Arch. Biochem. Biophys. 315 41–47. [DOI] [PubMed] [Google Scholar]
- 15.Sullards M. C., J. C. Allegood, S. Kelly, E. Wang, C. A. Haynes, H. Park, Y. Chen, and A. H. Merrill, Jr. 2007. Structure-specific, quantitative methods for analysis of sphingolipids by liquid chromatography-tandem mass spectrometry: “inside-out” sphingolipidomics. Methods Enzymol. 432 83–115. [DOI] [PubMed] [Google Scholar]
- 16.Merrill, A. H., Jr. 2008. Sphingolipids. In Biochemistry of Lipids, Lipoproteins and Membranes. 5th edition. D. E. Vance and J. E. Vance, editors. Elsevier, Amsterdam. 363–398.
- 17.Beauchamp E., D. Goenaga, J. Le Bloc'h, D. Catheline, P. Legrand, and V. Rioux. 2007. Myristic acid increases the activity of dihydroceramide Delta4-desaturase 1 through its N-terminal myristoylation. Biochimie. 89 1553–1561. [DOI] [PubMed] [Google Scholar]
- 18.Sakuma S., K. Usa, and Y. Fujimoto. 2006. The regulation of formation of prostaglandins and arachidonoyl-CoA from arachidonic acid in rabbit kidney medulla microsomes by linoleic acid hydroperoxide. Prostaglandins Other Lipid Mediat. 79 271–277. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay S., R. Zhan, Y. Wang, S. K. Pai, S. Hirota, S. Hosobe, Y. Takano, K. Saito, E. Furuta, M. Iiizumi, et al. 2006. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 66 5934–5940. [DOI] [PubMed] [Google Scholar]
- 20.Menendez J. A., and R. Lupu. 2007. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 7 763–777. [DOI] [PubMed] [Google Scholar]
- 21.Rosendal J., and J. Knudsen. 1992. A fast and versatile method for extraction and quantitation of long-chain acyl-CoA esters from tissue: content of individual long-chain acyl-CoA esters in various tissues from fed rat. Anal. Biochem. 207 63–67. [DOI] [PubMed] [Google Scholar]
- 22.Sohlenius-Sternbeck A. K. 2006. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol. In Vitro. 20 1582–1586. [DOI] [PubMed] [Google Scholar]
- 23.Zia A., E. H. Kolodny, and G. M. Pastores. 2007. Very long chain acyl-CoA dehydrogenase deficiency in a pair of mildly affected monozygotic twin sisters in their late fifties. J. Inherit. Metab. Dis. 30 817. [DOI] [PubMed] [Google Scholar]
- 24.Bell T. A., 3rd, M. D. Wilson, K. Kelley, J. K. Sawyer, and L. L. Rudel. 2007. Monounsaturated fatty acyl-coenzyme A is predictive of atherosclerosis in human apoB-100 transgenic, LDLr−/− mice. J. Lipid Res. 48 1122–1131. [DOI] [PubMed] [Google Scholar]
- 25.Heimerl S., C. Moehle, A. Zahn, A. Boettcher, W. Stremmel, T. Langmann, and G. Schmitz. 2006. Alterations in intestinal fatty acid metabolism in inflammatory bowel disease. Biochim. Biophys. Acta. 1762 341–350. [DOI] [PubMed] [Google Scholar]
- 26.Gregersen N., P. Bross, and B. S. Andresen. 2004. Genetic defects in fatty acid beta-oxidation and acyl-CoA dehydrogenases. Molecular pathogenesis and genotype-phenotype relationships. Eur. J. Biochem. 271 470–482. [DOI] [PubMed] [Google Scholar]
- 27.Vessey D. A., M. Kelley, and R. S. Warren. 2004. Characterization of triacsin C inhibition of short-, medium-, and long-chain fatty acid: CoA ligases of human liver. J. Biochem. Mol. Toxicol. 18 100–106. [DOI] [PubMed] [Google Scholar]
- 28.Gomez F. E., D. E. Bauman, J. M. Ntambi, and B. G. Fox. 2003. Effects of sterculic acid on stearoyl-CoA desaturase in differentiating 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 300 316–326. [DOI] [PubMed] [Google Scholar]
- 29.Kridel S. J., F. Axelrod, N. Rozenkrantz, and J. W. Smith. 2004. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 64 2070–2075. [DOI] [PubMed] [Google Scholar]
- 30.Pemble C. W. T., L. C. Johnson, S. J. Kridel, and W. T. Lowther. 2007. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by orlistat. Nat. Struct. Mol. Biol. 14 704–709. [DOI] [PubMed] [Google Scholar]
- 31.Kuhajda F. P., E. S. Pizer, J. N. Li, N. S. Mani, G. L. Frehywot, and C. A. Townsend. 2000. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA. 97 3450–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg I., J. R. Walker, and K. Bloch. 1973. Inhibition of lipid synthesis in Escherichia coli cells by the antibiotic cerulenin. Antimicrob. Agents Chemother. 3 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weis B. C., A. T. Cowan, N. Brown, D. W. Foster, and J. D. McGarry. 1994. Use of a selective inhibitor of liver carnitine palmitoyltransferase I (CPT I) allows quantification of its contribution to total CPT I activity in rat heart. Evidence that the dominant cardiac CPT I isoform is identical to the skeletal muscle enzyme. J. Biol. Chem. 269 26443–26448. [PubMed] [Google Scholar]
- 34.Wenz A., C. Thorpe, and S. Ghisla. 1981. Inactivation of general acyl-CoA dehydrogenase from pig kidney by a metabolite of hypoglycin A. J. Biol. Chem. 256 9809–9812. [PubMed] [Google Scholar]
- 35.Miyake Y., Y. Kozutsumi, S. Nakamura, T. Fujita, and T. Kawasaki. 1995. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 211 396–403. [DOI] [PubMed] [Google Scholar]
- 36.Mano N., M. Uchida, H. Okuyama, I. Sasaki, S. Ikegawa, and J. Goto. 2001. Simultaneous detection of cholyl adenylate and coenzyme A thioester utilizing liquid chromatography/electrospray ionization mass spectrometry. Anal. Sci. 17 1037–1042. [DOI] [PubMed] [Google Scholar]