

Abstract
We evaluated the morphological and functional features of hepatic cyst epithelium in adult autosomal dominant polycystic kidney disease (ADPKD). In six ADPKD patients, we investigated the morphology of cyst epithelium apical surface by scanning electron microscopy and the expression of estrogen receptors (ERs), insulin-like growth factor 1 (IGF1), IGF1 receptors (IGF1-R), growth hormone receptor, the proliferation marker proliferating cell nuclear antigen, and pAKT by immunohistochemistry and immunofluorescence. Proliferation of liver cyst-derived epithelial cells was evaluated by both MTS proliferation assay and [3H]thymidine incorporation into DNA. The hepatic cyst epithelium displayed heterogeneous features, being normal in small cysts (<1 cm), characterized by rare or shortened cilia in 1- to 3-cm cysts, and exhibiting the absence of both primary cilia and microvilli in large cysts (>3 cm). Cyst epithelium showed marked immunohistochemical expression of ER, growth hormone receptor, IGF1, IGF1-R, proliferating cell nuclear antigen, and pAKT. IGF1 was 10-fold more enriched in the hepatic cyst fluid than in serum. Serum-deprived liver cyst-derived epithelial cells proliferated when exposed to 17β-estradiol and IGF1 and when exposed to human cyst fluid. ER or IGF1-R antagonists inhibited the proliferative effect of serum readmission, cyst fluid, 17β-estradiol, and IGF1. Our findings could explain the role of estrogens in accelerating the progression of ADPKD and may suggest a potential benefit of therapeutic strategies based on estrogen antagonism.
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most prevalent human genetic diseases caused by mutations in the PKD genes.1,2,3,4 More than 50% of ADPKD patients display hepatic cysts, which are the most prevalent extrarenal clinical manifestation of the disease and which account for a significant morbidity.1,2,3,4 Hepatic cysts are lined by epithelial cells featuring phenotypical and functional characteristics of biliary epithelium with enhanced secretory and proliferative activities.5,6,7 Fluid secretion, extracellular matrix remodeling, proliferation of the lining epithelial cells, and neovascularization each are considered important steps in promoting cyst growth.8 Very recently, a number of growth factors and cytokines that are increased in serum or cystic fluid have been shown to drive cyst growth and expansion.8,9
In the last few years, important advances have been achieved in the pathophysiology of intrahepatic biliary epithelium.10 Recent experimental reports have outlined the relevance of primary cilia as mechanosensory organelles capable, when bended by bile flow, of modulating the intracellular levels of cAMP and Ca++, two key intracellular mediators of cholangiocyte secretory and proliferative activities.11,12 Unfortunately, no information exists on the morphological and functional characteristics of primary cilia in ADPKD patients. Furthermore, recent evidence indicates that the intrahepatic biliary epithelium is the target of GH/insulin-like growth factor 1 (IGF1) axis13 and is particularly sensitive to the proliferative effects of IGF113 and estrogens.14 Estrogens exert their proliferative effects either directly or by synergizing growth factors including nerve growth factor and IGF1.13,14,15,16,17,18 In addition, estrogens induce the expression and secretion of growth factors (IGF1, vascular endothelial growth factor) by epithelial cells.13,19 A number of clinical observations suggest that estrogens play a role in development and progression of hepatic cysts in ADPKDf. 1) Women with ADPKLD have more possibility to develop hepatic cysts with respect to men.20 2) Nulliparae have less probability to develop hepatic cysts with respect to pluripare in which there is a tight correlation between number and size of the hepatic cysts and number of pregnancies.20,21 3) Oral contraceptives or postmenopausal hormonal substitutive therapy increases the number and size of the hepatic cysts.22 No information exists on the role and mechanism by which estrogens and IGF1 modulate the functions of cyst epithelium in ADPKD.
The aim of our study was to investigate the morphological and functional features of hepatic cyst epithelium in ADPKD with specific focus on the morphology of primary cilia and on the involvement of estrogens and IGF1.
Materials and Methods
Human Liver Samples
We studied six patients (four females, two males, 61 to 78 years of age) with a diagnosis of ADPKD disease based on standard international criteria.23 Three ADPKD patients were submitted to liver transplantation and three patients received surgical resection. As controls, we investigated liver biopsies with a normal histology from patients submitted to laparotomy (n = 4, two females, age 59 to 75 years of age) and liver fragments from two female liver donors (45 and 52 years of age). The study protocol was in line with the ethical guidelines of the 1975 Declaration of Helsinki.
Light Microscopy and Immunohistochemistry
Common liver histology was performed in 3- to 4-μm sections stained with hematoxylin and eosin and Masson’s trichromic stains. For immunohistochemistry, glass slides were deparaffinated, and endogenous peroxidase activity was blocked by a 30-minute incubation in methanolic hydrogen peroxide (2.5%). The endogenous biotin was then blocked by a biotin blocking system (code X0590; DAKO, Copenhagen, Denmark) according to the instructions supplied by the vendor. Sections were then hydrated in graded alcohol and rinsed in phosphate-buffered saline (PBS, pH 7.4) before applying the primary antibody. Sections were incubated overnight at 4°C with polyclonal antibodies for estrogen receptor (ER)-α (a cocktail of three monoclonal antibodies, 33% each, 1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), ER-β (1:100 dilution, Santa Cruz Biotechnology), IGF-I (1:70 dilution, Santa Cruz Biotechnology), IGF1-R (1:80 dilution, Santa Cruz Biotechnology), phosphorylated-(p)-AKT (1:100 dilution, Santa Cruz Biotechnology), GH-R (rabbit anti-human, sc-20747; Santa Cruz Biotechnology), and proliferating cell nuclear antigen (PCNA) (PC10, 1:100 dilution; DAKO). Samples were then rinsed with PBS for 5 minutes, incubated for 10 minutes at room temperature with secondary biotinylated antibody (LSAB Plus system, DAKO, Milan, Italy), then with DAKO ABC (LSAB Plus system), and finally developed with 3–3′ diaminobenzidine. To demonstrate the specificity of immunoreaction, negative and positive controls were performed for all immunoreactions. For negative controls, the primary antibody was replaced (same dilution) with normal serum from the same species. For positive controls, the following tissues were tested: human breast carcinoma for ER-α, ER-β, IGF1, and IGF1-R; human kidney for pAKT; and human placenta for GH-R. Sections were analyzed in a coded manner using BX-51 light microscopy (Olympus, Tokyo, Japan) with a video cam (Spot Insight; Diagnostic Instrument, Inc., Sterling Heights, MI) and processed with an Image Analysis System (Delta Sistemi, Rome, Italy).
Immunofluorescence
After deparaffinization, sections were hydrated in graded alcohol and rinsed in phosphate-buffered saline and 0.1% Triton X (PBS-T) for 15 minutes and then incubated with 10% normal blocking serum in PBS for 30 minutes. After washing, slides were incubated overnight at 4°C with IGF1-R primary antibody (polyclonal goat anti-human, sc-7144; Santa Cruz Biotechnology) diluted in PBS with 1.5% normal blocking serum. Samples were rinsed in PBS-T with three changes and incubated for 45 minutes at room temperature with bovine anti-rabbit biotin-conjugated secondary antibody (sc-2363, Santa Cruz Biotechnology) diluted in PBS with 1.5% normal blocking serum (normal bovine serum and normal donkey serum, both from Santa Cruz Biotechnology). After washing, the slides were incubated with fluorescein-conjugated streptavidin (Jackson ImmunoResearch Laboratories, West Grove, PA) and donkey anti-goat Texas red-conjugated secondary antibody (sc-2783, Santa Cruz Biotechnology) for 20 minutes at room temperature in a dark chamber. Then the samples were washed in buffer and mounted with UltraCruz mounting medium (sc-24941, Santa Cruz Biotechnology). Pictures were taken by DM4500B light microscopy (Leica, Wetzlar, Germany).
Scanning Electron Microscopy (SEM)
Polycystic liver fragments were prepared by fixation at 4°C with 2.5% glutaraldehyde in 0.1 mol/L Sorensen’s phosphate buffer [0.067 mol/L Na2HPO4 plus 0.067 mol/L KH2PO4 (80:20, v/v), pH 7.4] and then washed in H2O for 30 minutes. Postfixation with OsO4 for 10 minutes at room temperature was performed, and the samples were washed again in phosphate buffer for 20 minutes, dehydrated in graded alcohol, and placed for critical point drying with liquid CO2. The samples were glued on stubs by Silver Dag (Achenson 1415M; Agar Scientific, Essex, UK) and covered with gold in a S150 sputter coat (Edwards, London, UK) and were examined with a Hitachi S4000 field emission SEM (Hitachi Ltd., Tokyo, Japan) operating at 8 to 10 kV.
Measurement of IGF1 in the Serum and Cyst Fluid of ADPKD Patients
IGF1 in the serum and cyst fluid of ADPKD patients and in the supernatant of liver cyst-derived epithelial (LCDE) cells was measured by an enzyme-linked immunosorbent assay kit (Diagnostic Systems Laboratories, Inc., Houston, TX). IGF1 was also measured in bile samples collected from patients (three females and two males, 62 ± 9 years of age) with external bile drainage by Kehr T-tube, submitted to surgery for benign biliary pathologies (biliary stones, common bile duct stenosis, and cholangitis). In these patients, bile samples were collected when serum liver enzymes returned normal. 17β-Estradiol levels in serum and cyst fluid were analyzed by a commercial radioimmunoassay kit (Cis-Diagnostici, Vicenza, Italy).
LCDE Cell Line
In vitro studies were performed on an immortalized cell line obtained from the epithelium lining the hepatic cysts from patients with ADPKD.7 LCDE cells were maintained in hormonally supplemented medium consisting of Dulbecco’s modified Eagle’s medium-Ham’s F-12 nutrient mixture (3:1) (Cambrex Bio Science, Walkersville, MD) supplemented with 1.8 × 10−4 mol/L adenine (LKT Laboratories, St. Paul, MN), 5 μg/ml insulin, 5 μg/ml transferrin (Calbiochem Biochemicals, Darmstadt, Germany), 2 × 10−9 mol/L triiodothyronine, 1.1 × 10−6 mol/L hydrocortisone, 1.64 × 10−6 human epidermal growth factor, 5.5 × 10−6 epinephrine, 10% fetal bovine serum (Gibco/BRL, Life Technologies, Italia srl., Milan, Italy), 100 U/ml of penicillin, and 100 μg/ml of streptomycin in a 5% CO2 atmosphere at 37°C. To evaluate the effect of 17β-estradiol or IGF1 on proliferation, LCDE cells, cultured in the appropriate medium containing 10% fetal bovine serum, were deprived of serum for 24 hours. Then, cells were maintained in serum-deprived conditions for an additional 4 hours for [3H]thymidine incorporation into DNA or for an additional 12 hours for MTS proliferation assay (controls) or exposed to serum, 17β-estradiol, IGF1, and/or specific antagonist for ER (Ici 182,780; Astra Zeneca, Basiglio, Italy)24 and IGF1-R (αIR3, Calbiochem Biochemicals)25 for an additional 4 to 12 hours. In detail, cell medium was replaced with a fresh serum-free medium without hormone supplementation and without phenol red but added with the tested agent. 17β-Estradiol was dissolved in dimethyl sulfoxide, IGF1 and αIR3 in saline, and Ici 182,780 in ethanol as stock solution, which was then added (dilution, 1:10,000) to serum-free culture medium. In selected experiments, the effect of cyst fluid on the proliferation of LCDE cell line was also evaluated. For this purpose, as described by others,26 cyst fluid pooled from five ADPKD patients was diluted (v/v) with LCDE culture medium to 20% final concentration. Proliferation of LCDE cells was evaluated by [3H]thymidine incorporation into DNA as described above.
Cell Proliferation Assays
Cell proliferation was assessed, as previously described27 by both MTS proliferation assay and [3H]thymidine incorporation into DNA. For MTS proliferation assay, we used a commercially available colorimetric cell proliferation assay (CellTiter 96 aqueous nonradioactive cell proliferation assay, MTS Kit; Promega, Madison, WI), following the manufacturer’s instructions. Proliferation index was calculated as the ratio (multiplied × 100) between cell numbers in unstimulated and stimulated cultures as described.27 For [3H]thymidine incorporation into DNA, methyl [3H]thymidine (25 Ci/mmol; Amersham Biosciences Europe GmbH, Cologno Monzese, Italy) was added into the culture medium (1 μCi/ml), and the incubation with [3H]thymidine continued for 2 hours, corresponding to the last 2 hours of treatment with serum, 17β-estradiol, IGF1, and/or receptor antagonists. Cell extraction and radioactivity measurement were performed as described.27 The carrier of 17β-estradiol (ie, dimethyl sulfoxide) and of Ici 182,780 (ie, ethanol) showed no effect (1:10,000 dilution) on MTS proliferation assay (n = 6) or on [3H]thymidine incorporation into DNA (n = 6) of LCDE cells as compared with controls in the absence of these two additives.
Western Blot Analyses
For Western blot analysis, after each treatment, LCDE cells were harvested, washed with PBS, and homogenized by sonicating in lysis buffer (15 mmol/L Tris-HCl, pH 7.4, 5 mmol/L ethylenediaminetetraacetic acid, 100 mmol/L NaCl, Igepal 1%, 2 mmol/L phenylmethyl sulfonyl fluoride, 2 mmol/L benzamidine, and 1% aprotinin) on ice for 30 to 60 seconds. After centrifugation at 14,000 × g for 60 seconds at 4°C, the supernatant was recovered and protein concentration determined with protein assay-dye reagent (Bio-Rad Laboratories GmbH, München, Germany). Cell extracts (20 μg) were diluted in 6× Laemmli sample buffer (100 mmol/L Tris-HCl, pH 6.8, 20% glycerol, 4% sodium dodecyl sulfate, 5% 2-mercaptoethanol, 0.1% bromophenol blue) and resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western blotting was performed as described10,11,12,13,14,15 by using the following primary antibodies (Santa Cruz Biotechnology): anti-IGF1, rabbit polyclonal antibody (1:100 dilution), anti-IGF1-Rβ, rabbit polyclonal antibody (1:400 dilution), anti-ER-α rabbit polyclonal antibody (1:200 dilution), and anti-ER-β rabbit polyclonal antibody (1:200 dilution). As secondary antibodies, anti-mouse IgG peroxidase conjugated (1:2000 dilution) or anti-rabbit IgG peroxidase conjugated (1:10,000) were used (Santa Cruz Biotechnology).
Statistical Analysis
Data are presented as arithmetic mean ± SE. Statistical analysis was conducted by using one-way analysis of the variance with pairwise comparison by the Fisher’s protected-least-significant-difference test. Only P ≤ 0.05 was considered significant.
Results
Human Liver Samples
Figure 1 shows the SEM study of apical surface of intrahepatic bile ducts in normal liver (interlobular ducts of 20 to 100 μm diameter examined in six normal livers) and of the epithelium lining hepatic cysts of different size in six ADPKD patients. The normal biliary epithelium (Figure 1A) and the epithelium lining cysts of small size (<1 cm maximum diameter, five cysts examined; Figure 1Ba) showed a similar apical surface carpeted by dense and regular microvilli and a typical solitary, cylindrical primary cilium projecting from the surface of almost each cell, with a length longer than 4 μm. In contrast, marked morphological abnormalities of apical surface were observed in larger cysts. In cysts with a maximum diameter between 2 and 3 cm (12 cysts examined, Figure 1Bb), microvilli were well represented, but clear areas were frequently seen. Most importantly, in these cysts (2 to 3 cm), cilia were rare (1/200 cells), shortened (1.25 ± 0.29 μm), and often showed alteration of the apical end (Figure 1Bb). Finally, cysts with a maximal diameter higher than 3 cm showed (Figure 1Bc) an epithelial apical surface completely lacking of both microvilli and primary cilia, and this was a consistent finding in all cysts examined (five hepatic cysts investigated, three different surface areas observed for each cyst) regardless of location in the different hepatic lobes.
Figure 1.

SEM. of luminal surface of normal bile duct (A) and hepatic cystic epithelium of ADPKD liver (B). A: Microvilli and long cilia are present in the luminal surface of normal cholangiocytes. B: a: Similar to the surface of normal cholangiocytes, the epithelium lining small cyst (<1 cm maximum diameter) shows a carpet of regular microvilli and typical primary cilia. Inset: At higher magnification the normal morphology of the cilium is evident. b: the epithelial surface of a cyst with a 2 to 2.5 cm maximum diameter shows less dense microvilli and clear areas become visible. The cilium may be absent or, when present, it is short (2 to 3 μm) and often showed alteration of the apical zone. Inset: At higher magnification a short cilium is evident together with an area clear of microvilli. c: The epithelial surface of a large cyst (>3 cm), appeared completely smooth and extremely thin (black arrows). CT, subepithelial connective tissue. Scale bars: 4 μm (A); B: 8 μm (a, b, inset in a); 20 μm (c), 4 μm (inset in b).
As previously described,28,29 cholangiocytes of normal intrahepatic bile ducts (20 to 100 μm) were negative at the immunohistochemical analysis of ER-α and -β (Figure 2, A and E). In ADPKD, the hepatic cyst epithelium showed a positive staining for ER-α (Figure 2, B–D) and a uniform and strong positivity for ER-β (Figure 2, F–H) involving both nucleus and cytoplasm, and this was constantly found in all cysts examined independently from size and location in hepatic segments. In ADPKD livers, reactive bile ducts close to the cysts are also positive for ER-α and -β (Figure 2, C and G) as also described for reactive ducts associated with other liver pathologies.10 In contrast, intrahepatic bile ducts with normal morphology, in ADPKD livers, were negative for ER-α and -β (Figure 2, B and F).
Figure 2.

Immunohistochemistry for ER-α and ER-β in normal liver and ADPKD hepatic cysts. In normal (A, E) livers, cholangiocytes lining intrahepatic bile ducts (yellow arrows) are negative for ER-α (A) and ER-β (E). In the ADPKD liver, cholangiocytes lining intrahepatic bile ducts with normal morphology remain negative for ER-α (B, yellow arrow) and ER-β (F, yellow arrow) whereas, cholangiocytes of reactive bile ducts close to the cysts are positive for both ER-α (C) and ER-β (G). The epithelium lining small (C, G) and large cysts (D, H) showed strong positivity for ER-α (C, D) and ER-β (G, H) located at both cytoplasmic and nuclear levels (red arrows). Scale bars: 40 μm (A, B, E–G); 20 μm (C, D, H).
The immunohistochemistry for IGF1 and IGF1-R in cholangiocytes lining intrahepatic bile ducts from normal livers appeared negative (<1% cholangiocytes showed a weak positivity; Figure 3, A and D). In contrast, IGF1 (Figure 3, B and C) and IGF1-R (Figure 3, E and F) were strongly positive in all of the epithelial cells lining hepatic cysts of different diameter and hepatic location. As shown in Figure 3F (inset), IGF1-R was localized in the apical pole of the cystic epithelium other than in the basolateral pole, and this was also confirmed by immunofluorescence (Figure 3G). Because experimental studies showed that the intrahepatic biliary epithelium is a target of the growth hormone (GH)/IGF1 axis,13 we investigated by immunohistochemistry the expression of the receptor (GH-R) in the cyst epithelium. In the normal liver, both hepatocytes and cholangiocytes were positive for GH-R (Figure 4A), and the positivity was maintained in the cystic epithelium (Figure 4B).
Figure 3.

Immunohistochemistry for IGF1 and IGF1-R in normal liver and ADPKD hepatic cysts and immunofluorescence for IGF1-R in hepatic cyst. In normal livers cholangiocytes are negative for IGF1 (A) and IGF1-R (D) (arrows, bile ducts). In ADPKD livers, both IGF1 (B, C) and IGF1-R (E, F) showed positive immunolocalization in the epithelium of small (B, E) and large cysts (C, F). At higher magnification the prevalent apical immunolocalization of IGF1-R is evident, by both immunohistochemistry (F, inset) and immunofluorescence (G, yellow arrows), in the epithelium of a large cyst (asterisk indicates the negativity of subepithelial connective tissue). Scale bars: 40 μm (A–F); 4 μm (G); 6 μm (inset in F).
Figure 4.

Immunohistochemistry for GH-R in normal liver and ADPKD hepatic cyst. A: In normal liver, cholangiocytes lining intrahepatic bile ducts (yellow arrows) and hepatocytes were positive for GH-R. B: In the ADPKD liver, the epithelium lining a large cyst showed strong positivity for GH-R. Scale bars: 40 μm (A); 30 μm (B).
The PI3-kinase/AKT pathway is the intracellular pathway activated by IGF1 in target cells including cholangiocytes.13 We evaluated the immunohistochemical expression of pAKT in normal and ADPKD biliary epithelium. Although less than 1% of cholangiocytes lining intrahepatic bile ducts of 20 to 100 μm displayed a thin positivity for pAKT (Figure 5A), the epithelium lining hepatic cysts showed an intense and diffuse cytoplasmic and nuclear pAKT positivity (Figure 5B) suggesting activation of the IGF1 system that together with estrogens probably sustains the enhanced proliferative activities characterizing cyst epithelium.5,6,7 In fact, the cyst epithelial cells showed an intense staining for PCNA (Figure 5D), a marker of cell proliferation, involving almost all cells, whereas in the normal intrahepatic biliary epithelium PCNA was virtually negative (Figure 5C), consistent with the quiescent status of normal cholangiocytes.10
Figure 5.

Immunohistochemistry for p-AKT and PCNA in normal liver and ADPKD hepatic cyst. In the normal liver cholangiocytes are negative for both p-AKT (A) and PCNA (C) (arrow, bile ducts). In the ADPKD liver, a strong and specific immunoreactivity for pAKT (B) and PCNA (D) of cystic epithelium is evident. At higher magnification (D, inset) the nuclear PCNA positivity is evident. Scale bars = 40 μm.
Concentration of IGF1 and 17β-Estradiol in Serum and Cyst Fluid of ADPK Patients
IGF1 concentration in the cyst fluid aspirated from hepatic cysts of five ADPKD patients was found to be 10-fold higher (766 ± 113 ng/ml) than that measured in the serum of the same patients (79 ± 9 ng/ml; normal range of healthy patients = 49 to 314 ng/ml) and 20-fold higher than that measured in bile samples collected from Kehr T-tube of five patients submitted to surgery for benign biliary pathologies (bile IGF1 = 38 ± 8 ng/ml, n = 5). The concentration of 17β-estradiol in the serum of six ADPKD patients was 33 ± 7 pg/ml (normal values menopause = 1 to 30 pg/ml) whereas, in the cyst fluid it was more than fourfold lower (7.5 ± 3.2 pg/ml, n = 5, P < 0.01 versus serum).
Effect of Estrogens and IGF1 on Cholangiocyte Proliferation
As evaluated by Western blot, LCDE cells constitutively expressed ER-α and -β, IGF1 and IGF1-R (Figure 6). To evaluate the effects of estrogens and IGF1 on proliferation, LCDE cells were starved without serum for 24 hours and then left without serum (controls) or exposed to serum, 17β-estradiol, IGF1, and/or receptor antagonist for an additional 12 hours for MTS proliferation assay and for an additional 4 hours for [3H]thymidine incorporation into DNA. When serum-deprived LCDE cells were exposed to the ER antagonists Ici 182,780 (1 μmol/L), or to the IGF1-R blocking antibody, αIR3 (1 μg/ml), no significant changes in proliferation were seen, as evaluated by both MTS proliferation assay (n = 5) and [3H]thymidine incorporation into DNA (n = 5) (not shown). Readmission of serum (n = 8) induced a marked (P < 0.02) increase of proliferation (MTS proliferation assay and [3H]thymidine) and a similar effect was found when serum-deprived (24 hours) LCDE cells were exposed to 10 nmol/L 17β-estradiol (n = 8, P < 0.02) or 10 ng/ml (1.3 nmol/L) IGF1 (n = 8, P < 0.02; Figure 7, A and B). The effect of serum readmission on proliferation of LCDE cells was partially blocked by the ER-specific antagonist, Ici 182,780 (P < 0.05 versus serum readmission or controls; Figure 8, A and B) or by the IGF1-R blocking antibody αIR3 (P < 0.05 versus serum readmission or controls; Figure 8, A and B) but totally blocked when ER and IGF1-R antagonists were added together (Figure 8, A and B). In fact, when LCDE cells deprived of serum for 24 hours were exposed to serum in the presence of Ici 182,780 plus αIR3, both proliferation index (MTS proliferation assay) and [3H]thymidine incorporation into DNA were similar with respect to serum-deprived cells (controls; Figure 8, A and B). This indicates that most of the proliferative effect of serum readmission in serum-deprived LCDE cells depends on ER and IGF1-R.
Figure 6.
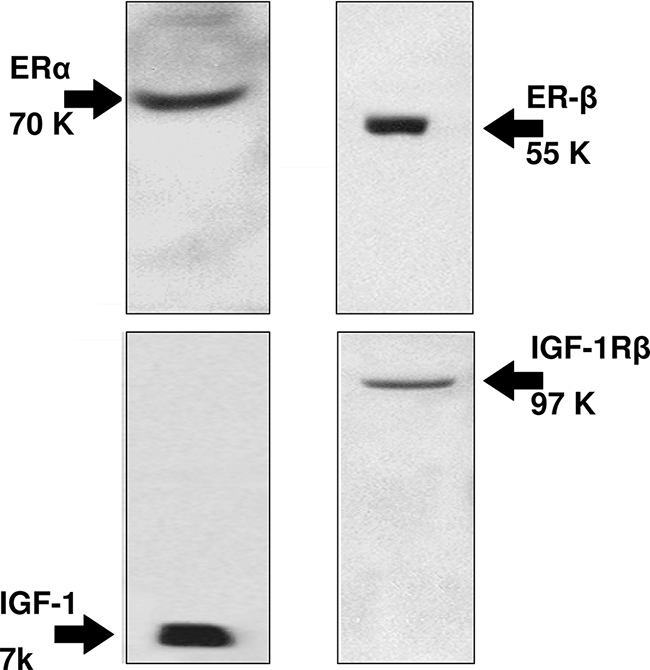
Protein expression (Western blot) of ER-α, ER-β, IGF1, and IGF1-R in LCDE cell line. LCDE cells express ER-α, ER-β, IGF1, and IGF1-R. Representative of five similar experiments.
Figure 7.

Effect of serum, 17β-estradiol, and IGF1 on proliferation of LCDE cell line. A: MTS proliferation assay. LCDE cells cultured in the appropriate medium containing 10% fetal bovine serum were deprived of serum and hormone supplementation (see Materials and Methods) for 24 hours. Then, cells were maintained in serum-deprived conditions for further 12 hours (controls) or exposed to serum, 17β-estradiol (17β-E, 10 nmol/L), IGF1 (10 ng/ml, 1.3 nmol/L) for further 12 hours. Proliferation index was calculated as the ratio (multiplied × 100) between cell number (MTS assay) in stimulated and unstimulated (control) cultures. *P < 0.02 versus controls (analysis of variance). Data were expressed as mean ± SE of n = 8. B: [3H]thymidine incorporation into DNA. [3H]thymidine was added into the culture medium (1 μCi/ml) for the last 2 hours of each treatment. LCDE cells cultured in the appropriate medium containing 10% fetal bovine serum were deprived of serum and hormone supplementation (see Materials and Methods) for 24 hours. Then, cells were maintained in serum-deprived conditions for further 4 hours (controls) or exposed to serum, 17β-estradiol (17β-E), or IGF1 for further 4 hours. Results were expressed as dpm/mg protein. *P < 0.02 versus controls (analysis of variance). Data were expressed as mean ± SE of n = 8.
Figure 8.

Effect of ER and IGF1-R antagonists on serum-induced proliferation of serum-deprived LCDE cells. A: MTS proliferation assay. HuH-28 cells cultured in the appropriate medium containing 10% fetal bovine serum were deprived of serum and hormone supplementation (see Materials and Methods) for 24 hours. Then, cells were maintained in serum-deprived conditions for further 12 hours (controls) or exposed to serum ± ER antagonist Ici 182,780 (1 μmol/L) or IGF1-R antagonist αIR3 (1 μg/ml) for 12 hours. Proliferation index was calculated as the ratio (multiplied × 100) between cell number (MTS assay) in stimulated and unstimulated (control) cultures. *P < 0.05 versus other columns; &P < 0.05 versus controls, serum readmission or serum plus Ici 182,780 plus αIR3 (analysis of variance). Data were expressed as mean ± SE of n = 10 independent experiments. B: [3H]thymidine incorporation into DNA. *P < 0.02 versus other columns; &P < 0.02 versus controls, serum readmission or serum plus Ici 182,780 plus αIR3 (analysis of variance). Data were expressed as mean ± SE of n = 8 independent experiments.
As evaluated by both MTS proliferation assay and [3H]thymidine incorporation into DNA, the effect of 10 nmol/L 17β-estradiol on the proliferation of LCDE cells (n = 8 to 10) was partially blocked by the ER-specific antagonist, Ici 182,780 but also by the IGF1-R antagonist, αIR3 (P < 0.05 versus 17β-estradiol alone, n = 8 to 10; Figure 9, A and B). The proliferative effect of 17β-estradiol was completely inhibited only in the presence of Ici 182,780 and αIR3 together (Figure 8, A and B; P < 0.05 versus serum plus Ici 182,780 or serum plus αIR3, n = 8 to 10; Figure 9, A and B). Because the specificity of αIR3 (specific blocking antibody) for IGF1-R is absolute,25 these findings indicate that the 17β-estradiol-induced LCDE cell proliferation involves the activation of both IGF1-R and ER. Conversely, the effect of IGF1 on the proliferation of LCDE cells was totally blocked by the IGF1-R antagonist αIR3 (n = 8 to 10, P < 0.05 versus IGF1 alone; Figure 10, A and B) but also partially blocked by the ER-specific antagonist Ici 182,780 μmol/L (n = 8 to 10, P < 0.05 versus IGF1 alone; Figure 10, A and B).
Figure 9.

Effect of ER and IGF1-R antagonists on 17β-estradiol-induced proliferation of serum-deprived LCDE cells. A: MTS proliferation assay. LCDE cells cultured in the appropriate medium containing 10% fetal bovine serum were deprived of serum and hormone supplementation (see Materials and Methods) for 24 hours. Then, cells were maintained in serum-deprived conditions for further 12 hours (controls) or exposed to serum, 17β-estradiol (17β-E, 10 nmol/L) in the absence or presence of ER antagonist Ici 182,780 (1 μmol/L) or IGF1-R antagonist αIR3 (1 μg/ml) for 12 hours. Proliferation index was calculated as the ratio (multiplied × 100) between cell number (MTS assay) in stimulated and unstimulated (control) cultures. *P < 0.05 versus other columns; &P < 0.05 versus 17β-estradiol alone or 17β-estradiol plus Ici 182,780 plus αIR3 (analysis of variance). Data are mean ± SE of n = 10. B: [3H]thymidine incorporation into DNA. *P < 0.05 versus other columns; &P < 0.05 versus 17β-estradiol alone or 17β-estradiol plus Ici 182,780 plus αIR3 (analysis of variance). Data were expressed as mean ± SE of n = 8.
Figure 10.

Effect of ER and IGF1-R antagonists on IGF1-induced proliferation of serum-deprived LCDE cells. A: MTS proliferation assay. LCDE cells cultured in the appropriate medium containing 10% fetal bovine serum were deprived of serum and hormone supplementation (see Materials and Methods) for 24 hours. Then, cells were maintained in serum-deprived conditions for further 12 hours (controls) or exposed to serum, IGF1 (10 ng/ml, 1.3 nmol/L) in the absence or presence of ER antagonist, Ici 182,780 (1 μmol/L), or IGF1-R antagonist αIR3 (1 μg/ml) for 12 hours. Proliferation index was calculated as the ratio (multiplied × 100) between cell number (MTS assay) in stimulated and unstimulated (control) cultures. *P < 0.05 versus other columns; &P < 0.05 versus IGF1 alone or IGF1 plus αIR3 plus Ici 182,780. Data were expressed as mean ± SE of n = 10. B: [3H]thymidine incorporation into DNA. *P < 0.05 versus other columns; &P < 0.05 versus IGF1 alone or IGF1 plus αIR3 plus Ici 182,780 (analysis of variance). Data were expressed as mean ± SE of n = 8.
Effect of Cyst Fluid on Proliferation of LCDE Cells
To evaluate the effect on LCDE proliferation, cyst fluid pooled from five cysts of ADPKD patients was diluted (v/v) with LCDE culture medium to 20% final concentration. When serum-deprived (24 hours) LCDE cells were exposed to cyst fluid for an additional 4 hours, proliferation of LCDE cells was significantly activated (P < 0.02 versus serum-deprived, n = 8; Figure 11), as evaluated by [3H]thymidine incorporation into DNA. The effect of cyst fluid was decreased by ∼55% by the IGF1-R antagonist, αIR3 (n = 8, P < 0.05 versus cyst fluid alone; Figure 11) and by 28% by the ER antagonist Ici 182,780 (n = 8; P < 0.05 versus cyst fluid alone or versus cyst fluid plus αIR3; Figure 11).
Figure 11.

Effect of cyst fluid on proliferation of LCDE cells. Cyst fluid pooled from five cysts of ADPKD patients was diluted (v/v) with LCDE culture medium to 20% final concentration. Serum-deprived (24 hours) LCDE cells were left without serum for an additional 4 hours (controls) or exposed to cyst fluid ± αIR3 or Ici 182,780 for 4 hours. [3H]Thymidine incorporation into DNA was significantly enhanced by cyst fluid indicating activation of cell proliferation. The effect of cyst fluid was decreased by 55% by the IGF1-R antagonist αIR3 and by 28% by the ER antagonist, Ici 182,780. Data were expressed as mean ± SE of n = 8. *P < 0.02 versus controls; &P < 0.05 versus cyst fluid alone; £P < 0.05 versus cyst fluid alone and versus cyst fluid plus αIR3 (analysis of variance).
Effect of Estrogens on the Secretion of IGF1 on the Supernatant of LCDE Cells
To evaluate whether estrogens stimulate IGF1 secretion, LCDE cells were incubated with or without 10 nmol/L 17β-estradiol for 4 hours at 37°C, and IGF1 was measured in the supernatant. IGF1 concentration in the supernatant was significantly higher after 4 hours of incubation of LCDE cells with 10 nmol/L 17β-estradiol than in control cells incubated with the carrier dimethyl sulfoxide (0.42 ± 0.03 ng/ml versus 0.11 ± 0.02 ng/ml, P < 0.01, n = 6). The effect of 17β-estradiol on IGF1 secretion in the supernatant was only partially blocked by the ER-specific antagonist Ici 182,780 (0.24 ± 0.02 ng/ml, n = 6, P < 0.05 versus 17β-estradiol or dimethyl sulfoxide controls), suggesting that IGF1 secretion is a partially ER-independent effect of 17β-estradiol.
Discussion
Our study firstly focused on liver cysts of patients with ADPKD, a diagnosis made by internationally accepted criteria.23 Large liver specimens were obtained from patients submitted to liver resection or transplantation, and this allowed accurate SEM and immunohistochemistry evaluation. The SEM study demonstrated that apical surface of cyst epithelium showed heterogeneous abnormalities depending on cyst size. The epithelial cells lining small cysts (<1 cm) showed an apical surface with adequately represented cilia and microvilli and which is similar to normal biliary epithelium investigated in this and previous studies.30,31 In contrast, the apical surface of medium (2 to 3 cm) cysts showed areas clear of microvilli with rare and shortened cilia. These features further worsen in large (>3 cm) hepatic cysts that totally lack microvilli and primary cilia. We cannot definitively establish whether these morphological abnormalities of primary cilia and microvilli represent a mechanic consequence of enhanced endoluminal pressure during cyst growth or a primary feature. The fact that these features progressively worsen with increasing cyst size should suggest a mechanic event, probably caused by enhanced endoluminal pressure. However, the possibility that the cyst growth occurs only in those cysts displaying cilia derangement or disappearance and that the rate of cyst growth strictly depends on how much cilia are impaired cannot completely be excluded. ADPKD is caused by mutations in PKD1 or PKD2, and at a cellular level it appears to be recessive where somatic second hits in the normal allele of cells containing the germ line mutation initiate or accelerate the formation of cysts.32,33,34,35 Several reports have shown that loss of heterozygosity and acquired somatic second hits may account, at least partly, for the heterogeneity of the disease, and, furthermore, the existence of other modifying loci has also been hypothesized.32,33,34,35 Reports regarding a trans-heterozygous model for cystogenesis add further complexity to the molecular mechanisms that can lead to pathogenesis.33,35 In fact, in cystic DNA from a kidney of an ADPKD1 patient, somatic mutations not only in the PKD1 gene of certain cysts, but also in the PKD2 gene of others, have been demonstrated, thus generating a trans-heterozygous state with mutations in both genes.35 With this background, the phenotypic heterogeneity of the apical surface of the hepatic cyst epithelium shown in this study is not surprising.
Recent experimental reports outlined the relevance of primary cilia in the modulation of cholangiocyte pathophysiology.11,12 Specifically, it was shown that cholangiocyte cilia express a mechanoreceptor, polycystin-1 (PC-1), a Ca++ channel, polycystin-2 (PC-2, or transient receptor potential P2 according to the transient receptor potential nomenclature) and the Ca++-inhibitable adenylyl cyclase isoform 6 (AC6).11 Perfusion of rat isolated bile duct units at flow rates of sufficient magnitude to bend cilia resulted in increased [Ca2+]i and in decreased forskolin-stimulated [cAMP]i levels, which was significantly reduced or abolished when cholangiocyte cilia were removed by chloral hydrate or when PC-1, PC-2, and AC6 were individually down-regulated by siRNAs.11 A possible suggestion is that cilia movement determines a threshold of intracellular Ca++ where proliferation is inhibited and that the absence of cilia in ADPKD could cause an impaired Ca++-dependent counter regulation of cAMP, the latter being a key determinant of cholangiocyte proliferation.10,29 Several observations demonstrate that the epithelium of both hepatic and kidney cysts displays features of aberrant proliferation including overexpression of growth factor receptors and activation of related intracellular pathways.1,2,3,4,5,6,7,8,9 In our study, the intense PCNA staining (Figure 5) confirms the enhanced proliferative activity of cyst epithelium, and the overexpression of pAKT, ER, and IGF1-R (Figures 2 to 5) are additional features. Strategically located within the cilia, polycystin 1 and 2 do interact, perhaps as part of a larger complex involving also other proteins, and work as important mediators of ciliary mechanosensation.11,12,30,31,36 Loss of this important function because of mutational changes in PKD1 or PKD2 occurring in ADPKD leads to loss of normal control over cellular proliferation, resulting in cyst formation.37,38,39 The disappearance or shortening of primary cilia has been so far described only in renal cysts but not in hepatic cysts of ADPKD patients.31,40,41 Only recently marked morphological abnormalities of primary cilia, resembling those described by us in cysts (>2 cm diameter) of ADPKD patients, have been reported in experimental models of polycystic liver.11,12,36 Therefore, structural derangement of primary cilia and the morphological heterogeneity among cysts of different sizes, phenotypically characterize the ADPKD epithelium at both renal and hepatic levels. Nevertheless, as described in a number of previous studies, the hepatic cyst epithelium displays immunohistochemical, structural, and functional features of cholangiocytes, including response to secretin and somatostatin, but with enhanced proliferative and secretory activities.5,6,7,8 Previous experimental and clinical studies have shown that proliferating cholangiocytes show an enhanced response to hormones, neuropeptides, growth factors, and cytokines and secrete large amounts of different agents,9,10,13,14,15,16,17,18,42,43,44 which through autocrine mechanisms sustain the cell proliferative machinery and exert anti-apoptotic effects. Among these agents, estrogens and IGF110,13,14,15,16,17,18,42,43,44 play a crucial role in sustaining proliferation of rat and human cholangiocytes by acting on specific receptors. They exert additive proliferative effects at both receptor and postreceptor levels in which survival pathways are activated.10,13,14,15,16,17,18,42,43,44 These studies and the clinical observations showing a strict estrogen sensitivity of cyst formation and progression in ADPKD patients20,21,22 represented the background for our study. First of all, the immunohistochemical studies indicate that the epithelial layer of hepatic cysts stains intensely for ER-β, GH-R, IGF1, and IGF1-R, and this occurs in all cysts examined whereas the staining for ER-α was less evident. The expression of GH-R, IGF1, and IGF1-R in cyst epithelium is consistent with recent studies from our group showing that rodent biliary epithelium is a target of GH/IGF1 axis.13 This has important clinical implications because recent studies demonstrated that octreotide, an inhibitor of GH/IGF1 axis, suppresses hepatic cyst growth.45 As far as estrogens are concerned, consistent with the benign nature of its proliferation, the cyst epithelium showed similarities, in terms of ER expression, with respect to the reactive proliferation associated with chronic cholestatic liver diseases (ie, less immunohistochemistry expression of ER-α than ER-β)28 whereas the neoplastic proliferation (ie, cholangiocarcinoma) is characterized by relatively much higher ER-α expression.27 In addition, we showed that epithelial cells lining hepatic cysts showed a strong immunohistochemistry positivity for IGF1, IGF1-R, and pAKT, the latter indicating activation of the PI3-kinase pathway, which is the main intracellular signal activated during IGF1-induced proliferation other than a main survival pathway.13 In the hepatic cyst fluid, IGF1 was found to be 10-fold more concentrated than in the serum of the same ADPKD patients and 20-fold more concentrated than in human bile. As suggested for other growth factors and cytokines concentrated in the cyst fluid,8 IGF1 may participate in inducing cell proliferation by acting on its receptor located on the cell apical pole (Figure 3, F and G). Our experiments with LCDE cells showing that cystic fluid-induced proliferation is sensitive to IGF1-R-specific blocker, αIR3, are consistent with this hypothesis, and the lack of a complete inhibition of cell proliferation by αIR3 indicates that IGF1 is only one of the possible players present in the cyst fluid. In contrast with IGF1, estrogens are fourfold less concentrated in cyst fluid than in the serum of the same patients, suggesting that estrogens mainly act through the cell basolateral pole.
To evaluate directly the role of estrogens, IGF1 and their receptors in the modulation of proliferation, we used an immortalized cell line (LCDE) obtained from the epithelium of liver cysts of ADPKD patients. LCDE cells constitutively express ER-α and -β, IGF1, and IGF1-R (Figure 6). In LCDE cells starved without serum for 24 hours, serum readmission induces a marked activation of cell proliferation as evaluated by two distinct methods. Interestingly, at the same experimental conditions, 17β-estradiol and IGF1 induce a rate of proliferation similar with respect to serum readmission, which was completely inhibited by two highly specific ER (Ici 182,780) and IGF1-R (α-IR3) antagonists.24,25 On the other hand, Ici 182,780 and α-IR3 showed no effect in the absence of serum, thus excluding nonspecific toxic effects. This indicates that proliferation of LCDE cells induced by serum readmission requires intact ER and IGF1-R and that these receptors play a major role in the complex loop of agents modulating cell proliferation. Although IGF1-induced proliferation was completely inhibited by the specific IGF-R-blocking antibody (α-IR3), that induced by 17β-estradiol was only partially inhibited by ER antagonist, Ici 182,780, which also partially blocked the proliferative effect of IGF1. Conversely, 17β-estradiol-induced proliferation was also partially inhibited by the IGF1-R antagonist and completely blocked only when LCDE cells were exposed to both ER and IGF1-R blockers. This indicates that IGF1-R is directly involved in estrogen-induced proliferation because the specificity of αIR3 for IGF1 is absolute25 and nonspecific interference of this blocking antibody could be excluded. Our findings indicate a sort of interplay between estrogen and IGF1 receptors in modulating LCDE proliferation. This is consistent with previous reports in other estrogen-sensitive cells, where IGF1 and estrogens display a number of different mechanisms of cooperation in modulating cell differentiation and proliferation.46,47,48,49 An additional mechanism of cooperation is the IGF1 secretion induced by estrogens, an effect only partially inhibited by ER antagonist, which has been described by us in rat proliferating cholangiocytes13 and now confirmed in LCDE cells. This could explain the marked enrichment of IGF1 found in the cyst fluid of ADPKD. The lack of a complete inhibitory effect of Ici 182,780 on both 17β-estradiol-induced proliferation and IGF1 secretion on LCDE cannot be attributed to the dose of Ici 182,780 used (100-fold higher than the agonist) because this dose fully antagonized ER.24 Rather, the estrogen-induced IGF1 secretion, as a partially receptor-independent effect, already described in other cell types, should be taken into consideration.46,47,48,49
In conclusion, we showed that the hepatic cyst epithelium of ADPKD patients display marked but heterogeneous abnormalities of primary cilia, and this could represent an additional example of a link between cilia abnormalities and disease pathogenesis that have been previously suggested for PKD, nephronophthisis, Senior-Loken (renal-retinal) syndrome, retinitis pigmentosa, anosmia, and laterality disturbances.50 We have also shown that ADPKD epithelium is sensitive to the proliferative effects of estrogens and IGF1. Estrogens act not only directly but also by promoting the synthesis and release of growth factors from the cyst epithelium. Our study furnishes the scientific background for the clinical observations showing how the formation and progression of hepatic cysts is highly sensitive to changes in the estrogen status in the body.
Acknowledgments
We thank F. Lucarelli and C. Tesse for technical assistance in immunoblotting; and C. N. De Cecco, G. Tisone, and G. Battisti for technical assistance in liver sample collection.
Footnotes
Address reprint requests to Domenico Alvaro, M.D., Division of Gastroenterology, Department of Clinical Medicine, University of Rome “Sapienza,” via R. Rossellini 51, 00137 Rome, Italy. E-mail: domenico.alvaro@uniroma1.
Supported by the Scott and White Hospital (grant award to G.A.), the Texas A&M University System (to G.A.), the Veterans Administration (merit award and research scholar award to G.A.), the National Institutes of Health (grants DK 58411 and DK062975 to G.A. and P30 DK-34928Y to D.M.J.), MIUR (Italian Ministry of Education, University and Research) (PRIN 2005 grants 2005069739_003 and 2005067975_002 to D.A. and A.F.A), Cofin 2003 (to E.G. and P.O.), Cofin 2005 (to E.G. and P.O.), Biomedicina (cluster C04, progetto n.ro 5, and ex 60% to E.G. and P.O.), the Fondazione San Martino (to M.S.), Bergamo (to M.S.), and Yale University (recruitment grant to M.S.).
References
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- Ramos A, Torres VE, Holley KE, Offord KP, Rakela J, Ludwig J. The liver in autosomal dominant polycystic kidney disease. Implications for pathogenesis. Arch Pathol Lab Med. 1990;114:180–184. [PubMed] [Google Scholar]
- Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65:1020–1025. doi: 10.1016/s0025-6196(12)65165-9. [DOI] [PubMed] [Google Scholar]
- Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. 2005;100:2569–2582. doi: 10.1111/j.1572-0241.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- Everson GT, Emmett M, Brown WR, Redmond P, Thickman D. Functional similarities of hepatic cystic and biliary epithelium: studies of fluid constituents and in vivo secretion in response to secretin. Hepatology. 1990;11:557–565. doi: 10.1002/hep.1840110406. [DOI] [PubMed] [Google Scholar]
- Perrone RD, Grubman SA, Rogers LC, Lee DW, Moy E, Murray SL, Torres VE, Jefferson DM. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol. 1995;269:G335–G345. doi: 10.1152/ajpgi.1995.269.3.G335. [DOI] [PubMed] [Google Scholar]
- Perrone RD, Grubman SA, Murray SL, Lee DW, Alper SL, Jefferson DM. Autosomal dominant polycystic kidney disease decreases anion exchanger activity. Am J Physiol. 1997;272:C1748–C1756. doi: 10.1152/ajpcell.1997.272.5.C1748. [DOI] [PubMed] [Google Scholar]
- Nichols MT, Gidey E, Matzakos T, Dahl R, Stiegmann G, Shah RJ, Grantham JJ, Fitz JG, Doctor RB. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- Fabris L, Cadamuro M, Fiorotto R, Roskams T, Spirli C, Melero S, Sonzogni A, Joplin RE, Okolicsanyi L, Strazzabosco M. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Mancino MG, Glaser S, Gaudio E, Marzioni M, Francis H, Alpini G. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology. 2007;132:415–431. doi: 10.1053/j.gastro.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Isolation and characterization of cholangiocyte primary cilia. Am J Physiol. 2006;291:G500–G509. doi: 10.1152/ajpgi.00064.2006. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Drudi Metalli V, Alpini G, Onori P, Franchitto A, Barbaro B, Glaser SS, Francis H, Cantafora A, Blotta I, Attili AF, Gaudio E. The intrahepatic biliary epithelium is a target of the growth hormone/insulin like growth factor 1 axis. J Hepatol. 2005;43:875–883. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Alpini G, Onori P, Perego L, Svegliati Baroni G, Franchitto A, Baiocchi L, Glaser SS, Le Sage G, Folli F, Gaudio E. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology. 2000;119:1681–1691. doi: 10.1053/gast.2000.20184. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Alpini G, Onori P, Franchitto A, Glaser SS, Le Sage G, Folli F, Attili AF, Gaudio E. Alfa and beta estrogen receptors and the biliary tree. Mol Cell Endocrinol. 2002;193:105–108. doi: 10.1016/s0303-7207(02)00103-x. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Alpini G, Onori P, Franchitto A, Glaser S, Le Sage G, Gigliozzi A, Attili AF, Gaudio E. Effect of ovariectomy on the proliferative capacity of intrahepatic biliary epithelium. Gastroenterology. 2002;123:336–344. doi: 10.1053/gast.2002.34169. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Onori P, Metalli VD, Svegliati-Baroni G, Folli F, Franchitto A, Alpini G, Mancino MG, Attili AF, Gaudio E. Intracellular pathways mediating estrogen-induced cholangiocyte proliferation in the rat. Hepatology. 2002;36:297–304. doi: 10.1053/jhep.2002.34741. [DOI] [PubMed] [Google Scholar]
- Gigliozzi A, Alpini G, Svegliati Baroni G, Marucci L, Drudi Metalli V, Glaser SS, Francis H, Mancino MG, Ueno Y, Barbaro B, Benedetti A, Attili AF, Alvaro D. Nerve growth factor modulates the proliferative capacity of the intrahepatic biliary epithelium in experimental cholestasis. Gastroenterology. 2004;127:1198–1209. doi: 10.1053/j.gastro.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Koduri S, Goldhar AS, Vonderhaar BK. Activation of vascular endothelial growth factor (VEGF) by the ER-alpha variant, ERdelta3. Breast Cancer Res Treat. 2005;3:1–7. doi: 10.1007/s10549-005-9028-4. [DOI] [PubMed] [Google Scholar]
- Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10:24–30. doi: 10.1053/jarr.2003.50005. [DOI] [PubMed] [Google Scholar]
- Gabow P, Johnson A, Kaehny W, Manco-Johnson M, Duley I, Everson G. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–1037. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, Everson GT. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease. Lancet. 1994;343:824–827. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- Diel P, Smolnikar K, Michna H. The pure antiestrogen Ici 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Res Treat. 1999;58:87–97. doi: 10.1023/a:1006338123126. [DOI] [PubMed] [Google Scholar]
- Surmacz E. Growth factor receptors as therapeutic targets: strategies to inhibit the insulin-like growth factor I receptor. Oncogene. 2003;2:6589–6597. doi: 10.1038/sj.onc.1206772. [DOI] [PubMed] [Google Scholar]
- Ye M, Grant M, Sharma M, Elzinga L, Swan S, Torres VE, Grantham JJ. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J Am Soc Nephrol. 1992;3:984–994. doi: 10.1681/ASN.V34984. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Barbaro B, Franchitto A, Onori P, Glaser S, Alpini G, Francis H, Marucci L, Sterpetti P, Ginanni-Corradini S, Onetti Muda A, Dostal D, De Santis A, Attili AF, Benedetti A, Gaudio E. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–888. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro D, Invernizzi P, Onori P, Franchitto A, De Santis A, Crosignani A, Sferra R, Ginanni-Corradini S, Mancino MG, Maggioni M, Attili AF, Podda M, Gaudio E. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. 2004;41:905–912. doi: 10.1016/j.jhep.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Gigliozzi A, Attili AF. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33:333–340. doi: 10.1016/s0168-8278(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Motta P, Fumagalli G. Scanning electron microscopy demonstration of cilia in rat intrahepatic bile ducts. Anat Embryol (Berl) 1974;145:223–226. doi: 10.1007/BF00519634. [DOI] [PubMed] [Google Scholar]
- Wheatley DN. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–228. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med. 2001;52:93–123. doi: 10.1146/annurev.med.52.1.93. [DOI] [PubMed] [Google Scholar]
- Koptides M, Deltas CC. Autosomal dominant polycystic kidney disease: molecular genetics and molecular pathogenesis. Hum Genet. 2000;107:115–126. doi: 10.1007/s004390000347. [DOI] [PubMed] [Google Scholar]
- Wu G, Somlo S. Molecular genetics and mechanism of autosomal dominant polycystic kidney disease. Mol Genet Metab. 2000;69:1–15. doi: 10.1006/mgme.1999.2943. [DOI] [PubMed] [Google Scholar]
- Koptides M, Mean R, Demetriou K, Pierides A, Deltas CC. Genetic evidence for a trans-heterozygous model for cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2000;12:447–452. doi: 10.1093/hmg/9.3.447. [DOI] [PubMed] [Google Scholar]
- Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter P, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Delmas P, Padilla F, Osorio N, Coste B, Raoux M, Crest M. Polycystins, calcium signaling, and human diseases. Biochem Biophys Res Commun. 2004;322:1374–1383. doi: 10.1016/j.bbrc.2004.08.044. [DOI] [PubMed] [Google Scholar]
- Delmas P. Polycystins: polymodal receptor/ion-channel cellular sensors. Pflugers Arch. 2005;451:264–276. doi: 10.1007/s00424-005-1431-5. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- Ong AC, Wheatley DN. Polycystic kidney disease—the ciliary connection. Lancet. 2003;361:774–776. doi: 10.1016/S0140-6736(03)12662-1. [DOI] [PubMed] [Google Scholar]
- Marzioni M, Glaser SS, Francis H, Phinizy JL, LeSage G, Alpini G. Functional heterogeneity of cholangiocytes. Semin Liver Dis. 2002;22:227–240. doi: 10.1055/s-2002-34501. [DOI] [PubMed] [Google Scholar]
- LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte proliferation. Liver. 2001;21:73–80. doi: 10.1034/j.1600-0676.2001.021002073.x. [DOI] [PubMed] [Google Scholar]
- Onori P, Alvaro D, Floreani A, Mancino M, Franchitto A, Guido M, Carpino G, De Santis A, Angelico M, Attili AF, Gaudio E. Activation of IGF1 system characterizes cholangiocyte survival during the progression of primary biliary cirrhosis (PBC). J Histochem Cytochem. 2007;55:327–334. doi: 10.1369/jhc.6R7125.2006. [DOI] [PubMed] [Google Scholar]
- Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- Chaurasia OP, Marcuard SP, Seidel ER. Insulin-like growth factor I in human gastrointestinal exocrine secretions. Regul Pept. 1994;50:113–119. doi: 10.1016/0167-0115(94)90026-4. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;75:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- Adesanya OO, Zhou J, Samathanam C, Powell-Braxton L, Bondy CA. Insulin-like growth factor 1 is required for G2 progression in the estradiol-induced mitotic cycle. Proc Natl Acad Sci USA. 1999;96:3287–3291. doi: 10.1073/pnas.96.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann V, Huber C, Kogianni G, Collins F, Noble B. The antioxidant effect of estrogen and selective estrogen receptor modulators in the inhibition of osteocyte apoptosis in vitro. Bone. 2007;40:674–684. doi: 10.1016/j.bone.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Rosenbaum JL. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 2002;12:551–555. doi: 10.1016/s0962-8924(02)02410-8. [DOI] [PubMed] [Google Scholar]