

Abstract
Prenatal viral infection has been associated with development of schizophrenia and autism. Our laboratory has previously shown that viral infection causes deleterious effects on brain structure and function in mouse offspring following late first trimester (E9) administration of influenza virus. We hypothesized that late second trimester infection (E18) in mice may lead to a different pattern of brain gene expression and structural defects in the developing offspring. C57BL6J mice were infected on E18 with a sublethal dose of human influenza virus or sham-infected using vehicle solution. Male offsping of the infected mice were collected at P0, P14, P35 and P56, their brains removed and prefrontal cortex, hippocampus and cerebellum dissected and flash frozen. Microarray, qRT-PCR, DTI and MRI scanning, western blotting and neurochemical analysis were performed to detect differences in gene expression and brain atrophy. Expression of several genes associated with schizophrenia or autism including Sema3a, Trfr2 and Vldlr were found to be altered as were protein levels of Foxp2. E18 infection of C57BL6J mice with a sublethal dose of human influenza virus led to significant gene alterations in frontal, hippocampal and cerebellar cortices of developing mouse progeny. Brain imaging revealed significant atrophy in several brain areas and white matter thinning in corpus callosum. Finally, neurochemical analysis revealed significantly altered levels of serotonin (P14, P35), 5-Hydroxyindoleacetic acid (P14) and taurine (P35). We propose that maternal infection in mouse provides an heuristic animal model for studying the environmental contributions to genesis of schizophrenia and autism, two important examples of neurodevelopmental disorders.
Keywords: schizophrenia, autism, viral model, mouse, DNA microarray, brain
1. Introduction
Schizophrenia is a major debilitating disease with a lifetime prevalence of 1% throughout the world (American Psychiatric Association, 1994). The familial transmission of this disorder is multifactorial and heterogeneous (Asherson et al, 1994). Not all cases of schizophrenia can be accounted for by genetic causes. There is robust epidemiologic evidence indicating that environmental contributions, such as infections prenatally, may lead to genesis of schizophrenia (Fatemi, 2005).
Recent serologic evidence also points to multiple prenatal exposures to various viruses as causative factors in rise of schizophrenic births (Brown et al, 2004). Several groups, including our laboratory, have shown evidence for viral infections and/or immune challenges being responsible for production of abnormal brain structure and function in rodents where mothers were exposed to viral insults throughout pregnancy (Fatemi et al, 2005a; Meyer et al, 2006).
Recent evidence has shown that the time of prenatal insult may provide distinct changes in the exposed offspring. In a recent series of experiments by Meyer et al (2006) using the viral mimic polyribocytidilic acid (PolyI:C) at E9 (which corresponds to midpregnancy) and E17 (which corresponds to late pregnancy) there were distinct behavioral deficits, neuropathological differences and acute cytokine responses (Meyer et al, 2006). Adult mice that were exposed on E9 displayed reduced exploratory behavior while those exposed on E17 displayed perseverative behavior (Meyer et al, 2006). At P24, mice that were exposed on E9 displayed a more pronounced reduction of Reelin immunoreactivity in hippocampus than mice exposed at E17 (Meyer et al, 2006). In contrast, mice exposed at E17 displayed an increase in apoptosis as visualized by immunoreactivity of caspase-3, a key enzyme involved in apoptosis (Rami et al, 2003), in the dorsal dentate gyrus (Meyer et al, 2006). Finally, Meyer et al (2006) found that late gestational immune challenge uniquely stimulated increased IL-10 and TNF-α in fetal brain (Meyer et al, 2006). Taken together, these results provide evidence that the time of prenatal insult results in important differences that are persistent through adulthood.
Previous work by our group showed that influenza infection at E9 of pregnancy in mice leads to abnormal corticogenesis, pyramidal cell atrophy, and alterations in levels of several neuroregulatory proteins, such as Reelin, and GFAP, in the exposed mouse progeny (Fatemi et al, 1999; Fatemi et al, 2002a; Fatemi et al, 2002b; Shi et al, 2003). We hypothesized that late second trimester infection (E18) in mice may lead to a different pattern of brain gene expression and structural defects in the developing offspring. Here, we present extensive genetic, imaging, and neurochemical data showing that a similar sublethal dose of human influenza virus (H1N1) in C57BL6J mice at E18, also leads to altered expression of many brain genes which may underlie brain atrophy and neurochemical dysregulation at puberty in the exposed mouse offspring.
2. Methods
2.1. Viral infection
All experimental protocols used in this study were approved by the Institute for Animal Care and Use and Institutional Biosafety Committees at the University of Minnesota. Influenza A/NWS/33 (H1N1) virus was obtained from R.W. Cochran, University of Michigan (Ann Arbor). A virus pool was prepared in Maden Darby canine kidney (MDCK) cells; the virus was ampuled and frozen at -80°C until used. Data were expressed as log10 cell culture infectious doses (CCID50)/ml by the method of Reed and Muench (Reed and Muench, 1938). By this titration, it was determined that at a dilution of 10-4.5, none of the mice died of the infection but displayed a mean lung consolidation scores and mean lung weights similar to those obtained by Fatemi et al (2002b) and had a mean virus titer of 105.25 CCID50/ml, indicating that a moderate but sublethal infection had been induced. This was the virus dose selected for use in the pregnant mouse study. On day 18 of pregnancy, C57BL6J mice (Charles River, Wilmington, MA) were anesthetized using 200 μl isoflurane, and intranasally (i.n.) administered a dilution of 10-4.5 of 6.5 log 10 (CCID50) per 0.1 ml human influenza virus A/NWS/33 in 90 μl of minimum essential medium (MEM). E18 was chosen as a time point as it corresponds to the second trimester in humans, a period when prenatal infection has been linked to the development of schizophrenia later in life (reviewed in Arndt et al., 2005; Libbey et al., 2005; Spear, 2000,2004). Sham infected mothers were treated identically but administered i.n. 90 μl MEM. After being infected, their drinking water contained 0.006% oxytetracycline (Pfizer, New York, NY) to control possible bacterial infections. Pregnant mice were allowed to deliver pups. The day of delivery was considered day 0. Groups of infected and sham-infected neonates were deeply anesthetized and killed on P0, P14, P35 and P56. Offspring were weaned from mothers at P21, and males and females were caged separately in groups of 2-4 littermates.
2.2. Brain collection and dissection
Pregnant mice were allowed to deliver pups. Day of delivery was considered as postnatal day P0. Male litter mates from P0, 14, 35, and 56 were obtained. Groups of infected (N=10 litters) and sham-infected male neonates (N=10 litters) were deeply anesthetized. The choice of ten animals per group was based on earlier calculations that yield an average effect size of 1.45sd between infected and control animals. With alpha = 0.05, and a sample size of 10 per group, power is = 0.798. Brains were removed from skull cavities following perfusion with phosphate-buffered 4% paraformaldehyde (pH 7.4) with later immersion in the same fixative for 14 days at 4°C and used for all imaging studies. Alternatively, unfixed cryopreserved brains were snap-frozen by immersion in liquid nitrogen and stored at -85°C for all gene array, western blotting and HPLC studies.
The areas of interest were dissected as follows: Frontal association area was identified using coronal images from the stereotaxic mouse brain atlas (Franklin and Paxinos, 1997) using the following coordinates: starting at +7.0mm to +6.6mm from interaural line and +3.2mm to +2.8mm from bregma. Moreover, the ventral and medial borders were identified by the corpus callosum and the external capsule. The ventrolateral border was defined by rhinal fissure. Hippocampus was identified using coronal images from the stereotaxic mouse brain atlas (Franklin and Paxinos, 1997). The boundaries of dorsal, lateral, and medial regions are readily identifiable. The boundaries to the entorhinal cortex and amygdala were identified by extrapolating the lateral boundary defined by alveus of hippocampus. We only sampled ventral hippocampus using the following coordinates: starting at +2.74mm to +1.62mm from interaural line (just before the start of dorsal hippocampal fissure) and from -1.06mm to -2.18mm from bregma. Additionally, Zapala et al (2005) provided step-by-step diagrams in their online supporting appendix 2. Cerebellum was identified using coronal images derived from the stereotaxic mouse brain atlas (Franklin and Paxinos, 1997) and using the following coordinates: starting at -1.40mm to -3.68mm from interaural line and from -5.20mm to -7.48mm from bregma. Additionally, sagitally, cerebellum is easily recognized using the boundary between the brainstem and the lateral recess of the 4th ventricle to the anterolateral edge of cerebellum.
2.3. Microarray
Tissues (N=3 control and N=3 infected for each of the three brain regions (cerebellum, hippocampus, and prefrontal cortex) at each of three time points (P0 (birth), P14 (childhood), P56 (adulthood)) were homogenized with a Polytron in 1 ml TRIzol (Invitrogen). After 5 min, 200 μl chloroform was added, tubes were shaken by hand, incubated, and centrifuged at 12,000 g for 15 min (4°C). The aqueous phase was transferred to a new tube and RNA was precipitated with 500 μl isopropyl alcohol. The pellet was washed by resuspension in 75% ethanol and centrifugation at 7,500 g for 5 min (4°C). The pellet was dried in air and dissolved in RNase-free water. RNA concentration and quality were determined by UV absorbance (Nanodrop) and ribosomal RNA integrity (Agilent 2100 Bioanalyzer). The RNA was then cleaned and concentrated with the Qiagen Rneasy Micro Kit which included a DNase digestion according to the manufacturer’s recommended procedure. The final preparation was stored at -80°C.
RNA was amplified and labeled with biotin using the Ovation kit (NuGEN) according to the manufacturer’s procedure. Briefly, this involved reverse transcription with a proprietary DNA/RNA chimeric primer that hybridized with the 5′ portion of the polyA tail, which left a unique RNA sequence at the 5′ end of the cDNA strand. The mRNA within the mRNA/cDNA double strand was fragmented to create priming sites for generation of cDNA with the unique RNA sequence at the 5′ end of the antisense strand. An isothermal linear amplification procedure was employed, in which RNAse H removed this RNA sequence, exposing the complementary strand for hybridization of a different RNA/DNA chimeric primer that initiated synthesis of an antisense cDNA strand by DNA polymerase. In the presence of RNAse H, this primer was degraded, again exposing the priming site for additional rounds of cDNA synthesis. Continuing synthesis of cDNA displaced the previously-synthesized strands, yielding single-stranded cDNA as the amplification product. cDNA yield was determined by UV absorbance (Nanodrop). The cDNA was then fragmented (average length 50-100 bases) enzymatically and labeled with biotin. The product was cleaned with a Centrisep column (Princeton Separations), and concentration of the cDNA in the final eluate was determined by UV absorbance (Nanodrop).
2.3.1. External Controls
Standard Affymetrix biotinylated cRNAs were spiked into the hybridization solution at the following final concentrations: 1.5 pM bioB, 5 pM bioC, 25 pM bioD, 100 pM cre.
2.3.2. Hybridization Procedure
The fragmented, biotinylated cDNA was used for hybridization at 45°C for 17 hr with Affymetrix U133 Plus 2.0 GeneChip arrays. The hybridization solution contained 0.0125 μg/μl cDNA, 50 pM oligonucleotide B2, 1.5 pM bioB cRNA, 5 pM bioC cRNA, 25 pM bioD cRNA, 100 pM cre cRNA, 100 mM MES, 1 M Na+, 20 mM EDTA, and 0.01% Tween-20, 0.1 mg/ml herring sperm DNA, and 0.5 mg/ml acetylated BSA. Arrays were washed and stained using an Affymetrix Fluidics Station 450 according to the standard Affymetrix protocol for eukaryotic expression arrays (see GeneChip Expression Analysis Technical Manual Rev. 5 available at www.affymetrix.com). This involved washing with low stringency Buffer A (6X SSPE, 0.01% Tween-20), then with stringent Buffer B (100 mM MES, 0.1 M Na+, 0.01% Tween-20), staining with phycoerythrin-streptavidin (SAPE) solution (10 μg/ml SAPE, 2 mg/ml BSA, 100 mM MES, 1 M Na+, 0.05% Tween-20), washing with Buffer A, staining with anti-streptavidin biotinylated antibody (3 μg/ml antibody, 0.1 mg/ml goat IgG, 2 mg/ml BSA, 100 mM MES, 1 M Na+, 0.05% Tween-20) then SAPE solution, and washing with Buffer A. All washing and staining steps were done at 25°C except for the buffer B wash (50°C) and the final wash in Buffer A (30°C).
2.3.3. Measurement of Data and Specifications
Arrays were scanned with the GeneChip Scanner 3000 7G. GeneChip Operating Software (GCOS, Affymetrix) was used for initial processing of the scanner data, including generation of cel files that were used for computation of GCRMA gene expression scores with GeneSifter software. The GCRMA procedure involved quantile normalization, and was described by Wu et al (2004) (www.bepress.com/jhubiostat/paper1).
2.3.4. Array Design
Mouse Genome 430 2.0 array was obtained from Affymetrix. The array provided comprehensive coverage of the transcribed mouse genome on a single array analyzing 39,000 transcripts and variants for over 14,000 genes. The oligonucleotide sequences, their locations on the array, and other annotations are available in files that can be viewed or downloaded from www.netaffx.com.
2.4. qRT-PCR
Quantitative RT-PCR was performed for selected genes from our microarray data that have an association with schizophrenia or autism or genes that are involved in influenza mediated RNA processing using the TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) and a GAPDH Endogenous control assay. Each TaqMan Gene Expression Assay is pre-formulated consisting 2 unlabeled PCR primers at a final concentration of 900 nM and 1 FAM™ dye-labeled TaqMan® MGB of 250 nM final concentration. Sequences of the primers and probe are proprietary information.
For each sample (N=3 for control and N=3 for infected), between 0.6ug and 2.5ug total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). 2ul of a 1:5 dilution for cDNA was combined with TaqMan Universal PCR Master Mix No AmpErase UNG (Applied Biosystems) and the Taqman Gene Expression Assay in a 10ul reaction set up by the CAS-1200 liquid handling system. The real-time RT-PCR amplfications were run on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Universal thermal cycling conditions were as follows: 10 minutes at 95°C, 40 cycles of denaturation at 95°C for 15 seconds, and annealing and extension at 60°C for 1 minute. Amplifications efficiencies were close to 100% for all assays according to analyses of a number of different dilutions of the cDNA. Calculations were done assuming that 1 delta Ct equals a 2-fold difference in expression. Significance values were determined using unpaired t-tests.
2.5. DTI and MR Scanning
2.5.1. Brain tissue acquisition for MR scanning
Brains from C57BL/6 male neonates born to infected and sham infected E18 mothers at P0, P14, P35 (which corresponds to adolescence) and P56 (N=4 infected, N=3 control, 1 male/litter per group) were perfusion fixed in 4% paraformaldehyde in PBS buffer (pH 7.4) and subjected to DTI and MR scanning in PBS at room temperature (Mori et al, 2001).
2.5.2. Imaging and data processing
We used an 11.8 T Bruker scanner equipped with a triple-axis gradient unit (80G/cm). The FSE (fast spin-echomultiple spin-echo)-based imaging sequence was used. Mouse brains were placed in home-built plastic tubes. Inside the tubes, fixed mouse brains were immersed in formalin (MRI-invisible liquid) solution to keep the brain from dehydrating. A custom-made solenoid volume coil was used as both RF coil and signal receiver. To maximize the SNR, it was important to use the smallest coil and sample chamber. We developed a probe with a detachable coil head where sample chambers are made from plastic shaped for individual brain sizes.
Native imaging resolution matrix was 144 × 80 × 60, which was zero filled to 256 × 128 × 128. The field-of-view was 12 × 9 × 9 mm for P0 and 20 × 15 × 15 mm for P56 brains. Six echoes were acquired after each excitation and the last two echoes were used for the navigator echoes. With the repetition time (TR) of 0.8s and two signal averaging, imaging time was 2 hours for each image. For the DTI studies, six to seven images with different diffusion weighting were obtained with TE = 40 ms and b = 1,200 s/mm2. The combination of the x, y, and z gradients were x ± y, x ± z, and y ± z, in addition to a low diffusion weighting image (b = 150 s/mm2). Once images with 6 different diffusion weightings were obtained (total 10.5 hours), an isotropically diffusion-weighted image was obtained by adding all 6 diffusion weighted images. 6 elements of diffusion tensor were determined by multivariable least square fitting using the equation;
Where is a 3×3 symmetric tensor, is a gradient vector, and S and S0 are signal intensities with and without diffusion weighting. The tensor was diagonalized to obtain three eigenvalues (λ1-3) and eigenvectors (v1-3). Anisotropy was measured by calculating fractional anisotropy (FA);
Color maps were created by assigning FA to its intensity level and RGB values to the x, y, and z component of v1. T2-weighted images will be obtained using the same imaging parameters except for TE = 120 ms [relatively short T1 (600 - 800 ms), and small T1 differences among tissues in 11.7T allow to use short TR]. T2 maps can be calculated using the least diffusion-weighted image (TE = 40 ms) as a short TE data point. In addition, two more DTI-based contrasts were generated; namely isotropically diffusion-weighed images (iDWI) and apparent diffusion constant (ADC, average of diffusion tensor diagonal elements) maps. All imaging was performed at 25°C.
2.5.3. Data analysis - Image alignment
All 3D volume data were aligned using our in-house landmark-based alignment program. It required identification of 5 anatomical landmarks at the mid-saggital level (anterior commissure, posterior commissure, optic chiasm, genu and splenium of corpus callosum) and automatically aligned a target brain to a standard brain by minimizing the distances.
2.5.4. Volume measurement
Using a series of axial sections (256 sections), the hemisphere and the ventricles were manually segmented. The delineation was based on the color-coded orientation maps that can clearly reveal the anatomical units of interest at each developmental stage. Once the segmentation was done, it was straightforward to calculate the number of pixels within each segmentation, from which the volume was obtained.
2.6. SDS-PAGE and Western Blotting
Brain tissue per mouse was prepared as previously described (Fatemi et al, 2006). Samples were mixed with denaturing SDS sample buffer (20% glycerol, 100 mM Tris, pH 6.8, 0.05% w/v Bromophenol blue, 2.5% SDS (w/v), 5% β-mercaptoethanol) and denatured by heating at 100°C for 5 minutes. Either 6% (Foxp2) or 10% SDS-polyacrylamide gels (β-actin) were prepared with standard Laemmli solutions (BioRad). Sixty μg of protein per lane was loaded onto the gel and electrophoresed, initially for 15 min at 75 V and then for 50 min at 150 V at RT. The proteins were electroblotted onto nitrocellulose paper for 2 hours at 300 mA at 4°C. Protein blots were blocked either 1 hour at RT (Foxp2) or overnight at 4°C (β-actin) with 0.2% I-Block (Tropix) in PBS with 0.3% Tween-20. The blots were then incubated with primary antibody overnight at 4°C (Foxp2 Abcam ab16046, 1:2,000) or for 1 hour at RT (mouse anti-β-actin, Sigma A5441, 1:5000) followed by secondary antibody incubation for 1 hour at RT (goat anti-mouse HRP conjugated or goat anti-rabbit HRP conjugated, 1:80,000, both from Sigma). Between each step, the immunoblots were rinsed with PBS-T. The immune complexes were visualized using the ECL Plus detection system (Amersham Pharmacia Biotech) and exposed to CL-Xposure film (Pierce). Sample densities were analyzed using a Bio-Rad densitometer and Quantity One software.
Results obtained are based on between two and eight independent experiments with N=3 infected and N=3 control mice per gel. For each experiment, control and infected samples were run on the same gel and processed simultaneously to avoid variability due to intragel differences.
2.7. Neurochemical Analyses
For neurochemical analyses, frozen cerebellum samples (N=3 control and N=3 infected) were weighed and homogenized by ultrasonication in 20 volumes of 0.1 N perchloric acid at 4 °C. 100 μl of the homogenates was added to equal volumes of 1 N sodium hydroxide for measurement of protein contents. Cerebellum was selected as recent evidence has emerged that it is important for cognition and memory (Gottwald et al., 2004; Schmahmann and Caplan, 2006). The remaining homogenates were centrifuged at 17,000 g and 4 °C for 10 min. Aliquots of the supernatants were added to equal volumes (50 μl) of 0.5 M borate buffer and stored at -80° for subsequent analyses of amino acids. The remaining supernatants were used for immediate measurement of monoamines and their metabolites. The levels of monoamines (dopamine and serotonin [5-hydroxytryptamine; 5-HT] and their metabolites 3, 4-dihydroxyphenylacetic acid (DOPAC) and 5-hydroxyindoleacetic acid (5-HIAA)) were measured by high performance liquid chromatography (HPLC) with electrochemical detection as previously described (Felice et al, 1978; Sperk et al, 1981; Sperk, 1982). Amino acids (glutamate, gamma-amino butyric acid (GABA), and the metabolite glutamine) as well as taurine were precolumn derivatized with o-phthalaldehyde-2-mercaptoethanol using a refrigerated autoinjector and then separated on a HPLC column (ProntoSil C18 ace-EPS) at a flow rate of 0.6 ml/min and a column temperature of 40 °C. The mobile phase was 50 mM sodium acetate pH = 5.7 in a linear gradient from 5% to 21% acetonitrile. Derivatized amino acids were detected by their fluorescence at 450 nm after excitation at 330 nm (Piepponen and Skujins, 2001).
2.8. Statistical Analysis
All statistical analyses were performed using SPSS. Differences of the normalized mRNA expression levels of selected genes between drug treated and saline-treated rats were assayed using two-tailed student’s t-test. Significant differences are defined as those with a p value < 0.05.
For western blots, differences of the protein levels of FOXP2 between the progeny of virally-exposed and sham-infected mice were normalized against β-actin and assayed using a two-tailed student’s t-test. Significant differences are defined as those with a p value < 0.05.
For neurochemical analyses data were statistically analyzed using SigmaStat version 3.0. A one or two-way analysis of variance (ANOVA) was used for all comparisons (treatment, testing day) and the Holm-Sidak method was used for post hoc analysis when indicated. A probability level (p) of less than 0.05 was considered statistically significant.
3. Results
We used Affymetrix microarrays, qRT-PCR, HPLC, and MRI-based imaging to evaluate our results. C57BL6J pregnant mice were infected with a sublethal dose of influenza A/NWS/33 (H1Nl) virus at E18 of pregnancy. Frontal, hippocampal, and cerebellar cortices (N=20 per group; 1 mouse/litter/group) were dissected at postnatal (P) days P0 (birth), P14 (childhood), P35 (adolescence) and P56 (adulthood) from exposed and control mouse progeny. Brains were also collected following intracardiac perfusion with 4% paraformaldahyde and prepared for MRI imaging (N=4 litters per group per postnatal time period). Frozen brain areas were prepared for RNA isolation, DNA microarray, and qRT-PCR measurements for all mRNAs of interest. All DNA microarray values, qRT-PCR data, and MRI morphometric values were subjected to statistical analysis with p<0.05 for statistical significance.
Gene expression data showed a significant (p<0.05) at least 1.5 fold up- or downregulation of genes in frontal (43 upregulated and 29 downregulated at P0; 16 upregulated and 17 downregulated at P14; and 86 upregulated and 24 downregulated at P56), hippocampal (129 upregulated; 46 downregulated at P0; 9 upregulated and 12 downregulated at P14; and 45 upregulated and 17 downregulated at P56), and cerebellar (120 upregulated and 37 downregulated at P0; 11 upregulated and 5 downregulated at P14; and 74 upregulated and 22 downregulated at P56) areas of mouse offspring (Supporting Table 1 online). Several genes, which have have been previously implicated in etiopathology of schizophrenia, were shown to be affected significantly (p<0.05) by DNA microarray and whose direction and magnitude of change were validated by qRT-PCR included: rho guanine nucleotide exchange factor 9 (Arhgef9), v-erb-B2 avian erythroblastic leukemia viral oncogene (Erbb4), semaphorin 3A (Sema3a), ephrin B2 (Efnb2), myelin transcription factor 1-like (Mytll), transferrin receptor 2 (Tfr2), sry-box 2 (Sox2), very-low density lipoprotein receptor (Vldlr), deadbox polypeptide 3 y-linked (Dby), neurexophilin 2 (Nxph2), ATPase, Na+/K+ transporting, beta 2 polypeptide (Atp1b2), ubiquitously transcribed tetratricopeptide repeat gene on Y chromosome (Uty), and death associated protein kinase 1 (Table 1). There were also several genes which are known to be involved in influenza-mediated RNA processing which were upregulated in all three brain areas and continued to be present at day 0, e.g. NS1 influenza binding protein and aryl hydrocarbon receptor nuclear translocator genes (Table 1).
Table 1.
Microarray and qRT-PCR Results for Selected Affected Genes in E18 Infected Mice
| Gene | Symbol | Area | Day | Microarray fold change | Microarray p-value | Gene relative to normalizer (qRT-PCR) | QPCR p-value |
|---|---|---|---|---|---|---|---|
| Cdc42 guanine nucleotide exchange factor (GEF) 9 (Collybistin) | Arhgef9 | PFC | P0 | 2.787 | 0.0229 | 1.251 | 0.026 |
| Aryl hydrocarbon receptor nuclear translocator | Arnt | CER | P0 | * | * | 1.505 | 0.024 |
| Arnt | HIP | P0 | * | * | 2.082 | 0.021 | |
| Arnt | PFC | P0 | * | * | 1.447 | 0.006 | |
| Death associated protein kinase 1 | Dapk1 | CER | P0 | 2.180 | 0.0051 | 1.220 | 0.013 |
| DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, Y-linked | Dby | CER | P56 | 3.996 | 0.0436 | 6.711 | 0.005 |
| Ephrin B2 | Efnb2 | HIP | P0 | 2.334 | 0.0345 | 2.413 | 0.021 |
| V-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) | Erbb4 | HIP | P0 | 3.434 | 0.0055 | 2.097 | 0.048 |
| Influenza virus NS1A binding protein | Ivns1abp | CER | P0 | * | * | 1.215 | 0.0004 |
| Ivns1abp | HIP | P0 | * | * | 1.91 | 0.079 | |
| Ivns1abp | PFC | P0 | * | * | 1.02 | 0.8603 | |
| Myelin transcription factor 1-like | Myt1l | HIP | P0 | 2.355 | 0.0387 | 2.034 | 0.051 |
| Neurexophilin 2 | Nxph2 | HIP | P0 | 4.146 | 0.0006 | 3.504 | 0.010 |
| Sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3A | Sema3a | HIP | P0 | 3.517 | 0.0184 | 3.489 | 0.013 |
| SRY-box containing gene 2 | Sox2 | CER | P0 | 2.164 | 0.0083 | 1.299 | 0.010 |
| Transferrin receptor 2 | Trfr2 | CER | P0 | 2.266 | 0.0449 | 1.565 | 0.026 |
| Ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome | Uty | CER | P56 | 3.681 | 0.0333 | 5.345 | 0.018 |
Not altered in microarray analysis
Forkhead box P2 (Foxp2), a gene implicated in both autism (Gong et al, 2004) and schizophrenia (Sanjuan et al, 2006), has previously been demonstrated to be upregulated in P35 cerebellum of exposed mice of Balb/c dams infected on E9 (Fatemi et al, 2007a). Western blotting experiments revealed that Foxp2 protein was upregulated significantly in P0 (p<0.050) and P35 cerebellum (p<0.032) of exposed mouse progeny (Figure 1, Table 2). Foxp2 was also found to be significantly upregulated in P35 (p<0.036) and P56 (p<0.022) hippocampi of exposed mice (Figure1, Table 2).
Figure 1.

Mean Foxp2/ β-actin ratios for the progeny of infected (filled histogram bars) and sham-infected mice are shown. Levels of Foxp2/ β-actin were significantly increased in P0 (a) and P35 (b) cerebellum (p=0.050 and 0.032 respectively) and in P35 (c) and P56 (d) hippocampus (p=0.036 and 0.022 respectively). Foxp2 bands from P0 and P35 cerebellum and P35 and P56 hippocampus homogenates (60 μg per lane) of representative progeny from sham-infected and infected mice are shown. **p < 0.05 vs. control.
Table 2.
Foxp2/ β-actin Ratios for Infected and Control Progeny
| Brain Region | Postnatal Day | Infected | Control | P Value |
|---|---|---|---|---|
| Cerebellum | P0 | 0.757 ± 0.18 | 0.444 ± 0.074 | p<0.050 |
| P14 | 0.34 ± 0.07 | 0.314 ± 0.075 | p<0.67 | |
| P35 | 0.513 ± 0.056 | 0.387 ± 0.071 | p<0.032 | |
| P56 | 0.237 ± 0.028 | 0.244 ± 0.031 | p<0.76 | |
| Hippocampus | P14 | 0.152 ± 0.067 | 0.231 ± 0.138 | p<0.35 |
| P35 | 0.22 ± 0.048 | 0.127 ± 0.034 | p<0.036 | |
| P56 | 0.229 ± 0.029 | 0.157 ± 0.018 | p<0.022 | |
| Prefrontal Cortex | P14 | 0.494 ± 0.114 | 0.636 ± 0.261 | p<0.37 |
| P35 | 0.489 ± 0.04 | 0.459 ± 0.154 | p<0.72 | |
| P56 | 0.428 ± 0.071 | 0.342 ± 0.108 | p<0.26 | |
Analysis of brain and lateral ventricular volume areas in postnatal brains showed significant atrophy of the brain volume by ≈4% (p<0.05) in P35 offspring of exposed mice (Figure2; Table 3). The ventricular values did not vary significantly between the two groups (data not shown). Fractional anisotropy of corpus callosum revealed white matter atrophy on P35 offspring (p<0.0082) of exposed mice (Table 4).
Figure 2.
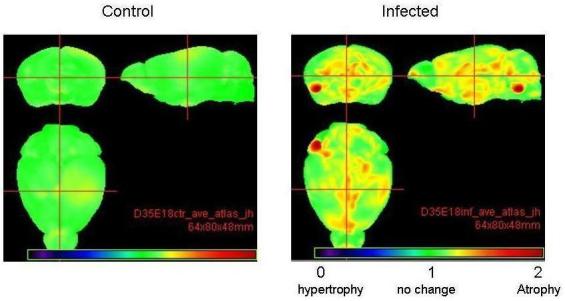
MRI imaging reveals significant (p<0.05) brain atrophy in multiple brain areas of the 35 day old virally infected mouse offspring (right panel) as compared to sham infected mice (left panel).
Table 3.
E18 Brain Volume Measurements
| Postnatal Day | Infected | Control | P Value |
|---|---|---|---|
| P0 | 89.09 ± 4.94 | 88.73 ± 1.93 | p<0.91 |
| P14 | 357.18 ± 15.32 | 366.95 ± 8.44 | p<0.37 |
| P35 | 407.22 ± 7.08 | 425.72 ± 5.63 | p<0.014 |
| P56 | 420.39 ± 2.52 | 413.30 ± 26.57 | p<0.61 |
Table 4.
E18 Fractional Anisotropy of Corpus Callosum
| Postnatal Day | Infected | Control | P Value |
|---|---|---|---|
| P0 | 0.73 ± 0.13 | 0.76 ± 0.19 | p<0.87 |
| P14 | 0.68 ± 0.04 | 0.70 ± 0.01 | p<0.48 |
| P35 | 0.82 ± 0.04 | 0.91 ± 0.02 | p<0.0082 |
| P56 | 0.88 ± 0.02 | 0.86 ± 0.01 | p<0.11 |
Levels of monoamines (DA and 5-HT), monoamine metabolites (DOPAC and 5-HIAA), and amino acids (glutamate, GABA, glutamine), were determined in the cerebella of mice virally exposed at embryonic day E18 and controls as previous work has demonstrated altered levels of neurotransmitters in schizophrenia (reviewed by Sawa and Snyder, 2002) and autism (reviewed by Zimmerman et al, 2006). Absolute concentrations of neurotransmitters are given in Table 5. Post hoc test revealed significant differences between groups in 5-HT and 5-HIAA at postnatal day P14 (Figure 3a, b; Table 5) and in 5-HT and taurine at postnatal day P35 (Figure 3c, d; Table 5).
Table 5.
Neurotransmitter Content in μM/ g Protein
| CONTROLS | VIRALLY EXPOSED | |||||||
|---|---|---|---|---|---|---|---|---|
| P0 | P14 | P35 | P56 | P0 | P14 | P35 | P56 | |
| Dopamine (DA) | 14.2+/-1.0 bP35,P56 | 5.8+/-0.8 bP35,P56 | 1.1+/-0.2 bP0,P14 | 1.0+/-0.1 bP0,P14 | 15.9+/-0.8 bP14,P35,P56 | 3.2+/-0.1 bP0,P35,P56 | 1.0+/-0.2 bP0,P14 | 1.2+/-0.3 bP0,P14 |
| 3, 4-dihydroxyphenylacetic acid (DOPAC) | 2.4+/-0.2 bP14,P35,P56 | 2.0+/-0.4 bP0,P35,P56 | 5.0+/-0.3 bP0,P14 | 4.3+/-0.02 bP0,P14 | 2.7+/-0.2 bP14,P35,P56 | 1.2+/-0.1 bP0,P35,P56 | 4.3+/-0.2 bP0,P14 | 4.5+/-0.1 bP0,P14 |
| Serotonin (5-HT) | 56.5+/-4.6 bP35 | 51.8+/-1.0 bP35,P56 | 73.2+/-2.3 bP0,P14 | 70.2+/-6.9 bP14 | 56.1+/-3.3 bP14 | 37.8+/-2.6 a, bP0,P35,P56 | 62.0+/-6.9 a, bP14 | 61.1+/-1.7 bP14 |
| 5-hydroxy indole acetic acid (5-HIAA) | 351.8+/-41.5 bP14,P35,P56 | 103.6+/-5.9 bP0 | 57.1+/-7.0 bP0 | 37.9+/-2.9 bP0 | 299+/-102.1 bP14,P35,P56 | 74.6+/-4.8 a, bP0 | 49.2+/-4.3 bP0 | 42.6+/-5.0 bP0 |
| Glutamate (GLU) | 39.3+/-2.5 bP14,P35,P56 | 61.8+/-2.4 bP0,P35,P56 | 76.6+/-1.7 bP0,P14 | 75.8+/-2.3 bP0,P14 | 47.8+/-2.3 bP14,P35,P56 | 64.3+/-0.3 bP0,P56 | 68.6+/-1.5 bP0,P56 | 76.2+/-1.9 bP0,P14,P35 |
| Glutamine (GLN) | 57.9+/-2.5 bP14,P35,P56 | 40.1+/-1.6 bP0 | 39.2+/-0.2 bP0 | 37.1+/-1.6 bP0 | 56.2+/-5.0 bP14,P35,P56 | 38.6+/-0.3 bP0,P35 | 30.5+/-0.7 bP0,P14 | 36.6+/-0.9 bP0 |
| Gamma amino butyric acid (GABA) | 22.4+/-0.9 | 24.8+/-1.6 | 24.1+/-1.6 | 19.8+/-1.2 | 24.5+/-1.5 | 20.3+/-1.2 | 16.9+/-0.7 | 20.9+/-0.6 |
| Taurine (TAU) | 424.2+/-4.4 bP14,P35,P56 | 145.3+/-8.1 bP0,P35,P56 | 59.5+/-3.0 bP0,P14 | 60.0+/-3.0 bP0,P14 | 277.8+/-3.1 bP14,P35,P56 | 139.4+/-4.6 bP0,P35,P56 | 47.6+/-2.8 a, bP0,P14 | 60.8+/-1.2 bP0,P14 |
Values (μM per g protein) are the means+/-SEM from 3 mice each. Statistical significance of difference analyzed by one or two way analysis of variance followed by Holm Sidak post hoc test. ap<0.05 vs. control; b p<0.05 vs. other postnatal days P(0, 14, 35, 56)
Figure 3.

Neurochemical analysis of offspring of mice infected on E18. Shown are mean values +/- SEM of the mean for serotonin (5-HT, a and c), 5-HIAA (b), and taurine (d). Statistical significance of difference analyzed by one way analysis of variance followed by Holm Sidak post hoc test. **p < 0.05 vs. control.
4. Discussion
Analysis of some of the virally-regulated brain genes in the exposed progeny show positive associations with schizophrenia including the following genes: 1) Arhgef9 is a Rho GTPase that has been shown to activate cell division cycle 42 (Cdc42) (Reid et al, 1999) which in turn is involved in elongation of dendritic spines. Cdc42 is reduced in prefrontal cortex of schizophrenics (Hill et al, 2006; Nishimura et al, 2006). Expression of mRNA for Arhgef9 was significantly increased (p<0.025) in frontal cortex of day 0 mice potentially in response to altered Cdc42 levels in the virally exposed brain tissue; 2) Erbb4 gene codes for a protein which serves as receptor for neuregulin 1. This gene is involved in neuron and glial proliferation, differentiation, and migration processes. Binding of Erbb4 and neuregulin1 lead to NMDA receptor current propagation, a process which is apparently defective in schizophrenia (Hahn et al, 2006). A recent report shows that polymorphisms in neuregulin 1 are associated with grey and white matter alterantions in childhood onset schizophrenia (Addington et al, 2007), a striking similarity seen in our viral model of schizophrenia, where brain atrophy also occurs in puberty in the exposed mice (Fig. 2, Table 3). The significant increase in Erbb4 mRNA seen here may be due to decreases in levels of neuregulin 1 in the exposed mice; 3) Sema3a is an important gene involved in axonal guidance and white matter development. Interestingly, levels of this mRNA is elevated in cerebella of schizophrenic subjects (Eastwood et al, 2003) akin to significant (p<0.013) 3.5 fold elevations in hippocampi of offspring of exposed mice at P0; 4) Efnb2 is a gene important in neural crest development (Irie and Yamaguchi, 2004; Ivanov et al, 2005) and is known to become upregulated by exposure to LPS (Ivanov et al, 2005) and is a ligand for Nurr protein (Calo et al, 2005). It is also upregulated by hypoxia (Vihanto et al, 2005) and is involved in NMDA dependent synaptic function (Henderson et al, 2001); 5) Tfr2 levels are known to be elevated in plasma of schizophrenic and bipolar patients (Maes et al, 1995; Tsai et al, 2003) and may serve as a potential receptor for viral activity in the exposed mouse brain; 6) Sox2 is involved in neuronal patterning with mutations in Sox2 being responsible for neurological symptoms (Kelberman et al, 2005). Sox10 mRNA has been shown to be reduced in hippocampus and anterior cingulate cortex of subjects with schizophrenia (Dracheva et al, 2006); 7) mRNA levels for Vldlr, a receptor for Reelin was elevated by two fold in the hippocampal tissues of offspring of infected mice and showed a near significant trend. This important gene is also upregulated in autism (Fatemi et al, 2005b) and was recently considered important in regulation of sensorimotor gating which is abnormal in our viral model (Shi et al, 2003) and in schizophrenia and autism (Braff et al, 2001; Perry et al, 2007); 8) Dby was elevated by seven fold (p<0.0052) in the infected frontal cortex of exposed day 56 mice and is involved in RNA splicing, ribosome assembly, cell growth and differentiation (Bartholomew and Parks, 2007). This gene codes for a RNA helicase and may in fact connote direct action of influenza viral activity in our exposed virally infected brains, since it is known to also be affected by the influenza virus (Pichlmair et al, 2006). Additionally, T. Cruzi infection also leads to increases in RNA helicase activity (Diaz Añel et al, 2000).
Foxp2 is a putative transcription factor containing a polyglutamine tract and a forkhead DNA binding domain. Foxp2 is associated with language and speech deficits (Hurst et al, 1990; Lai et al, 2000). Lai et al (2000), studied a three-generation pedigree with a severe speech and language disorder and found a point mutation in the forkhead domain segregated with affected status. Gong et al (2004) found a significant association between an SNP of Foxp2 and autism (Gong et al, 2004). Additionally, a significant genetic association has also been found between Foxp2 polymorphisms and subjects with schizophrenia (Sanjuan et al, 2006). Our results have shown an upregulation of Foxp2 protein product in both cerebellum (P0, P35) and hippocampus (P35, P56) of the progeny of virally exposed mice suggesting that this candidate gene for autism is affected by our animal model and may serve as an important gene to study in schizophrenia. Furthermore, prenatal viral infection of E9 Balb/c mice caused upregulation of Foxp2 gene in cerebella of day 35 mouse progeny pointing to specificity of viral insult in upregulation of this gene in various embryogenic stages (E9 vs. E18) and in two strains of mouse (C57BL6J vs. Balb/c) (Fatemi et al, 2007a).
Analysis of brain volume in the exposed mouse progeny showed a significant (p<0.05) ∼ 4.3% decrease at postnatal day 35 (Fig. 2). Quite interestingly, this atrophy has been seen in adolescent children with schizophrenia and been associated with abnormalities in the neuregulin 1 gene function, a ligand for receptor Erbb4 which is significantly elevated in frontal cortex of day 56 exposed mouse progeny. Previous work had also shown pyramidal cell atrophy and thinning of cortex and hippocampus in E9 infected mouse progeny (Calo et al, 2005). Fractional anisotropy of the corpus callosum revealed white matter atrophy at postnatal day 35, suggesting a decrease in myelin and axonal density. This finding is relevant to our study since numerous reports show white matter thinning in brains of subjects with schizophrenia, especially in corpus callosum (Ardekani et al, 2003; Kubicki et al, 2005; Wolkin et al, 1998).
Neurochemical measurements in cerebella of virally exposed mice provided a very intriguing pattern i.e., levels of serotonin were reduced significantly in postnatal days 14 and 35. Serotonin receptor abnormalities have been demonstrated in patients with schizophrenia (Penas-Lledo et al, 2007). The level of serotonin metabolite 5HIAA was also reduced significantly in postnatal day 14 and nonsignificantly in postnatal day 35. Serotonin also has neurotrophic effects during embryogenesis (Sodhi and Sanders-Bush, 2004) and reductions in serotonin in critical periods of brain development could contribute to brain atrophy observed in this study. Finally, levels of taurine, a non-proteinogenic amino acid abundantly found in brain (Albrecht and Schousboe, 2005) was reduced significantly in day 35 virally exposed mouse cerebella. Taurine has an essential role in development and survival of neurons. Taurine deficiency in development leads to abnormal cerebellar organization and function. Reduced levels of CSF taurine lave been demonstrated in drug-free schizophrenic patients (Do et al, 1995).
Additionally, previous work has shown that prenatal viral infection in mice leads to several behavioral abnormalities (deficits in prepulse inhibition (PPI), decreased exploratory behavior in both open-field and novel-object tests and deficits in social interaction) observed in subjects with schizophrenia (Shi et al, 2003). PPI deficits can be restored following acute administration of clozapine and chlorpromazine, adding to the predicative validity of this model (Shi et al, 2003).
The current study provides a chance to compare infection early in pregnancy (E9) with infection late in pregnancy (E18) (Table 6). Our microarray data for E9 was not as systematized as E18 (whole brain in E9 vs. three areas in E18), however the comparing totality of genes between two groups, far fewer genes displayed changed expression at E9 than at E18 (Table 6). Thus, it appears that the effect of viral infection early in pregnancy is not nearly as dramatic as at later time points. Mice infected at E9 displayed behavioral abnormalities as described above (Shi et al., 2003). We have not tested the offspring of mice infected at E18 for behavioral abnormalities, this will be investigated in future experiments. Morphological comparisons between the two infection time points varied in terms of technique. For E9 exposed offspring we used cell counting and serial sectioning which revealed pyramidal cell atrophy, increased numbers of nonpyramidal and pyramidal cells at P14 weeks, and early thinning of cortex and hippocampus at P0 (Fatemi et al., 2002b). Adults displayed increased brain area and decreased ventricular area (Fatemi et al., 2002b). For E18, we did not use cell counting but rather MRI and DTI which revealed brain atrophy at P35 and fractional anisotropy revealed thinning white matter in corpus callosum. Finally, it appears that expression of different sets of genes and proteins are altered depending on time of infection. At E9, we observed alterations in gene and protein expression related to neurodevelopment (Reelin, SNAP-25), glutamate/GABAergic system (GAD 65/67, nNOS), immune system dysregulation and astroglial activation (GFAP), glial-neuronal communication (aquaporin 4 and connexin 43), (Fatemi et al, 1998a, Fatemi et al, 1998b; Fatemi et al, 1999; Fatemi et al, 2000; Fatemi et al, 2002b; Fatemi et al, 2004; Fatemi et al, 2007b). We did not find these genes to be altered in the current study. Thus, clear but different patterns are apparent with E9 infection vs. E18 infection. Interestingly, Foxp2 expression is altered following insult at both E9 and E18.
Table 6.
Comparison of the Effects of Viral Infections at E9 and E18 on Exposed Mouse Offspring
| E9 | E18 | |||||||
|---|---|---|---|---|---|---|---|---|
| Morphology | P0 a | ↑Pyramidal cell density, ↓Pyramidal cell nuclear size | P35 a | Brain atrophy and thinning white matter in corpus callosum | ||||
| P98 a | ↑Pyramidal cell density, ↑Nonpyramidal cell density, ↓Pyramidal cell nuclear size, ↑Brain area, ↓Ventricular area | |||||||
| Microarray | P0 b | Whole Brain | ↑21 | ↓18 | P0 a | PFC | ↑43 | ↓29 |
| P35 b | Neocortex | ↑50 | ↓21 | Hippocampus | ↑129 | ↓46 | ||
| Cerebellum | ↑103 | ↓102 | Cerebellum | ↑120 | ↓37 | |||
| P56 b | Neocortex | ↑13 | ↓11 | P14 a | PFC | ↑16 | ↓17 | |
| Cerebellum | ↑27 | ↓23 | Hippocampus | ↑9 | ↓12 | |||
| Cerebellum | ↑11 | ↓5 | ||||||
| P56 a | PFC | ↑86 | ↓24 | |||||
| Hippocampus | ↑45 | ↓17 | ||||||
| Cerebellum | ↑74 | ↓22 | ||||||
| Behavior | P42-P56 a, b | ↓Prepulse inhibition, ↓Statle response, ↓Exploratory behavior, ↓Social interactions | ND | |||||
| Genes/Proteins | P0 | ↑nNOS a, ↑SNAP-25 a, ↓Reelin a, ↑GFAP b | P0 a | ↑Arhgef9, ↑Arnt, ↑Dapk1, ↑Efnb2, ↑Erbb4, ↑Ivns1abp, ↑Myt1l, ↑Nxph2, ↑Sema3a, ↑Sox2, ↑Trfr2, ↑Foxp2 | ||||
| P14 | ↑GFAP a, ↑GAD65 b | |||||||
| P35 | ↑nNOS a, ↑GFAP a, ↑GAD65 b, ↑GAD67 b, ↑Foxp2b | P35 a | ↑Foxp2 | |||||
| P56 | ↓nNOS a, ↑GAD67 b | P56 a | ↑Dby, ↑Uty, ↑Foxp2 | |||||
| Neurotransmitters | ND | P14 a | ↓5HT, ↓5HIAA | |||||
| P35 a | ↓5HT, ↓Taurine | |||||||
ND, not determined; ↑, increase; ↓, decrease
C57BL6J mice
Balb/c mice.
Data from Fatemi et al, 1998a; Fatemi et al, 1998b, Fatemi et al, 1999; Fatemi et al, 2000; Fatemi et al, 2002a; Fatemi et al, 2002b, Fatemi et al, 2003; Shi et al, 2003; Fatemi et al, 2004; Fatemi et al, 2005; Fatemi et al, 2007a; Fatemi et al, 2007b.
Comparison of our animal model with other models reveals similarities in terms of outcomes, particularly regarding infection on E9 (for a review see Meyer et al., 2007). As mentioned previously, infection on E9 resulted in decreased Reelin (Fatemi et al., 1999), which was similarly observed by Meyer et al. (2006) following insult with PolyI:C on E9. Infection on E9 in our model led to defective corticogenesis (Fatemi et al, 1999) which has also been observed by Meyer et al. (2006) following insult on E9 and E17. The current study has not examined corticogenesis following insult on E18; this should be examined in future experiments, however, presence of brain atrophy and thinning of white matter do occur in P35 exposed offspring. Finally, infection on E9 resulted in gliosis in our model (Fatemi et al, 2002a). Similar results have been obtained by Samuelsson et al (2006) following administration of IL-6 to rats at mid-pregnancy (E8, E10, E12) and late pregnancy (E16, E18, E20) (Samuelsson et al, 2006) and by treatment of rats throughout gestation with lipopolysaccharide (Borrell et al, 2002).
While caution should be used in comparing our animal model with viral infection and the possibility of subsequent development of autism or schizophrenia in humans, comparing E9 to E18, the implication is that infection at different times during pregnancy may result in differing pathologies. Infection on E18 may be a better model for schizophrenia based on the changes in brain at P35 or adolescence. Previous reports have shown that an acceleration of gray matter loss is observed with very early onset schizophrenia (Rapoport et al., 1999; Thompson et al., 2001) similar to the brain volume atrophy of exposed offspring of mice infected on E18 display at P35. Infection on earlier time points such as E9 may be a better model for autism with early thinning and later enlargement of the brain. Several epidemiological reports point to 1st trimester infection for autism (reviewed by Arndt et al, 2005; Libbey et al., 2005; Spear, 2000, 2004) vs. some reports supporting 2nd trimester infection for schizophrenia (Limosin et al., 2003; Suvisaari et al., 1999).
In conclusion, genetic, morphometric, and neurochemical data presented here, support the notion that prenatal influenza infection can indeed precipitate altered patterns of gene expression, and neuroanatomic abnormalities in the developing brain. This model mimics several biochemical, genetic, behavioral, and neuroanatomic characteristics of schizophrenia and may shed light on mechanisms responsible for genesis of this severe disorder.
Supplementary Material
Acknowledgments
Grant support by National Institute of Child Health and Human Development (#1R01 HD046589-01A2) to SHF is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addington AM, Gornick MC, Shaw P, Seal J, Gogtay N, Greenstein D, Clasen L, Coffey M, Gochman P, Long R, Rapoport JL. Neuregulin 1 (8p12) and childhood-onset schizophrenia: susceptibility haplotypes for diagnosis and brain developmental trajectories. Mol. Psychiatry. 2006;12(2):195–205. doi: 10.1038/sj.mp.4001906. [DOI] [PubMed] [Google Scholar]
- Albrecht J, Schousboe A. Taurine interaction with neurotransmitter receptors in the CNS: an update. Neurochem. Res. 2005;30(12):1615–1621. doi: 10.1007/s11064-005-8986-6. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) APA; Washington D.C.: 1994. [Google Scholar]
- Ardekani BA, Nierenberg J, Hoptman MJ, Javitt DC, Lim KO. MRI study of white matter diffusion anisotropy in schizophrenia. NeuroReport. 2003;14(16):2025–2029. doi: 10.1097/00001756-200311140-00004. [DOI] [PubMed] [Google Scholar]
- Arndt TL, Stodgell CJ, Rodier PM. The teratology of autism. Int. J. Dev. Neurosci. 2005;23(23):189–199. doi: 10.1016/j.ijdevneu.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Asherson P, Mane R, McGiffin P. Genetics and schizophrenia. In: Mirsch SR, Weinberger DR, editors. Schizophrenia. Blackwell Scientific; Boston: 1994. pp. 253–274. [Google Scholar]
- Bartholomew RA, Parks JE. Identification, localization, and sequencing of fetal bovine VASA homolog. Anim. Reprod. Sci. 2007;101(34):241–251. doi: 10.1016/j.anireprosci.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Borrell J, Vela JM, Arévalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26(2):204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156(23):234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry. 2004;61(8):774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Calo L, Spillantini M, Nicoletti F, Allen ND. Nurr1 co-localizes with EphB1 receptors in the developing ventral midbrain, and its expression is enhanced by the EphB1 ligand, ephrinB2. J. Neurochem. 2005;92(2):235–245. doi: 10.1111/j.1471-4159.2004.02853.x. [DOI] [PubMed] [Google Scholar]
- Diaz Añel AM, Rossi MS, Espinosa JM, Guida C, Freitas FA, Kornblihtt AR, Zingales B, Flawia MM, Torres NH. mRNA encoding a putative RNA helicase of the DEAD-box gene family is up-regulated in trypomastigotes of Trypanosoma cruzi. J. Eukaryot. Microbiol. 2000;47(6):555–560. doi: 10.1111/j.1550-7408.2000.tb00089.x. [DOI] [PubMed] [Google Scholar]
- Do KQ, Lauer CJ, Schreiber W, Zollinger M, Gutteck-Amsler U, Cuenod M, Holsboer F. gamma-Glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J. Neurochem. 1995;65(6):2652–2662. doi: 10.1046/j.1471-4159.1995.65062652.x. [DOI] [PubMed] [Google Scholar]
- Dracheva S, Davis KL, Chin B, Woo DA, Schmeidler J, Haroutunian V. Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiol. Dis. 2006;21(3):531–540. doi: 10.1016/j.nbd.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Law AJ, Everall IP, Harrison PJ. The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol. Psychiatry. 2003;8(2):148–155. doi: 10.1038/sj.mp.4001233. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Sidwell R, Akhter P, Sedgewick J, Thuras P, Bailey K, Kist D. Human influenza viral infection in utero increases nNOS expression in hippocampi of neonatal mice. Synapse. 1998a;29(1):84–88. doi: 10.1002/(SICI)1098-2396(199805)29:1<84::AID-SYN8>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Sidwell R, Kist D, Akhter P, Meltzer HY, Bailey K, Thuras P, Sedgwick J. Differential expression of synaptosome-associated protein 25 kDa [SNAP-25] in hippocampi of neonatal mice following exposure to human influenza virus in utero. Brain Res. 1998b;800(1):1–9. doi: 10.1016/s0006-8993(98)00450-8. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol. Psychiatry. 1999;4(2):145–154. doi: 10.1038/sj.mp.4000520. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Cuadra AE, El-Fakahany EE, Sidwell RW, Thuras P. Prenatal viral infection causes alterations in nNOS expression in developing mouse brains. Neuroreport. 2000;11(7):1493–1496. [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, Thuras P. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol. Psychiatry. 2002a;7(6):633–640. doi: 10.1038/sj.mp.4001046. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle J, Kanodia R, Kist D, Patterson P, Shi L, Sidwell RW. Prenatal viral infection causes macrocephaly and pyramidal cell atrophy in the developing mice. Cell. Mol. Neurobiol. 2002b;22(1):25–33. doi: 10.1023/a:1015337611258. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Araghi-Niknam M, Laurence JA, Stary JM, Sidwell RW, Lee S. Glial fibrillary acidic protein and glutamic acid decarboxylase 65 and 67 kDa proteins are increased in brains of neonatal BALB/c mice following viral infection in utero. Schizophr. Res. 2004;69(1):121–123. doi: 10.1016/S0920-9964(03)00175-0. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, editor. Neuropsychiatric Disorders and Infection. Taylor & Francis; London: 2005. [Google Scholar]
- Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: A potential animal model for schizophrenia and autism. Synapse. 2005a;57(2):91–99. doi: 10.1002/syn.20162. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Brooks AI, Pearce DA, Reutiman TJ, Lee S. Reelin signaling is impaired in autism. Biol. Psychiatry. 2005b;57(7):777–787. doi: 10.1016/j.biopsych.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Bell C, Nos L, Fried P, Pearce DA, Singh S, Siderovski DP, Willard FS, Fukuda M. Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology. 2006;31:1888–1899. doi: 10.1038/sj.npp.1301002. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Sidwell RW. The role of cerebellar genes in pathology of autism and schizophrenia. Cerebellum. 2007a doi: 10.1007/s12311-008-0017-0. In Press. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Sidwell RW. Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophr. Res. 2007b doi: 10.1016/j.schres.2007.09.031. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice LJ, Felice JD, Kissinger PT. Determination of catecholamines in rat brain parts by reverse-phase ion-pair liquid chromatography. J. Neurochem. 1978;31(6):1461–1465. doi: 10.1111/j.1471-4159.1978.tb06573.x. [DOI] [PubMed] [Google Scholar]
- Franklin BJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; San Diego, CA: 1997. [Google Scholar]
- Gong X, Jia M, Ruan Y, Shuang M, Liu J, Wu S, Guo Y, Yang J, Ling Y, Yang X, Zhang D. Association between the FOXP2 gene and autistic disorder in Chinese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004;127(1):113–116. doi: 10.1002/ajmg.b.20162. [DOI] [PubMed] [Google Scholar]
- Gottwald B, Wilde B, Mihajlovic Z, Mehdorn HM. Evidence for distinct cognitive deficits after focal cerebellar lesions. J. Neurol. Neurosurg. Psychiatry. 2004;75(11):1524–1531. doi: 10.1136/jnnp.2003.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006;12(7):824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC, Pawson T. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron. 2001;32(6):1041–1056. doi: 10.1016/s0896-6273(01)00553-0. [DOI] [PubMed] [Google Scholar]
- Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry. 2006;11(6):557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- Hurst JA, Baraitser M, Auger E, Graham F, Norel SV. An extended family with a dominantly inherited speech disorder. Dev. Med. Child. Neurol. 1990;32(4):352–355. doi: 10.1111/j.1469-8749.1990.tb16948.x. [DOI] [PubMed] [Google Scholar]
- Irie F, Yamaguchi Y. EPHB receptor signaling in dendritic spine development. Front. Biosci. 2004;9:1365–1373. doi: 10.2741/1325. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Steiner AA, Scheck AC, Romanovsky AA. Expression of Eph receptors and their ligands, ephrins, during lipopolysaccharide fever in rats. Physiol. Genomics. 2005;21(2):152–160. doi: 10.1152/physiolgenomics.00043.2004. [DOI] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J. Clin. Invest. 2006;116(9):2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicki M, Park H, Westin CF, Nestor PG, Mulkern RV, Maier SE, Niznikiewicz M, Connor EE, Levitt JJ, Frumin M, Kikinis R, Jolesz FA, McCarley RW, Shenton ME. DTI and MTR abnormalities in schizophrenia: analysis of white matter integrity. Neuroimage. 2005;26(4):1109–1118. doi: 10.1016/j.neuroimage.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Levy ER, Hodgson S, Fox M, Jeremiah S, Povey S, Jamison DC, Green ED, Vargha-Khadem F, Monaco AP. The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder. Am. J. Hum. Genet. 2000;67(2):357–368. doi: 10.1086/303011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Sweeten TL, McMahon WM, Fujinami RS. Autistic disorder and viral infections. J. Neurovirol. 2005;11(1):1–10. doi: 10.1080/13550280590900553. [DOI] [PubMed] [Google Scholar]
- Limosin F, Rouillon F, Payan C, Cohen JM, Strub N. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta. Psychiatr. Scand. 2003;107(5):331–335. doi: 10.1034/j.1600-0447.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- Maes M, Meltzer HY, Buckley P, Bosmans E. Plasma-soluble interleukin-2 and transferrin receptor in schizophrenia and major depression. Eur. Arch. Psychiatry Clin. Neurosci. 1995;244(6):325–329. doi: 10.1007/BF02190412. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. 2006;26(18):4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Yee BK, Feldon J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist. 2007;13(3):241–256. doi: 10.1177/1073858406296401. [DOI] [PubMed] [Google Scholar]
- Mori S, Ito R, Zhang J, Kaufmann WE, van Zijl PC, Solaiyappan M, Yarowski P. Diffusion tensor imaging of the developing mouse brain. Magn. Reson. Med. 2001;46(1):18–23. doi: 10.1002/mrm.1155. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Tokunaga A, Hara A, Hamaguchi T, Kato K, Iwamatsu M, Okano H, Kaibuchi K. Role of numb in dendritic spine development with a Cdc42 GEF intersectin and EphB2. Mol. Biol. Cell. 2006;17(3):1273–1285. doi: 10.1091/mbc.E05-07-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penas-Lledo EM, Dorado P, Caceres MC, de la Rubia A, Llerena A. Association between T102C and A-1438G polymorphisms in the serotonin receptor 2A (5-HT2A) gene and schizophrenia: relevance for treatment with antipsychotic drugs. Clin. Chem. Lab. Med. 2007;45(7):835–838. doi: 10.1515/CCLM.2007.181. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol. Psychiatry. 2007;61(4):482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljenstrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Piepponen TP, Skujins A. Rapid and sensitive step gradient assays of glutamate, glycine, taurine and gamma-aminobutyric acid by high-performance liquid chromatography-fluorescence detection with o-phthalaldehyde-mercaptoethanol derivatization with an emphasis on microdialysis samples. J. Chromatogr. B Biomed. Sci. Appl. 2001;757(2):277–283. doi: 10.1016/s0378-4347(01)00156-6. [DOI] [PubMed] [Google Scholar]
- Rami A, Jansen S, Giesser I, Winckler J. Post-ischemic activation of caspase-3 in the rat hippocampus: evidence of an axonal and dendritic localization. Neurochem. Int. 2003;43(3):211–223. doi: 10.1016/s0197-0186(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Giedd JN, Blumenthal J, Hamburger S, Jeffries N, Fernandez T, Nicolson R, Bedwell J, Lenane M, Zijdenbos A, Paus T, Evans A. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry. 1999;56(7):649–654. doi: 10.1001/archpsyc.56.7.649. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
- Reid T, Bathoorn A, Ahmadian MR, Collard JG. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 1999;274(47):33587–33593. doi: 10.1074/jbc.274.47.33587. [DOI] [PubMed] [Google Scholar]
- Samuelsson AM, Jennische E, Hansson HA, Holmäng A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;290(5):R1345–R1356. doi: 10.1152/ajpregu.00268.2005. [DOI] [PubMed] [Google Scholar]
- Sanjuan J, Tolosa A, Gonzalez JC, Aguilar EJ, Perez-Tur J, Najera C, Molto MD, de Frutos R. Association between FOXP2 polymorphisms and schizophrenia with auditory hallucinations. Psychiatr. Genet. 2006;16(2):67–72. doi: 10.1097/01.ypg.0000185029.35558.bb. [DOI] [PubMed] [Google Scholar]
- Sawa A, Snyder SH. Schizophrenia: diverse approaches to a complex disease. Science. 2002;296(5568):692–695. doi: 10.1126/science.1070532. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Caplan D. Cognition, emotion and the cerebellum. Brain. 2006;129(Pt 2):290–292. doi: 10.1093/brain/awh729. [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. A mouse model of mental illness: maternal influenza infection causes behavioral and pharmacological abnormalities in the offspring. J. Neurosci. 2003;23(1):297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodhi MS, Sanders-Bush E. Serotonin and brain development. Int. Rev. Neurobiol. 2004;59:111–174. doi: 10.1016/S0074-7742(04)59006-2. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci. Behav. Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Spear LP. Adolescent brain development and animal models. Ann. N.Y. Acad. Sci. 2004;1021:23–26. doi: 10.1196/annals.1308.002. [DOI] [PubMed] [Google Scholar]
- Sperk G, Berger M, Hortnagl H, Hornykiewicz O. Kainic acid-induced changes of serotonin and dopamine metabolism in the striatum and substantia nigra of the rat. Eur. J. Pharmacol. 1981;74(4):279–286. doi: 10.1016/0014-2999(81)90046-7. [DOI] [PubMed] [Google Scholar]
- Sperk G. Simultaneous determination of serotonin, 5-hydroxindoleacetic acid, 3,4-dihydroxyphenylacetic acid and homovanillic acid by high performance liquid chromatography with electrochemical detection. J. Neurochem. 1982;38(3):840–843. doi: 10.1111/j.1471-4159.1982.tb08708.x. [DOI] [PubMed] [Google Scholar]
- Suvisaari J, Haukka J, Tanskanen A, Hovi T, Lonnqvist J. Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am. J. Psychiatry. 1999;156(7):1100–1102. doi: 10.1176/ajp.156.7.1100. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, Toga AW, Rapoport JL. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc. Natl. Acad. Sci. USA. 2001;98(20):11650–11655. doi: 10.1073/pnas.201243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SY, Lee HC, Chen CC, Lee CH. Plasma levels of soluble transferrin receptors and Clara cell protein (CC16) during bipolar mania and subsequent remission. J. Psychiatr. Res. 2003;37(3):229–235. doi: 10.1016/s0022-3956(02)00103-6. [DOI] [PubMed] [Google Scholar]
- Vihanto MM, Plock J, Erni D, Frey BM, Frey FJ, Huynh-Do U. Hypoxia up-regulates expression of Eph receptors and ephrins in mouse skin. FASEB J. 2005;19(12):689–1691. doi: 10.1096/fj.04-3647fje. [DOI] [PubMed] [Google Scholar]
- Wolkin A, Rusinek H, Vaid G, Arena L, Lafargue T, Sanfilipo M, Loneragan C, Lautin A, Rotrosen J. Structural magnetic resonance image averaging in schizophrenia. Am. J. Psychiatry. 1998;155(8):1064–1073. doi: 10.1176/ajp.155.8.1064. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry R, Gentleman R, Murillo F, Spencer F. A model based background adjustment for oligonucleotide expression arrays. Johns Hopkins University, Department of Biostatistics Working Papers; Baltimore, MD: 2004. Technical report. [Google Scholar]
- Zapala MA, Hovatta I, Ellison JA, Wodicka L, Del Rio JA, Tennant R, Tynan W, Broide RS, Helton F, Stoveken BS, Winrow C, Lockhart DJ, Reilly JF, Young WG, Bloom FE, Lockhart DJ, Barlow C. Adult mouse brain gene expression patterns bear an embryologic imprint. Proc Natl Acad Sci USA. 2005;102(29):10357–10362. doi: 10.1073/pnas.0503357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman AW, Connors SL, Pardo CA. Neuroimmunology and neurotransmitters in autism. In: Tuchman R, Rapin I, editors. Autism: A Neurological Disorder of Early Brain Development. Mac Keith Press, International Child Neurology Association; London: 2006. pp. 141–159. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.