

Abstract
Protein solution nuclear magnetic resonance (NMR) can be conducted in a slightly anisotropic environment, where the orientational distribution of the proteins is no longer random. In such an environment, the large one-bond internuclear dipolar interactions no longer average to zero and report on the average orientation of the corresponding vectors relative to the magnetic field. The desired very weak ordering, on the order of 10−3, can be induced conveniently by the use of aqueous nematic liquid crystalline suspensions or by anisotropically compressed hydrogels. The resulting residual dipolar interactions are scaled down by three orders of magnitude relative to their static values, but nevertheless can be measured at high accuracy. They are very precise reporters on the average orientation of bonds relative to the molecular alignment frame, and they can be used in a variety of ways to enrich our understanding of protein structure and function. Applications to date have focused primarily on validation of structures, determined by NMR, X-ray crystallography, or homology modeling, and on refinement of structures determined by conventional NMR approaches. Although de novo structure determination on the basis of dipolar couplings suffers from a severe multiple minimum problem, related to the degeneracy of dipolar coupling relative to inversion of the internuclear vector, a number of approaches can address this problem and potentially can accelerate the NMR structure determination process considerably. In favorable cases, where large numbers of dipolar couplings can be measured, inconsistency between measured values can report on internal motions.
Structure determination by NMR traditionally has relied on the measurement of a large number of semiquantitative local restraints. The most important of these is the 1H-1H NOE, which provides distance information for pairs of protons separated by less than ∼5 Å. The accuracy of the NOE-derived distance usually decreases with the actual value of the distance because the precision of the measured intensity decreases with longer distance (weaker NOE), and the effect of indirect NOE magnetization transfer (spin diffusion) generally is worse for protons farther apart. Three-bond J couplings, either homonuclear 1H-1H, 13C-13C, or heteronuclear 13C-1H, 13C-15N, or 15N-1H, are related to the intervening dihedral angles via empirically parameterized Karplus relationships (Karplus 1959; Bystrov 1976; Hu and Bax 1997) and are also commonly used in structure determination. Numerous methods have been proposed in recent years for the measurement of such couplings (Bax et al. 1994; Biamonti et al. 1994; Vuister et al. 1999). It even has been shown possible to measure J coupling interaction through hydrogen bonds, permitting the hydrogen bond donor and acceptor atoms to be linked unambiguously (Dingley and Grzesiek 1998; Pervushin et al. 1998; Wang et al. 1999). Another relatively recent addition to the arsenal of experimental restraints includes cross-correlated relaxation (Reif et al. 1997; Yang et al. 1997; Pelupessy et al. 1999). In contrast to the other parameters mentioned above, cross-correlated relaxation can, at least in principle, report on the relative orientation of dipolar or CSA tensors anywhere in the molecule. However, in practice, the requirement to monitor the decay of a two-spin coherence limits this type of information to spatially proximate pairs of spins. Finally, chemical shifts provide yet another source of structural information: There are well characterized relationships between the polypeptide backbone angles φ and ψ and 1Hα, 13Cα, 13Cβ, and 13C′ chemical shifts, which have been exploited in a variety of ways to improve the quality of protein structures (Spera and Bax 1991; Wishart et al. 1991; Kuszewski et al. 1995; Sitkoff and Case 1998; Cornilescu et al. 1999), but again, all contain strictly local information. The main exception has been the paramagnetic shift, which can extend over distances as large as 15–20 Å and which has become the center of renewed interest in structure determination (Banci et al. 1997; Gochin 1998; Boisbouvier et al. 1999), precisely because it complements the short-range information contained in the other parameters mentioned above.
The present mini-review focuses on a different source of structural information: the direct magnetic dipole-dipole coupling between spin-½ nuclei (1H, 13C, 15N). Dipolar couplings contain information on the orientation of internuclear, usually one-bond vectors relative to the magnetic field, regardless of where in the protein this vector is situated. Besides constraining local geometry, dipolar couplings therefore also have a global ordering character as they restrain all bond orientations relative to a common frame. This therefore provides a highly needed complement to the strictly local NOE and J coupling restraints.
Dipolar couplings are potentially quite large interactions, caused by the magnetic flux lines of one nucleus affecting the magnetic field at the site of another nucleus (Fig. 1 ▶). Only the component parallel to the external, much stronger magnetic field (Bo) concerns us; the components orthogonal to the Bo magnetic field have a negligible effect on the total magnitude of the vector sum of the external and the dipolar field. So, the z component of the dipolar field of nucleus P will change the resonance frequency of nucleus Q by an amount that depends on the internuclear distance and on the orientation of the internuclear vector relative to Bo. For a fixed orientation of the vector, say parallel to Bo, nuclear spin P can increase or decrease the total magnetic field at nucleus Q, depending on whether P is parallel or antiparallel to Bo. In an ensemble of molecules, half of the P nuclei will be parallel to Bo, the other half antiparallel, and Q will show two resonances (doublet), separated in frequency by
Figure 1.

Magnetic dipole-dipole coupling, illustrated for a 15N-1H spin pair. 15N and 1H magnetic moments are aligned parallel (or antiparallel) to the static magnetic field, Bo. The total magnetic field in the Bo direction at the 15N position can increase or decrease relative to Bo, depending on the orientation of the 15N-1H vector and the spin state of the proton (parallel or antiparallel to Bo).
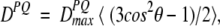 |
(1a) |
where θ is the angle between the internuclear vector and Bo, the 〈〉 brackets denote time or ensemble averaging, and
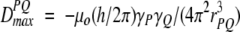 |
(1b) |
is the doublet splitting that applies for the case where θ = 0. The meanings of other symbols are: μo, magnetic permitivity of vacuum; h, Planck’s constant; γP, magnetogyric ratio of nucleus P; rPQ, the distance between nuclei P and Q. Equation 1a shows that the dipolar splitting, DPQ, provides direct information on the angle θ, i.e., on the orientation of the internuclear vector, and that it scales with the inverse of the cubed internuclear distance.
In isotropic solution, rotational Brownian diffusion rapidly averages the internuclear dipolar interaction of Equation 1a to exactly zero. As a result, the solution NMR spectrum shows narrow resonances, which can be assigned to individual nuclei in the protein but which no longer contain the valuable orientational information. In contrast, in solid-state NMR such averaging does not take place, and each nucleus couples with a very large number of other nuclei (each of which can point parallel or antiparallel to the magnetic field), resulting in unresolvable, very broad resonances. Mechanical rotation of the sample around an axis that makes an angle of 54.7° with the Bo field (magic angle spinning), together with radiofrequency irradiation, can then be used to average the dipolar interaction to zero and to reintroduce sharp features in the NMR spectrum (Mehring 1982).
The work described in this review concerns the intermediate case, where the protein is dissolved in a slightly anisotropic aqueous medium, where not all orientations of the protein are equally likely to occur. In this case, the alignment of the protein can be described by an alignment tensor, A′. The A′ tensor is a real, symmetric matrix and therefore can be diagonalized, and in the corresponding molecular frame its principal components A′xx, A′yy, and A′zz reflect the probabilities for the x, y, and z axes to be parallel to Bo. It is only the difference among A′xx, A′yy, and A′zz that contributes to dipolar coupling, and the alignment matrix is therefore commonly used in its traceless form, A. Defining |Azz| > |Ayy| > |Axx|, the dipolar coupling depends on the polar coordinates of the P-Q vector in the frame of the diagonalized alignment tensor (Fig. 2 ▶) as:
Figure 2.

Bands of allowed vector orientations, corresponding to a given value of a dipolar coupling, in the frame of the diagonalized alignment tensor. The width of the bands corresponds to a typical measurement uncertainty that is 3% of the total range of the couplings. The bottom cone corresponds to the inverse vector orientations relative to the top cone. Cones are distorted due to the nonzero rhombicity, R, in Equation 2B. Polar angles θ and φ are defined for the vector shown.
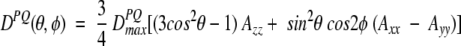 |
(2a) |
This equation is usually rewritten as
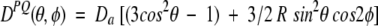 |
(2b) |
where Da = ¾DPQmaxAzz is referred to as the magnitude of the dipolar coupling tensor, commonly normalized to the N-H dipolar interaction, and R = ⅔(Axx − Ayy)/Azz is the rhombicity. Equation 2 indicates that for a given value of DPQ there is an entire cone of (θ,φ) solutions that correspond to this dipolar coupling. So, the dipolar coupling does not uniquely define the orientation but restricts it to be on the surface of a distorted cone (Fig. 2 ▶). Another important point to note is that the inverted vector orientation (QP) gives rise to the same coupling, and the inverted cone is therefore also included in the solutions to Equation 2.
As discussed later, in order to permit facile measurement of dipolar interactions it is essential that they are averaged to a very small fraction (typically ∼10−3) of their static value. The first dipolar coupling measurements in a solubilized protein were carried out by Prestegard and coworkers on paramagnetic myoglobin (Tolman et al. 1995). This work built on the pioneering studies by Bothner-By, Maclean, and coworkers (Gayathri et al. 1982), who had shown that the magnetic susceptibility of small molecules can be sufficient to yield very small but observable degrees of orientation, that scale with the square of the magnetic field strength. In Prestegard’s myoglobin study, the substantial paramagnetism of the iron resulted in an alignment strength that yielded one-bond 1H-15N dipolar interactions of up to several Hertz. Although this splitting is smaller than the natural resonance line width in proteins, it nevertheless can be measured with reasonable accuracy because it manifests itself as a field-dependent contribution to the one-bond 1JNH splitting (normally −94 Hz). So it gives rise to a small change in a well resolved splitting, rather than in an unresolvably small splitting.
For diamagnetic proteins the magnetically induced alignment usually is considerably smaller, making accurate measurement of dipolar contributions quite challenging (Tjandra et al. 1996). Nevertheless, in favorable systems such as a protein/DNA complex, where the parallel stacking of the nucleic acid basis in the helical B-form DNA causes their magnetic susceptibility tensors to co-add constructively, dipolar couplings of several Hertz can be obtained for the backbone amides and 13Cα-1Hα sites. Importantly, Tjandra developed a clever but simple procedure to incorporate these experimental dipolar couplings into the structure calculation (1997), and these early results clearly demonstrated the utility of the dipolar couplings. Not only did they substantially improve the percentage of backbone angles in the most favored region of the Ramachandran map (Tjandra et al. 1997), the structure also yielded considerably higher cross-validation statistics (Ottiger et al. 1997).
Although feasible, the magnetic susceptibility-induced alignment generally remains considerably smaller than optimal, making it difficult to measure dipolar couplings at high relative accuracy. The weakness of the alignment also limits the measurement of dipolar couplings to the largest interactions, such as one-bond 15N-1H or 13C-1H, and leaves many of the other potentially useful couplings, such as 13C-13C and 13C-15N, inaccessible.
Alignment by liquid crystals
Liquid crystal NMR has long been known as a method to study the structural details of small organic molecules at very high levels of precision. First demonstrated by Saupe and Englert in the early 1960’s, an organic molecule dissolved in a nematic liquid crystalline phase exhibits quite strong alignment when placed in an NMR magnet (Saupe and Englert 1963). The rapid translational diffusion eliminates any intermolecular dipolar interactions, but very large intramolecular dipolar couplings can typically be seen, permitting the measurement of molecular parameters such as the C-C-H angle in methyl groups at unprecedented accuracy (Wooton et al. 1979). For molecules bearing more than about half a dozen hydrogens, which all couple to one another, the spectra become intractably complex, however. Clearly, the use of such organic liquid crystalline phases, besides being incompatible with protein solubility requirements, makes the approach inapplicable to biological macromolecules.
Initially, while searching for media that could impose the required much weaker degree of order, we focused on mechanical alignment, such as that imposed by wound hydrated films, and the use of laminar flow in ultrathin capillaries. However, while watching the very weak degree of order imposed on water by a self-assembling phase of lipid-like molecules in a presentation by Olle Soderman at a 1997 Royal Society of Chemistry meeting, it became clear to me that such media could offer a technically far simpler and more practical way to weakly align proteins. Although Soderman’s highly charged system turned out to be inapplicable to the first few proteins we tried, other systems that have liquid crystalline phase behavior had been around for quite some time. One of these is the so-called bicelle phase, consisting of a mixture of long-chain phospholipids and detergent (Sanders and Schwonek 1992; Sanders et al. 1994). At the right molar ratio, these adopt an α-lamellar phase of highly porous bilayers that cooperatively order in the magnetic field, with the bilayer normal orthogonal to the magnetic field (Fig. 3 ▶; Gaemers and Bax 2001; Nieh et al. 2001). Originally, this system was developed by Sanders and Prestegard for the study of lipophilic molecules, anchored to the bilayers (Sanders and Prestegard 1990; Sanders et al. 1994). Due to the high degree of bilayer order, the anchored molecules become ordered equally strongly, resulting in very large dipolar couplings and intractable 1H NMR spectra. However, confinement of the aqueous phase by the bilayers was expected to be perfectly suitable for inducing a weak nonrandomness in the orientational distribution of water-soluble molecules.
Figure 3.

Morphology of liquid crystalline bicelles. (A) Cross-section orthogonal to the NMR sample cell shows domains of parallel lipid bilayers. Domain size is approximately 10 μm. (B) Section of an individual, highly porous bilayer. The total surface area of the pores is comparable to that of the lipid bilayers. (C) Expansion of a pore, with the rim lined with detergent (DHPC), and the planar surface consisting of DMPC.
Indeed, our initial experiments using the bicelle medium, at a volume fraction of ∼5%, were remarkably successful and showed that a variety of previously studied macromolecules, including the proteins ubiquitin and calmodulin, a protein/DNA complex, and a DNA dodecamer could all be aligned to the optimal degree of ∼10−3 (Tjandra and Bax 1997). Even though the viscosity of bicelle solutions is orders of magnitude higher than that for pure water (Struppe and Vold 1998), rotational diffusion of ubiquitin was shown to be completely unaffected by the bicelles (Bax and Tjandra 1997). The explanation for this behavior is that the macroscopic fluidity is restricted by the phospholipid bilayers, whereas the solute protein is essentially surrounded by pure water and only “once in a while” bounces into one of the bilayers.
Figure 4 ▶ shows an example of NMR data for ubiquitin, measured in different volume fractions of bicelles. At the higher bicelle concentration (8% w/v), 1H-1H dipolar couplings become larger than the 3JHH couplings, and line width in the 1H dimension rapidly increases, causing sensitivity and resonance overlap problems. However, at a 4.5% volume fraction, the 1H multiplet width is only slightly larger than that observed in the isotropic phase, whereas dipolar contributions to the 1JNH splittings are still easily measurable. Even the much smaller 13C-15N and 13C-13C dipolar interactions can accurately be measured on such samples (Ottiger and Bax 1998b).
Figure 4.

Small regions of the 600 MHz 15N-1H correlation spectra of ubiquitin, recorded in the absence of 1H decoupling in the 15N dimension, at three different levels of molecular alignment. (A) Isotropic spectrum, with the marked splitting corresponding to 1JNH. (B) Spectrum recorded in 4.5% (w/v) bicelles, consisting of a 30:10:1 molar ratio of DMPC, DHPC, and cetyl-trimethyl ammonium bromide (CTAB). (C) Spectrum recorded in 8% (w/v) bicelles. Marked splittings in panels B and C correspond to the sum of the 1JNH and dipolar coupling. The broadening in the 1H dimension, observed in panels B and C relative to A is caused by 1H-1H dipolar couplings.
Other liquid crystals
Although very useful, the bicelle medium also has its disadvantages. The phospholipids hydrolyze in a matter of weeks or days if the pH is not carefully kept close to 6.5, and the region of the phase diagram over which the system adopts liquid crystalline order is relatively narrow, in terms of its composition, ionic strength, and temperature (Ottiger and Bax 1998a). Numerous other user-friendly liquid crystalline media have since been shown to be compatible with protein NMR. Most widely used is probably the filamentous bacteriophage, Pf1, introduced by Pardi et al. (Hansen et al. 1998). It owes its popularity, in part, to being commercially available at reasonable cost, but it also is remarkably robust and allows adjustment of its concentration over more than an order of magnitude without sacrificing liquid crystallinity. Independently, Clore et al. demonstrated the utility of other rod-shaped viruses, namely tobacco mosaic virus and bacteriophage fd for inducing the desired degree of solute protein order (Clore et al. 1998). Of these viruses, with a length of 2 μm and a diameter of only 6.5 nm, Pf1 has the highest aspect ratio, causing it to remain liquid crystalline down to concentrations as low as a few mg/mL, depending on ionic strength (Zweckstetter and Bax 2001a).
Pf1 carries a substantial amount of net surface charge, ∼0.5e/nm2. As a result, electrostatic interaction between the phage and the solute protein commonly dominates the alignment. This actually makes Pf1 an ideal complement to the bicelle medium, where the alignment mechanism is steric in nature: the two different average alignment frames of a protein in the two media permit measurement of the orientation of internuclear vectors relative to these two frames, partially lifting the cone-type degeneracy shown in Figure 2 ▶ (Fig. 5 ▶). Contrary to undocumented but widespread perception, we find the Pf1 medium to be remarkably robust and insensitive to vortexing, pipetting, stirring, squeezing between the narrow layer separating plunger and glass wall in NMR microcells, and the like. However, a very slow decrease in alignment, on the order of about 1% per month, is commonly observed, which may reflect a very slow decay of the Pf1 structural integrity.
Figure 5.

Bands of allowed vector orientations for the Gln40 backbone amide 15N-1H bond vector in ubiquitin, in the frame of the crystal structure (1UBQ). Band A corresponds to the orientations compatible with the experimental dipolar coupling value measured in neutral bicelles. Band B corresponds to allowed orientations in bicelles charged with CTAB. The solid dot marks the orientation of the N-H vector when the proton is model-built into the crystal structure, assuming that HN is located on the line bisecting the C′-N-Cα angle. (Reprinted, with permission, from Ramirez and Bax 1998.)
Other useful liquid crystalline phases have been proposed as well. One consists of a mixture of cetylpyridinium halide and hexanol (Prosser et al. 1998; Barrientos et al. 2000). The halide can be either Cl− or Br−, each with distinctly different properties towards ionic strength tolerance. In contrast to Pf1 and fd, the cetylpyridinium carries positive surface charge and therefore generally yields yet a different alignment orientation for charged proteins.
A mixture of n-hexanol and alkylethylene glycol also forms a robust and convenient liquid crystalline alignment medium (Ruckert and Otting 2000). It carries little net surface charge, and solute alignment is therefore largely steric in nature. An advantage relative to bicelles is that its components are chemically inert and samples remain liquid crystalline for very long times, even years.
Anisotropic gels
As noted above, a wide variety of liquid crystals is available to date. However, there remain systems that are incompatible with all of these media. For example, the detergent used to solubilize certain proteins destructively interferes with most liquid crystalline media, or is absorbed on their surface. For others, only a single liquid crystalline medium works well, whereas, as described above, the use of multiple alignment media is strongly preferred to help alleviate the degeneracy problem.
A particularly useful and practical, non-liquid crystal method for inducing protein alignment relies on anisotropic compression of polyacrylamide gels. The method, referred to as strain-induced alignment in gel, or SAG, was developed independently by Tycko et al. (2000) and Grzesiek and coworkers (Sass et al. 2000). In its original implementation, a 6%–8% polyacrylamide gel is cast in a 3-mm cylinder. Subsequently, it is washed and immersed in a concentrated protein solution, which diffuses into the gel. The 3-mm gel is then transferred into an NMR tube with an inner diameter (ID) of ∼4 mm, and compressed with a plunger until it snugly fits inside the NMR tube, that is, with its diameter expanded to that of the ID of the NMR tube (Fig. 6B ▶). As a result, the “cavities” in the gel are no longer random in shape, but will have a slightly oblate character. When placed vertically in the magnet, proteins diffusing inside the aqueous phase of the gel will, on average, have a slight preference to have their long axis oriented orthogonal to the magnetic field.
Figure 6.


Anisotropic environment created by strained gels. (A) 3-mm gel, contained in a 4.5-mm inner diameter Shigemi sample cell, prior to compression by the plunger. (B) After compression by the plunger. (C) Device for achieving radial compression, resulting in axial stretching of the gel.
The method works remarkably well and appears to be applicable to all systems studied so far. The inertness of the acrylamide gel is key to the success of this approach, and an additional advantage is that it is very simple to recover the protein from the gel, simply by immersing it in a large volume of water, followed by concentrating the wash. The primary disadvantage of the gel approach is that it can inhibit the rotational diffusion rate of proteins, thereby increasing resonance line widths and decreasing NMR sensitivity (Sass et al. 2000). The severity of this effect steeply increases with gel density, and it therefore is desirable to use the lowest possible gel densities and largest possible compression factor (aspect ratio).
One convenient way to lower the required gel density is to stretch the gel in the direction parallel to the NMR tube axis. This causes the long axis of proteins to align parallel instead of orthogonal to the magnetic field, which approximately doubles the dipolar couplings obtained for a given gel density and aspect ratio. A simple way to stretch the slippery gel is to cast it originally in a cylinder with an ID (typically ∼6 mm) larger than that of the ID of the NMR sample tube, and then to use a funnel-like device to squeeze it into the narrower-ID, bottomless NMR sample tube, thereby stretching it in the axial direction (Fig. 6C ▶; Chou et al. 2001a).
In a first application of the stretched gel method to detergent-solubilized systems, we focused on a small helical fragment of the C-terminal domain of the HIV envelope protein gp41 (Chou et al. 2002). The peptide encompasses residues 282–304 and is completely insoluble in water. However, it is readily soluble in the zwitterionic detergent dihexanoyl phosphatidylcholine (DHPC), or in bicelles, consisting of a 1:4 molar ratio of dimyristoyl phosphatidylcholine (DMPC) and DHPC. This latter bicelle system is distinctly different from the liquid crystalline bicelles, mentioned earlier, in that their molar ratio of detergent to long-chain phospholipid is more than an order of magnitude larger. They adopt a small disk-like morphology, with the DMPC making up the planar region and the DHPC sequestered on the rim (Sanders and Prestegard 1990; Sanders and Schwonek 1992). The DMPC:DHPC molar ratio determines the size of the mixed micelle (Vold and Prosser 1996), which has been proposed to be a better membrane mimetic than simple spherical detergent micelles (Vold et al. 1997).
A 6% gel, stretched twofold in the NMR sample tube, yielded optimal alignment conditions for the gp41[282–304]-bicelle and permitted measurement of a nearly complete set of backbone 15N-1H, 15N-13C′, and 13Cα-13C′ one-bond dipolar couplings. 1H and 13C chemical shifts of the peptide indicate that it is α-helical for most of its length, and dipolar couplings then can readily be used to refine its structure (see below). Sidechain orientations in the perdeuterated peptide are readily established by measurement of 3JC′Cγ and 3JNCγ couplings. Comparison of the peptide structure in bicelles and DHPC micelles shows a nearly straight helix for the bicelle case, with its Trp and Leu residues anchored in the bilayer, whereas a strongly curved helix is observed for the micelle case (Fig. 7 ▶). This curvature presumably is induced by the natural shape of the small, spherical micelles. It is also interesting to note that the difference in curvature does not result in any sizable (>0.5 Å) changes in short interproton distances between any of the backbone hydrogens, and therefore would remain completely indetectible by conventional NOE-based NMR.
Figure 7.
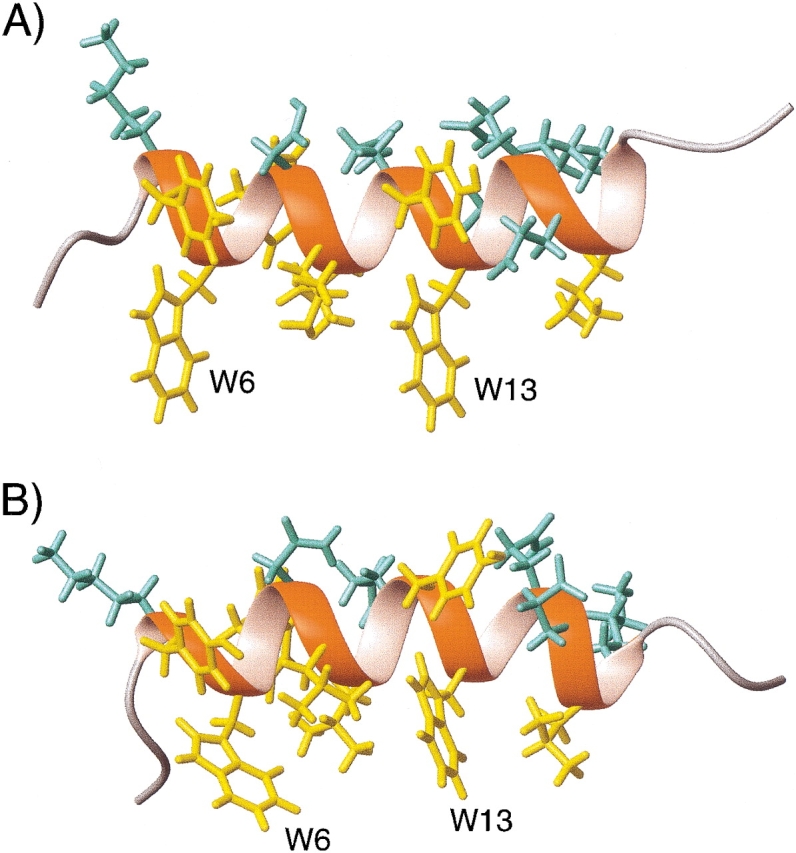
Structures of gp41[282–304] in (A) q = 0.25 bicelles and (B) DHPC micelles, determined from dipolar couplings. Sidechain χ1 angles are derived from 3JNCγ and 3JC′Cγ couplings. Hydrophobic and hydrophilic sidechains are shown in yellow and in aqua, respectively. (Reprinted, with permission, from Chou et al. 2002.)
Application to structure validation
Before discussing the application of dipolar couplings to structure determination, another important use of orientational restraints is highlighted: structure validation. Although various approaches have been described to validate NOE-based NMR structures (Gonzalez et al. 1991; Thomas et al. 1991; Withka et al. 1992; Brunger et al. 1993), none of these have become widely accepted. An intrinsic problem in validating such structures stems from the bootstrap nature in which the NOE restraints are collected, and from difficulties in accounting for indirect NOE contributions.
There are two quite similar ways to validate structures using parameters measured on the oriented protein: measurement of changes in chemical shift, and dipolar couplings. In the first such application, we focused on the small changes in 15N shift measured at 500 and 750 MHz for a complex between a GATA-binding domain and its cognate DNA oligomer (Tjandra et al. 1997) These small changes in 15N shift are caused by the magnetic field-induced alignment of the complex and the 15N chemical shift anisotropy (CSA), meaning that the chemical shift depends on the orientation of the peptide group relative to the magnetic field. Such shift changes range up to Azz times the width of the solid-state NMR CSA powder pattern, that is, up to about 150 ppb (0.15 ppm) for 15N and 13C′, and are readily measured at an accuracy of better than 5 ppb. The quality of the correlation between the predicted and observed changes in 15N shift reflects the accuracy at which the orientation of the peptide groups is known. Rather than reporting the correlation coefficient itself, which for reasonable structures always falls above 0.9, we defined a “quality factor”, Q (Cornilescu et al. 1998):
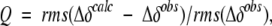 |
(3a) |
where Δδobs and Δδcalc are the observed change in chemical shift and the corresponding change predicted on the basis of the structure, respectively, and rms refers to the root-mean-square function. Even for a perfect structure, Q does not approach zero because the 15N chemical shift tensor is not known exactly and varies from site to site, resulting in a lower limit for Q of about 10% (Cornilescu and Bax 2000). There is a direct relation between Q and the Pearson’s correlation coefficient, RP, with RP = 0.9 about equivalent to Q = 42%, and RP = 0.99 equivalent to Q = 14% (Cornilescu et al. 1998).
The Q factor is more commonly used in terms of dipolar couplings:
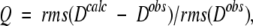 |
(3b) |
where Dobs and Dcalc are observed and calculated one-bond dipolar couplings. Here it is important to ensure that the couplings used in the evaluation are not used in the structure calculation process, which would make the validation essentially meaningless. The same, of course, applies to the Δδobs values in Equation 3a, and care must also be taken when interpreting Δδobs(15N) values when corresponding DNH dipolar couplings are used in the structure calculation, as these two types of parameters are partially correlated (Ottiger et al. 1997). This is much less of a problem when using Δδobs(13C′) which, on average, is nearly uncorrelated with DCαC′ and DC′N (Cornilescu and Bax 2000).
Figure 8A ▶ shows a correlation between DCαHα dipolar couplings measured in ubiquitin and values predicted on the basis of its crystal structure. In order to predict the dipolar couplings using, for example, Equation 2, the magnitude and orientation or the alignment tensor relative to the molecular frame needs to be known. There are two fundamentally different ways to obtain this. First, it can be predicted on the basis of its molecular shape (Zweckstetter and Bax 2000). However, to date this only works well if the alignment mechanism is entirely steric (see below), because accurate quantitative calculation of the electrostatic forces between charged liquid crystals and solute proteins remains quite challenging. Second, the alignment tensor may be obtained by searching for the tensor that best agrees with the experimental data. This is a linear fitting problem that can be solved by singular value decomposition (Losonczi et al. 1999). There are five independent parameters in such a fit, and it is therefore important in Q factor evaluations that the number of fitted dipolar couplings is very much larger than five. For NMR structures calculated with dipolar couplings, the alignment tensor used in the structure calculation may be used, provided that the subset used for Q factor evaluation was not included in the structure calculation process. The most reliable value of Q is obtained when the structure calculation is repeated many times, each time omitting a small, random fraction of the dipolar couplings that is then used for structure validation (Clore and Garrett 1999; Drohat et al. 1999).
Figure 8.

Correlation of experimental dipolar couplings, measured for ubiquitin in 5% (w/v) bicelles, and values predicted by the crystal and solution structures, after best fitting the alignment tensor to the 13Cα-1Hα couplings by singular value decomposition (Losonczi et al. 1999). (A) Plot of the 13Cα-1Hα couplings vs. values predicted by the X-ray structure (Vijay-Kumar et al. 1987). (B) The experimental sidechain dipolar couplings vs. the X-ray structure. (C) The experimental sidechain dipolar couplings vs. the lowest-energy NMR structure, calculated in the absence of sidechain dipolar couplings. (D) The sidechain dipolar couplings vs. the average sidechain dipolar couplings predicted for an ensemble of 50 NMR structures. Q factors for these correlations are (A) 25%, (B) 73%, (C) 67%, and (D) 47%.
For small proteins, there tends to be considerable clustering of internuclear vector orientations. For example, within an α-helix, all N-H vectors point roughly parallel to the helix axis, and in a single β-sheet, N-H vectors also span a limited range of orientations. This nonuniformity in bond vector orientations causes some undesirable variation in the denominator of Equation 3. In cases where measurement errors in the dipolar couplings are small, a better solution is to replace the denominator by (Clore et al. 1999):
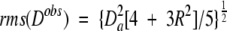 |
(4) |
where Da and the rhombicity, R, are defined under Equation 2. Even with this more robust definition of the Q factor, small variations of a few % are usually not meaningful, and the derived Q factor remains somewhat sensitive to the distribution of vector orientations used in the evaluation. For example, internuclear vectors oriented near the poles of the alignment tensor have small derivatives with respect to orientation and tend to yield lower Q values. The larger the protein, the smaller the effect of this nonrandomness generally becomes.
The Q factor offers a very straightforward and unambiguous way to evaluate structural quality. However, it also has some shortcomings. For example, it will not detect translational errors, such as occur if a helix is translated relative to a sheet while retaining its correct orientation. Although no cases of low Q factors for incorrect structures have surfaced to date, it is not inconceivable that such cases could occur, particularly if no dipolar couplings are available for a segment of the polypeptide chain.
The denominator in Equation 3 was introduced such that if the measurement error in the dipolar coupling becomes much larger than the couplings themselves, the Q factor approaches 100%. In another definition, Clore uses a denominator that is √2 larger than that of Equation 4, and refers to it as an R factor (Clore and Garrett 1999), which therefore is by definition √2 smaller than Q. In this latter definition, the R factor approaches 100% if the structure becomes essentially random, but the Da and rhombicity of the alignment tensor are known correctly.
The Q factor can be used to evaluate any type of structural model, regardless of how it was generated. High-resolution crystal structures typically score below 25%, with the very best structures yielding Q values in the 10%–15% range, a 1.8 Å structure yielding about 20%–25% and a 2.5 Å structure yielding about 40%. For example, evaluation of the 13Cα-1Hα couplings predicted by the 1.1-Å crystal structure of the third Igg-binding domain of protein G (Derrick and Wigley 1994) yields a Q factor of 10%. Further refinement of this structure, using dipolar interactions measured for 13Cα-13C′ and 13C′-15N, can bring this number down to about 6% (T. Ulmer and B.E. Ramirez, unpubl.). At this level, the residual differences between measured and predicted dipolar couplings are believed to be dominated by small deviations (∼2°) from ideal tetrahedral geometry at Cα, which are not accounted for in the NMR refinement procedure, but which are known to occur in proteins (Karplus 1996).
An implicit assumption when calculating a Q factor is that all dipolar interactions are affected to the same extent by internal dynamics. Although at first sight this appears a dramatic oversimplification, in practice this does not limit its utility very much. Generalized order parameters, as measured for the backbone 15N-1H interaction, typically correspond to S2 values in the 0.85–0.95 range. Dipolar couplings to a first approximation scale with the square root of this number, so the assumption of a uniform S value introduces errors of only a few percent, much smaller than the scatter usually observed. Only if there is very clear evidence from relaxation measurements or 15N line widths that a region is highly dynamic, such as often found at the polypeptide chain termini, is it recommended to exclude residues from the Q factor evaluation. In contrast to the very low Q factors observed for backbone interactions, sidechain dipolar couplings typically agree less well with predictions made on the basis of an individual structure. Figure 8B ▶ compares the sidechain 13C-1H dipolar couplings measured in ubiquitin with its crystal structure, yielding a rather poor correlation. A similarly poor correlation is obtained when comparing the lowest-energy NMR structure (out of an ensemble of 50), calculated on the basis of a very large number of NOEs and J couplings (Fig. 8C ▶). However, when comparing the measured dipolar couplings with the dipolar couplings predicted for the entire ensemble of NMR structures, the correlation becomes considerably better (Fig. 8D ▶), indicating that the ensemble provides a better description for the structure than any individual member. The reason for the poor correlation observed with any individual structure is that many sidechains undergo rotameric averaging. This is particularly true for the surface sidechains, but even when the correlation of Figure 8B ▶ is restricted to interior sidechains, it remains much poorer than that observed for the 13Cα-1Hα couplings (data not shown).
Understanding protein alignment
Tjandra et al. (1997) demonstrated that in a bicelle medium the principal axes of the molecular alignment tensor closely coincide with those of the rotational diffusion tensor (Tjandra and Bax 1997; de Alba et al. 1999). This shows that in this nearly neutral medium, alignment is defined by the solute’s shape. The alignment tensor can be modified by adding a net charge to the bicelles, by doping them with charged amphiphiles such as CTAB (+) or SDS (−) (Ramirez and Bax 1998). This demonstrates that electrostatic interactions can also play a role. In fact, for oriented media of strongly negatively charged, rod-shaped viral particles, or oriented purple membrane fragments, electrostatic interactions often dominate alignment of solute proteins.
A simple steric model has been proposed that quantitatively describes the relation between the solute’s shape and its alignment in a lyotropic liquid crystal (Zweckstetter and Bax 2000). So far, it has only been demonstrated for the case of (nearly) neutral particles, such as bicelles, but preliminary results indicate that the method can be extended to account for the effect of charge.
In the so-called steric-obstruction model, the solutes are simulated as a collection of randomly oriented, uniformly distributed molecules, from which the fraction that sterically clashes with the ordered array of liquid crystal particles is removed. For example, for a disk-shaped nematogen and a rod-shaped solute molecule, a larger fraction of molecules will be obstructed when oriented orthogonal to the disk surface than when oriented parallel, resulting in net ordering of the remaining, nonobstructed molecules. In an extension of this method, which also accounts for the effect of electrostatics, different weighting factors are given to each of the nonobstructed solute molecules, depending on the Boltzmann factor calculated when taking the electrostatic potential into account (M. Zweckstetter, unpubl.). Computationally simpler and faster methods that correlate shape and alignment have also been described (Fernandes et al. 2001; Almond and Axelsen 2002).
Figure 9 ▶ shows the correlation between the 15N-1H dipolar couplings measured for the Igg-binding domain of Streptococcal protein G, and that predicted from its 1.1-Å crystal structure (Derrick and Wigley 1994), using an alignment tensor that is not best-fitted to the data, but calculated on the basis of its shape. The excellent agreement seen testifies to the accuracy at which the alignment has been predicted. When ignoring electrostatics, the predicted alignment tensors for bicelle and phage media are very similar. However, the experimentally observed dipolar couplings in the two media are very different and, as expected, good agreement is only observed for the bicelle medium (Fig. 9 ▶). The poor agreement observed in the phage medium can be improved dramatically by including electrostatic terms in the calculations, but the agreement generally remains worse than what can be obtained for the neutral bicelle medium (M. Zweckstetter, unpubl.), reflecting the well known difficulties in accurately accounting for electrostatic forces in aqueous solution.
Figure 9.

Correlations between experimental 1DNH values and values calculated from the shape-predicted alignment tensor of the third immunoglobulin binding domain of protein G in (A) 5% w/v bicelle medium, and (B) 28 mg/mL fd medium. Dashed lines correspond to y = x. The poor correlation in panel B indicates that in phage medium the protein alignment is dominated by electrostatic interactions, which are ignored in the alignment tensor prediction, and not by steric interaction. (Adapted with permission from Zweckstetter and Bax 2000.)
The ability to predict the alignment tensor on the basis of the molecule’s shape has several interesting applications. First, it can be used to validate a structure determined by NMR or crystallography. For example, it is possible to distinguish between different oligomeric states, which sometimes can be difficult to identify by conventional NMR. Second, it permits selection of different relative orientations of the two halves in a homodimer. For example, work by Bewley et al. indicates that in solution, the average relative orientation of the two halves of the domain-swapped homodimeric form of cyanovirin-N is quite different from that seen in its X-ray structure (Bewley and Clore 2000). Thirdly, ongoing work indicates that the relation between shape and alignment can yield quantitative information on interdomain flexibility in multidomain systems.
Refinement of NMR structures
Although a dipolar coupling puts tight restrictions on the orientation of the corresponding internuclear vector, calculation of entire three-dimensional structures on the basis of this information is not straightforward. One major problem is the above-mentioned twofold degeneracy in orientation, that is, the inability to distinguish an isolated vector orientation from its inverse. In practice this means that if any of the backbone N-Cα or Cα-C′ bonds is nearly parallel to any of the three principal axes of the alignment tensor, a 180° rotation of all atoms following this bond will yield the same dipolar couplings, and dipolar couplings therefore cannot establish unambiguously the orientation of the fragment preceding and following this bond.
A second, possibly even more serious problem is that dipolar couplings tend to “compete” with one another when used in simulated annealing-type programs. With NOE restraints, this is usually not the case. For example, if A and B are atoms of residue X, and C and D belong to residue Y, two experimental NOE restraints between atoms A and C and between B and D help one another, that is, the A-C NOE already constrains the B-D distance. This results in a funnel-type energy landscape during the simulated annealing. With dipolar couplings, on the other hand, the opposite may occur. If, for example, an N-H bond is reoriented such that it satisfies the experimental DNH dipolar coupling, this frequently decreases the agreement for the adjoining N-C′ bond, unless the structure locally is already quite close to the true structure. Therefore, the energetic surface that includes the dipolar potential energy function tends to have a very large number of sharp local minima, and is not amenable to simulated annealing for finding the global structure that provides best agreement with the dipolar couplings. As a result, most initial applications of dipolar couplings have focused on refinement of NMR structures, where the initial global fold is determined using conventional NOE restraints.
A method for incorporating dipolar couplings into simulated annealing-type structure determination has first been developed for the program X-PLOR (Brunger 1993) by Tjandra et al. (1997). In brief, a tetra-atomic pseudomolecule OXYZ is defined to represent the alignment tensor, where the OX, OY, and OZ bond vectors are orthogonal to one another. The O atom of this molecule is defined at a fixed position in space, away from the protein. An energetic penalty function term Edip is defined which accounts for the difference between an observed dipolar coupling and the one predicted under the assumption that the orientation of the alignment tensor corresponds to that of OXYZ. As OXYZ freely reorients, it aligns itself to yield a best fit to the observed couplings during the simulated annealing process.
For a dipolar coupling between a pair of atoms P and Q, Edip is given by
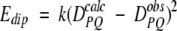 |
(5) |
If Edip is included in the regular simulated annealing protocol, the force constant k is increased exponentially during the cooling stage, typically starting at 10−4 kcal/Hz2 for N-H dipolar couplings and increased to 0.5 or 1 kcal/Hz2 at the final temperature. Force constants for other dipolar couplings are scaled according to (DPQmax)−2 (see Eq. 1b). If the relative experimental uncertainty for some of the intrinsically smaller 15N-13C or 13C-13C couplings is significant, smaller scale factors may be used, such that after refinement the fit to the experimental couplings does not become tighter than the measurement error. In general, use of too high a k value results in poor convergence (Clore and Garrett 1999).
Identification of folds and refinement of homology models
With rapid advances in gene sequencing, an enormous array of proteins is becoming available for structural studies. However, with the total number of unique folds being limited to an estimated 1000 (Chothia 1992), a structural homolog already exists in the PDB for an ever-increasing fraction of these new proteins. If backbone dipolar couplings can be measured, it is feasible to search the PDB database for structures that are compatible with this set of dipolar couplings (Aitio et al. 1999; Annila et al. 1999; Meiler et al. 2000), making it possible to find homologous structures even if they cannot be identified on the basis of their amino acid sequences.
Due to the nonlinear relationship between dipolar coupling and the orientation of the corresponding internuclear vector, it is difficult to quantitatively evaluate the degree of structural difference between a homology model (selected on the basis of dipolar coupling homology) and the structure under study, although on average a lower correlation between the experimental data and the PDB model indicates lower structural similarity. However, considerably closer agreement between the structure under study and the PDB-derived model can be obtained if the latter is first subjected to dipolar coupling refinement (see below). Similarly, dipolar couplings can define conformational rearrangements that may take place when sample conditions are altered.
For homologous proteins, the vast majority of corresponding backbone torsion angles have roughly similar values. The essence of the dipolar refinement approach is to keep the backbone torsion angles relatively close to their starting values (avoiding the multiple minima problem) while simultaneously employing a dipolar coupling term to force individual bond vectors into allowed orientations during a low-temperature simulated annealing protocol. The flow diagram of the method is shown in Figure 10 ▶. Torsion angles from the original model are used as relatively tight harmonic restraints in a first round of simulated annealing, but nevertheless, relative orientations of helices, for example, can change considerably if several of the intervening torsion angles all change by small amounts. In a second round, the annealing process is repeated but with the backbone torsion angles restrained to the output of the first round. Typically, two or three cycles of this protocol suffice to obtain a structure that satisfies the experimental couplings and remains relatively similar in fold to the starting model. Whether or not the refined model is of adequate quality can be evaluated either by using different starting models, or by the type of dipolar cross-validation described above. When using the cross-validation approach, it is necessary that the number of dipolar couplings available significantly exceeds the number of degrees of freedom in the structural model. Considering the variety of different backbone-related dipolar couplings that can easily be measured (Fig. 11 ▶), this usually does not present much of a problem. However, even if only a single type of dipolar coupling is available, say 13Cα-1Hα, the approach can be adapted to work by rigidly fixing the conformations of all elements of secondary structure, thereby greatly reducing the number of degrees of freedom, but simultaneously limiting the attainable goodness of fit to the data.
Figure 10.

Flow diagram of the approach used to refine a starting model and bring it in agreement with experimental dipolar couplings. The simulated annealing process is carried out at very low temperature (<200 K) in order to prevent backbone angles from jumping to false minima. Typically, two or three rounds of the annealing process suffice to obtain agreement with the dipolar couplings. (For a detailed description of the protocol, see Chou et al. 2000.)
Figure 11.

Protein backbone fragment, with dipolar interactions that can readily be measured marked by dashed lines. Although 1DNCα can be measured experimentally, its relative accuracy tends to be lower, and for transpeptide bonds its normalized value is very similar to that of 1DC′Cα of the preceding residue. 1DCαCβ is most useful in perdeuterated proteins, where no value for 1DCαHα can be measured, and where the slow transverse relaxation of the deuterated 13Cα permits its accurate measurement.
An example of the refinement approach is shown for the N-terminal domain of Ca2+-ligated calmodulin. Calmodulin’s crystal structure is available for a diverse set of organisms. It is interesting to note, however, that the best agreement between experimental dipolar couplings obtained from mammalian calmodulin and backbone structure was found for Paramecium tetraurelia calmodulin, for which a 1-Å resolution structure was recently solved (Wilson and Brunger 2000), despite a lower sequence identity (85%) compared to several other structures (98%–100%) solved at slightly lower resolution. Nevertheless, in all cases the agreement between dipolar couplings and structure was less good than expected on the basis of the resolution of the X-ray structures. Subsequent refinement of calmodulin’s two domains, which are flexibly linked in solution (Barbato et al. 1992) and align to different degrees in the liquid crystalline medium, shows a small rearrangement in the relative orientation of the helices in each domain, particularly noticeable for the N-terminal domain (Fig. 12 ▶). This change in helix orientation is independent of the starting model, be it the apo-calmodulin NMR structure, the Ca2+-ligated X-ray structure, or a parvalbumin-derived (27% identity) homology model, and results in essentially perfect agreement with dipolar couplings, even for subsets of dipolar couplings omitted from the calculations (Chou et al. 2001b). Considering the variation in these interhelical angles observed in crystal structures for calmodulin when complexed with various targets, the difference in interhelical angles observed between the solution and crystalline states is perhaps not surprising.
Figure 12.

Backbone ribbon diagrams of the Ca2+-CaM solution structure, shown in red, and the 1-Å crystal structure (1EXR), in blue. (A) For the N-terminal domain, the superposition is optimized for residues 29–54 (helices II and III), revealing the large difference in the orientation of helices I (27°) and IV (21°). (B) For the C-terminal domain, residues 102–127 (helices VI and VII) are superimposed, showing much smaller orientation differences of 15° and 6° for helices V and VIII, respectively. (Reprinted, with permission, from Chou et al. 2001.)
Protein structure by molecular fragment replacement
As mentioned above, addition of dipolar coupling terms to the NMR energy function during simulated annealing results in a highly rippled surface with innumerable sharp local minima. In the absence of long-range distance information from NOEs, it is usually not possible to find a structurally reasonable model by conventional simulated annealing methods. Mostly, such simulated annealing approaches remain stuck in local minima in the energy surface that correspond to very unfavorable local conformations and are typically far removed from the global minimum. However, if a starting model is used that is close to the true structure, convergence to the correct structure in a simulated annealing approach is much less of a problem. One simple method for obtaining such a starting model simply breaks the protein of interest into overlapping fragments of 7–10 residues in length. Then, the entire PDB or a representative subset is searched for fragments that provide the best fit to the experimental dipolar couplings (Delaglio et al. 2000). In this respect, it is important to note that in a rigid protein, all fragments by definition have the same values of Da and R (cf. Eq. 2b). Therefore, when searching the PDB for protein segments that would match the experimentally observed dipolar couplings, it is important that next to the goodness of the fit, the values of Da and R are also considered. If there is a high degree of consistency among the best hits, either the best fragment itself or the average backbone angles of the ensemble of best hits can be used for deriving a suitable starting model for the protein, which subsequently is refined either by a simple conjugate gradient procedure (Delaglio et al. 2000) or by a low-temperature simulated annealing protocol (Chou et al. 2000) that includes a radius of gyration term to ensure appropriate compactness of the final structure (Kuszewski et al. 1999). Recent improvements to this protocol were obtained by first subjecting each member of the set of best-fitting fragments to a short simulated annealing protocol that includes the dipolar terms, and selecting only the cluster of lowest energy fragments (G. Kontaxis, unpubl.). The partially overlapping fragments can then be strung together, while maintaining their correct orientation relative to the alignment tensor (Fig. 13 ▶). The approach appears reasonably robust, and when applied to the RecA-inactivating protein DinI, it yields a backbone structure that differs by less than 2 Å from the previously determined NMR structure (Ramirez et al. 2000). Differences between the two structures correspond primarily to small translational displacements of the two helices relative to one another and relative to the β-sheet, and in the relative position of the strands. These translational differences result from the accumulation of small errors in the individual fragments, and in the absence of NOE or hydrogen bonding information, they are not easily corrected.
Figure 13.

Assembly of the backbone of a protein by the molecular fragment replacement (MFR) approach, using a seven-residue fragment length. For each set of seven contiguous residues, the entire PDB is searched for fragments that are compatible with the experimental dipolar couplings and chemical shifts. The 20 best-fitting fragments are minimized in a short simulated annealing protocol, and if convergence is obtained, the cluster of lowest energy structures is averaged and subsequently minimized. If only a single liquid crystalline medium is used, the orientation of each fragment relative to the alignment tensor frame is fourfold degenerate and best fitting to the preceding, partially overlapping fragment is needed to resolve this ambiguity. If dipolar couplings have been measured in two or more media, the absolute orientation of the fragment is known. Assembly is illustrated for the backbone of the RecA-inactivating protein DinI (Voloshin et al. 2001).
A variant of the molecular fragment approach has been described by Rohl and Baker (2002). In their approach, no massaging of the individual fragments is used, but a powerful Monte Carlo simulated annealing program, Rosetta, shuffles the large number of fragments until a suitable solution is obtained. It is likely that the use of refined fragments in combination with the Rosetta program may yield even better results, while at the same time resolving ambiguities in the assembly process of our approach, which tend to occur for proteins with less complete dipolar coupling sets than those used in the above DinI example.
Concluding remarks
The ability to measure dipolar couplings in a weakly aligned protein by solution NMR provides unique new opportunities for the study of proteins. In most cases, the dipolar couplings can be measured at a very high degree of accuracy, typically better than a few percent of the total allowed range. This translates into very sharp measures for the orientation of the corresponding internuclear vectors and offers the opportunity to determine the local aspects of structure at unprecedented levels of detail, as well as to accurately define the relative orientations of different domains or different proteins in a complex (Clore 2000; Skrynnikov et al. 2000; Ulmer et al. 2002). The steepness of the relation between dipolar coupling and orientation is a two-sided sword, however. On the one hand, this steep dependence makes it a more precise measure of orientation, and on the other hand it complicates direct determination of protein structure from dipolar couplings. Strategies to deal with this structure determination problem are slowly emerging and are expected to increase the applicability of dipolar couplings also in the earliest stages of structure determination.
For moderate-size proteins, the dipolar coupling measurement may be integrated into the resonance assignment process, and potentially can greatly accelerate this remaining tedious step (Zweckstetter and Bax 2001b). Nevertheless, it is expected that in the longer term, dipolar couplings will be used in conjunction with a small subset of easily accessible NOEs to determine protein structures by NMR. The distance information contained in NOEs and the global orientational information contained in dipolar couplings ideally complement each other. Moreover, dipolar couplings provide a long-overdue tool for evaluating the quality of NMR structures in an objective manner.
As demonstrated for the sidechains in ubiquitin (Fig. 8 ▶), dipolar couplings are sensitive to internal motions. However, it is important to realize that the dipolar couplings calculated for a distribution of vectors around an average position only start deviating significantly from the average orientation once the distribution becomes wide, that is, cone angles larger than 20°. For rotameric averaging of sidechains, for example, where the difference in orientation between rotamers is about 110°, the effect is then quite large. In contrast, protein backbone fluctuations of moderate amplitude are not easily detected by dipolar couplings. As a consequence, when small discrepancies or inconsistencies between different types of backbone dipolar coupling measurements are interpreted in terms of dynamics, they frequently indicate amplitudes of motion that are larger than anticipated (Tolman et al. 1997; Peti et al. 2002). More data and further study will be needed to determine to what extent internal motions are indeed the primary cause for these inconsistencies.
Even for flexibly connected multidomain proteins, liquid crystal NMR can provide an accurate determination of the relative domain orientations. A primary concern in such studies is the possibility that the liquid crystal could skew the average distribution. Data available so far suggest that this is not the case. For example, a study by Kay and coworkers on the relative orientation of the two domains in a T4 lysozyme mutant finds the same answer in both bicelle and phage media (Goto et al. 2001), where the alignment mechanisms (steric and electrostatic) are quite different. Remarkably, the average is roughly halfway in between two clusters of X-ray structures found for this protein.
With the availability of a steadily increasing number of different alignment methods, virtually all systems amenable to conventional NMR are now also suitable for measurement of dipolar couplings. Even in the partially and fully denatured state, measurement of dipolar couplings offers intriguing new insights into the ensemble-averaged distribution of individual bond vectors (Shortle and Ackerman 2001).
In my view, measurement of dipolar couplings in solution offers a tremendous new wealth of previously inaccessible information. Only a very limited subset of the total range of applications, primarily taken from our own laboratory, have been discussed in this review. However, numerous other findings have already resulted from this technology and many other applications are anticipated.
Acknowledgments
A large group of people have contributed to the work described in this mini-review. I thank Aksel Bothner-By for inspiring us with his original alignment work; James Prestegard and coworkers for numerous discussions and his pioneering work on myoglobin; Jim Emsley, Zeev Luz, and Olle Soderman for pointing me toward liquid crystals as alignment mechanisms; and numerous colleagues—including Marius Clore, Angela Gronenborn, Gerhard Hummer, Attila Szabo, Dennis Torchia, and Robert Tycko—for help and encouragement. But most of all, I thank all of the members of my group, who played pivotal roles in the development of the weak-alignment technology. Foremost, Nico Tjandra, who spearheaded this effort and whose computational and experimental prowess were key to making the technology succeed, and also all others, including current and former group members Jerome Boisbouvier, Erin Cabello, James Chou, Gabriel Cornilescu, Frank Delaglio, Sander Gaemers, Bernd Koenig, Georg Kontaxis, John Marquardt, Marcel Ottiger, Ben Ramirez, Justin Wu, and Markus Zweckstetter, who are responsible for many of the ideas and applications described in this review.
Editor’s Note: Ad Bax received the 2002 Hans Neurath Award of the Protein Society at its Symposium in August 2002. This article is a summary of his Award address to the Society. Protein Science is most grateful to Dr. Bax for this fine contribution to the Society and to Protein Science.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.0233303.
References
- Aitio, H., Annila, A., Heikkinen, S., Thulin, E., Drakenberg, T., and Kilpelainen, I. 1999. NMR assignments, secondary structure, and global fold of calerythrin, an EF-hand calcium-binding protein from Saccharopolyspora erythraea. Protein Sci. 8 2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almond, A. and Axelsen, J.B. 2002. Physical interpretation of residual dipolar couplings in neutral aligned media. J. Am. Chem. Soc. 124 9986–9987. [DOI] [PubMed] [Google Scholar]
- Annila, A., Aitio, H., Thulin, E., and Drakenberg, T. 1999. Recognition of protein folds via dipolar couplings. J. Biomol. NMR 14 223–230. [Google Scholar]
- Banci, L., Bertini, I., Savellini, G.G., Romagnoli, A., Turano, P., Cremonini, M.A., Luchinat, C., and Gray, H.B. 1997. Pseudocontact shifts as constraints for energy minimization and molecular dynamics calculations on solution structures of paramagnetic metalloproteins. Proteins-Structure Function and Genetics 29 68–76. [DOI] [PubMed] [Google Scholar]
- Barbato, G., Ikura, M., Kay, L.E., Pastor, R.W., Bax, A. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: The central helix is flexible. Biochemistry 31 5269–5278. [DOI] [PubMed] [Google Scholar]
- Barrientos, L.G., Dolan, C., and Gronenborn, A.M. 2000. Characterization of surfactant liquid crystal phases suitable for molecular alignment and measurement of dipolar couplings. J. Biomol. NMR 16 329–337. [DOI] [PubMed] [Google Scholar]
- Bax, A. and Tjandra, N. 1997. High-resolution heteronuclear NMR of human ubiquitin in an aqueous liquid crystalline medium. J. Biomol. NMR 10 289–292. [DOI] [PubMed] [Google Scholar]
- Bax, A., Vuister, G.W., Grzesiek, S., Delaglio, F., Wang, A.C., Tschudin, R., and Zhu, G. 1994. Measurement of homonuclear and heteronuclear J-couplings from quantitative J-correlation. Meth. Enzymol. 239 79–105. [DOI] [PubMed] [Google Scholar]
- Bewley, C.A. and Clore, G.M. 2000. Determination of the relative orientation of the two halves of the domain-swapped dimer of cyanovirin-N in solution using dipolar couplings and rigid body minimization. J. Am. Chem. Soc. 122 6009–6016. [Google Scholar]
- Biamonti, C., Rios, C.B., Lyons, B.A., and Montelione, G.T. 1994. Multidimensional NMR experiments and analysis techniques for determining homo- and heteronuclear scalar coupling constants in proteins and nucleic acids. Adv. Biophys. Chem. 4 51–120. [Google Scholar]
- Boisbouvier, J., Gans, P., Blackledge, M., Brutscher, B., and Marion, D. 1999. Long-range structural information in NMR studies of paramagnetic molecules from electron spin-nuclear spin cross-correlated relaxation. J. Am. Chem. Soc. 121 7700–7701. [Google Scholar]
- Brunger, A.T. 1993. XPLOR: A system for x-ray crystallography and NMR. Yale University Press, New Haven, CT.
- Brunger, A.T., Clore, G.M., Gronenborn, A.M., Saffrich, R., and Nilges, M. 1993. Assessing the quality of solution nuclear-magnetic-resonance structures by complete cross-validation. Science 261 328–331. [DOI] [PubMed] [Google Scholar]
- Bystrov, V.F. 1976. Spin-spin couplings and the conformational states of peptide spin systems. Prog. Nucl. Magn. Reson. Spectrosc. 10 41–81. [Google Scholar]
- Chothia, C. 1992. Proteins—1000 families for the molecular biologist. Nature 357 543–544. [DOI] [PubMed] [Google Scholar]
- Chou, J.J., Li, S., and Bax, A. 2000. Study of conformational rearrangement and refinement of structural homology models by the use of dipolar couplings. J. Biomol. NMR 18 217–227. [DOI] [PubMed] [Google Scholar]
- Chou, J.J., Gaemers, S., Howder, B., Louis, J.M., and Bax, A. 2001a. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR 21 377–382. [DOI] [PubMed] [Google Scholar]
- Chou, J.J., Li, S., Klee, C.B., and Bax, A. 2001b. Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol. 8 990–997. [DOI] [PubMed] [Google Scholar]
- Chou, J.J., Kaufman, J.D., Stahl, S.J., Wingfield, P.T., and Bax, A. 2002. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J. Am. Chem. Soc. 124 2450–2451. [DOI] [PubMed] [Google Scholar]
- Clore, G.M. 2000. Accurate and rapid docking of protein-protein complexes on the basis of intermolecular nuclear Overhauser enhancement data and dipolar couplings by rigid body minimization. Proc. Natl. Acad. Sci. 97 9021–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M. and Garrett, D.S. 1999. R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc. 121 9008–9012. [Google Scholar]
- Clore, G.M., Starich, M.R., and Gronenborn, A.M. 1998. Measurement of residual dipolar couplings of macromolecules aligned in the nematic phase of a colloidal suspension of rod-shaped viruses. J. Am. Chem. Soc. 120 10571–10572. [Google Scholar]
- Clore, G.M., Starich, M.R., Bewley, C.A., Cai, M.L., and Kuszewski, J. 1999. Impact of residual dipolar couplings on the accuracy of NMR structures determined from a minimal number of NOE restraints. J. Am. Chem. Soc. 121 6513–6514. [Google Scholar]
- Cornilescu, G. and Bax, A. 2000. Measurement of proton, nitrogen, and carbonyl chemical shielding anisotropies in a protein dissolved in a dilute liquid crystalline phase. J. Am. Chem. Soc. 122 10143–10154. [Google Scholar]
- Cornilescu, G., Marquardt, J.L., Ottiger, M., and Bax, A. 1998. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 120 6836–6837. [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- de Alba, E., Baber, J.L., and Tjandra, N. 1999. The use of residual dipolar coupling in concert with backbone relaxation rates to identify conformational exchange by NMR. J. Am. Chem. Soc. 121 4282–4283. [Google Scholar]
- Delaglio, F., Kontaxis, G., and Bax, A. 2000. Protein structure determination using molecular fragment replacement and NMR dipolar couplings. J. Am. Chem. Soc. 122 2142–2143. [Google Scholar]
- Derrick, J.P. and Wigley, D.B. 1994. The 3rd Igg-binding domain from streptococcal protein-G—An analysis by x-ray crystallography of the structure alone and in a complex with Fab. J. Mol. Biol. 243 906–918. [DOI] [PubMed] [Google Scholar]
- Dingley, A.J. and Grzesiek, S. 1998. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide (2)J(NN) couplings. J. Am. Chem. Soc. 120 8293–8297. [Google Scholar]
- Drohat, A.C., Tjandra, N., Baldisseri, D.M., and Weber, D.J. 1999. The use of dipolar couplings for determining the solution structure of rat apo-S100B(ββ). Protein Sci. 8 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, M.X., Bernado, P., Pons, M., and de la Torre, J.G. 2001. An analytical solution to the problem of the orientation of rigid particles by planar obstacles. Application to membrane systems and to the calculation of dipolar couplings in protein NMR spectroscopy. J. Am. Chem. Soc. 123 12037–12047. [DOI] [PubMed] [Google Scholar]
- Gaemers, S. and Bax, A. 2001. Morphology of three lyotropic liquid crystalline biological NMR media studied by translational diffusion anisotropy. J. Am. Chem. Soc. 123 12343–12352. [DOI] [PubMed] [Google Scholar]
- Gayathri, C., Bothnerby, A.A., Vanzijl, P.C.M., and Maclean, C. 1982. Dipolar magnetic-field effects in NMR-spectra of liquids. Chem. Phys. Lett. 87 192–196. [Google Scholar]
- Gochin, M. 1998. Nuclear magnetic resonance characterization of a paramagnetic DNA-drug complex with high spin cobalt: Assignment of the H-1 and P-31 NMR spectra, and determination of electronic, spectroscopic and molecular properties. J. Biomol. NMR 12 243–257. [DOI] [PubMed] [Google Scholar]
- Gonzalez, C., Rullmann, J.A.C., Bonvin, A., Boelens, R., and Kaptein, R. 1991. Toward an NMR R factor. J. Magn. Reson. 91 659–664. [Google Scholar]
- Goto, N.K., Skrynnikov, N.R., Dahlquist, F.W., and Kay, L.E. 2001. What is the average conformation of bacteriophage T4 lysozyme in solution? A domain orientation study using dipolar couplings measured by solution NMR. J. Mol. Biol. 308 745–764. [DOI] [PubMed] [Google Scholar]
- Hansen, M.R., Mueller, L., and Pardi, A. 1998. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 5 1065–1074. [DOI] [PubMed] [Google Scholar]
- Hu, J.S. and Bax, A. 1997. Determination of phi and chi(1) angles in proteins from C-13-C- 13 three-bond J couplings measured by three-dimensional heteronuclear NMR. How planar is the peptide bond? J. Am. Chem. Soc. 119 6360–6368. [Google Scholar]
- Karplus, M. 1959. Contact electron-spin coupling of nuclear magnetic moments. J. Phys. Chem. 30 11–15. [Google Scholar]
- Karplus, P.A. 1996. Experimentally observed conformation-dependent geometry and hidden strain in proteins. Protein Sci. 5 1406–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski, J., Qin, J., Gronenborn, A.M., and Clore, G.M. 1995. The impact of direct refinement against C-13(alpha) and C- 13(beta) chemical-shifts on protein-structure determination by NMR. J. Magn. Reson. Ser B 106 92–96. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J., Gronenborn, A.M., and Clore, G.M. 1999. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J. Am. Chem. Soc. 121 2337–2338. [Google Scholar]
- Losonczi, J.A., Andrec, M., Fischer, M.W.F., and Prestegard, J.H. 1999. Order matrix analysis of residual dipolar couplings using singular value decomposition. J. Magn. Reson. 138 334–342. [DOI] [PubMed] [Google Scholar]
- Mehring, M. 1982. High resolution NMR in solids, 2nd ed. Springer, Berlin, Germany.
- Meiler, J., Peti, W., and Griesinger, C. 2000. DipoCoup: A versatile program for 3D-structure homology comparison based on residual dipolar couplings and pseudocontact shifts. J. Biomol. NMR 17 283–294. [DOI] [PubMed] [Google Scholar]
- Nieh, M.P., Glinka, C.J., Krueger, S., Prosser, R.S., and Katsaras, J. 2001. SANS study of the structural phases of magnetically alignable lanthanide-doped phospholipid mixtures. Langmuir 17 2629–2638. [Google Scholar]
- Ottiger, M. and Bax, A. 1998a. Characterization of magnetically oriented phospholipid micelles for measurement of dipolar couplings in macromolecules. J. Biomol. NMR 12 361–372. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. Determination of relative N-H, N-C′, Ca-C′, and Cα-Hα effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J. Am. Chem. Soc. 120 12334–12341. [Google Scholar]
- Ottiger, M., Tjandra, N., and Bax, A. 1997. Magnetic field dependent amide N-15 chemical shifts in a protein-DNA complex resulting from magnetic ordering in solution. J. Am. Chem. Soc. 119 9825–9830. [Google Scholar]
- Pelupessy, P., Chiarparin, E., Ghose, R., and Bodenhausen, G. 1999. Efficient determination of angles subtended by C-alpha-H-alpha and N-H-N vectors in proteins via dipole-dipole cross-correlation. J. Biomol. NMR 13 375–380. [DOI] [PubMed] [Google Scholar]
- Pervushin, K., Ono, A., Fernandez, C., Szyperski, T., Kainosho, M., and Wuthrich, K. 1998. NMR scalar couplings across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation optimized spectroscopy. Proc. Natl. Acad. Sci. 95 14147–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti, W., Meiler, J., Bruschweiler, R., and Griesinger, C. 2002. Model-free analysis of protein backbone motion from residual dipolar couplings. J. Am. Chem. Soc. 124 5822–5833. [DOI] [PubMed] [Google Scholar]
- Prosser, R.S., Losonczi, J.A., and Shiyanovskaya, I.V. 1998. Use of a novel aqueous liquid crystalline medium for high- resolution NMR of macromolecules in solution. J. Am. Chem. Soc. 120 11010–11011. [Google Scholar]
- Ramirez, B.E. and Bax, A. 1998. Modulation of the alignment tensor of macromolecules dissolved in a dilute liquid crystalline medium. J. Am. Chem. Soc. 120 9106–9107. [Google Scholar]
- Ramirez, B.E., Voloshin, O.N., Camerini-Otero, R.D., and Bax, A. 2000. Solution structure of DinI provides insight into its mode of RecA inactivation. Protein Sci. 9 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif, B., Hennig, M., and Griesinger, C. 1997. Direct measurement of angles between bond vectors in high-resolution NMR. Science 276 1230–1233. [DOI] [PubMed] [Google Scholar]
- Rohl, C.A. and Baker, D. 2002. De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J. Am. Chem. Soc. 124 2723–2729. [DOI] [PubMed] [Google Scholar]
- Ruckert, M. and Otting, G. 2000. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 122 7793–7797. [Google Scholar]
- Sanders, C.R. and Prestegard, J.H. 1990. Magnetically orientable phospholipid-bilayers containing small amounts of a bile-salt analog, chapso. Biophys. J. 58 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, C.R. and Schwonek, J.P. 1992. Characterization of magnetically orientable bilayers in mixtures of dihexanoylphosphatidylcholine and dimyristoylphosphatidylcholine by solid-state NMR. Biochemistry 31 8898–8905. [DOI] [PubMed] [Google Scholar]
- Sanders, C.R., Hare, B.J., Howard, K.P., and Prestegard, J.H. 1994. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Prog. Nucl. Magn. Reson. Spectrosc. 26 421–444. [Google Scholar]
- Sass, H.J., Musco, G., Stahl, S.J., Wingfield, P.T., and Grzesiek, S. 2000. Solution NMR of proteins within polyacrylamide gels: Diffusional properties and residual alignment by mechanical stress or embedding of oriented purple membranes. J. Biomol. NMR 18 303–309. [DOI] [PubMed] [Google Scholar]
- Saupe, A. and Englert, G. 1963. High-resolution nuclear magnetic resonance spectra of oriented molecules. Phys. Rev. Lett. 11 462–464. [Google Scholar]
- Shortle, D. and Ackerman, M.S. 2001. Persistence of native-like topology in a denatured protein in 8 M urea. Science 293 487–489. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D. and Case, D.A. 1998. Theories of chemical shift anisotropies in proteins and nucleic acids. Prog. Nucl. Magn. Reson. Spectrosc. 32 165–190. [Google Scholar]
- Skrynnikov, N.R., Goto, N.K., Yang, D.W., Choy, W.Y., Tolman, J.R., Mueller, G.A., and Kay, L.E. 2000. Orienting domains in proteins using dipolar couplings measured by liquid-state NMR: Differences in solution and crystal forms of maltodextrin binding protein loaded with beta-cyclodextrin. J. Mol. Biol. 295 1265–1273. [DOI] [PubMed] [Google Scholar]
- Spera, S. and Bax, A. 1991. Empirical correlation between protein backbone conformation and C-alpha and C-beta C-13 nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113 5490–5492. [Google Scholar]
- Struppe, J. and Vold, R.R. 1998. Dilute bicellar solutions for structural NMR work. J. Magn. Reson. 135 541–546. [DOI] [PubMed] [Google Scholar]
- Thomas, P.D., Basus, V.J., and James, T.L. 1991. Protein solution structure determination using distances from 2-dimensional nuclear overhauser effect experiments—Effect of approximations on the accuracy of derived structures. Proc. Natl. Acad. Sci. 88 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra, N. and Bax, A. 1997. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278 1111–1114. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Grzesiek, S., and Bax, A. 1996. Magnetic field dependence of nitrogen-proton J splittings in N-15-enriched human ubiquitin resulting from relaxation interference and residual dipolar coupling. J. Am. Chem. Soc. 118 6264–6272. [Google Scholar]
- Tjandra, N., Omichinski, J.G., Gronenborn, A.M., Clore, G.M., and Bax, A. 1997. Use of dipolar H1-N15 and H1-C13 couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4 732–738. [DOI] [PubMed] [Google Scholar]
- Tolman, J.R., Flanagan, J.M., Kennedy, M.A., and Prestegard, J.H. 1995. Nuclear magnetic dipole interactions in field-oriented proteins—Information for structure determination in solution. Proc. Natl. Acad. Sci. 92 9279–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman, J.R., Flanagan, J.M., Kennedy, M.A., and Prestegard, J.H. 1997. NMR evidence for slow collective motions in cyanometmyoglobin. Nat. Struct. Biol. 4 292–297. [DOI] [PubMed] [Google Scholar]
- Tycko, R., Blanco, F.J., and Ishii, Y. 2000. Alignment of biopolymers in strained gels: A new way to create detectable dipole-dipole couplings in high-resolution biomolecular NMR. J. Am. Chem. Soc. 122 9340–9341. [Google Scholar]
- Ulmer, T.S., Werner, J.M., and Campbell, I.D. 2002. SH3-SH2 domain orientation in Src kinases: NMR studies of Fyn. Structure 10 901–911. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar, S., Bugg, C.E., and Cook, W.J. 1987. Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol. 194 531–544. [DOI] [PubMed] [Google Scholar]
- Vold, R.R. and Prosser, R.S. 1996. Magnetically oriented phospholipid bilayered micelles for structural studies of polypeptides. Does the ideal bicelle exist? J. Magn. Reson. Ser. B 113 267–271. [Google Scholar]
- Vold, R.R., Prosser, R.S., and Deese, A.J. 1997. Isotropic solutions of phospholipid bicelles: A new membrane mimetic for high-resolution NMR studies of polypeptides. J. Biomol. NMR 9 329–335. [DOI] [PubMed] [Google Scholar]
- Voloshin, O.N., Ramirez, B.E., Bax, A., and Camerini-Otero, R.D. 2001. A model for the abrogation of the SOS response by an SOS protein: A negatively charged helix in DinI mimics DNA in its interaction with RecA. Genes & Dev. 15 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister, G.W., Tessari, M., Karimi-Nejad, Y., and Whitehead, B. 1999. Pulse sequences for measuring coupling constants. In Biological magnetic resonance (eds. R. Krishna and L. Berliner), Vol. 16, pp. 195–257. Kluwer, Dordrecht, The Netherlands.
- Wang, Y.X., Jacob, J., Cordier, F., Wingfield, P., Stahl, S.J., Lee-Huang, S., Torchia, D., Grzesiek, S., and Bax, A. 1999. Measurement of (3h)J(NC′) connectivities across hydrogen bonds in a 30 kDa protein. J. Biomol. NMR 14 181–184. [DOI] [PubMed] [Google Scholar]
- Wilson, M.A. and Brunger, A.T. 2000. The 1.0-Å crystal structure of Ca2+-bound calmodulin: An analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 301 1237–1256. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- Withka, J.M., Srinivasan, J., and Bolton, P.H. 1992. Problems with, and alternatives to, the NMR R-factor. J. Magn. Reson. 98 611–617. [Google Scholar]
- Wooton, J.B., Savitsky, G.B., Jacobus, J., Bayerlein, A.L., and Emsley, J.W. 1979. Methyl group geometry. J. Chem. Phys. 70 438–442. [Google Scholar]
- Yang, D.W., Konrat, R., and Kay, L.E. 1997. A multidimensional NMR experiment for measurement of the protein dihedral angle psi based on cross-correlated relaxation between (Hα-13Cα)-H1 dipolar and C-13′ (carbonyl) chemical shift anisotropy mechanisms. J. Am. Chem. Soc. 119 11938–11940. [Google Scholar]
- Zweckstetter, M. and Bax, A. 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J. Am. Chem. Soc. 122 3791–3792. [Google Scholar]
- ———. 2001a. Characterization of molecular alignment in aqueous suspensions of Pf1 bacteriophage. J. Biomol. NMR 20 365–377. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Single-step determination of protein substructures using dipolar couplings: Aid to structural genomics. J. Am. Chem. Soc. 123 9490–9491. [DOI] [PubMed] [Google Scholar]