

Abstract
The large number of uncharacterized genes emerging from genome sequencing projects has resulted in a need for quick and reliable screening methods for protein expression parameters. We have utilized the univector plasmid recombination system (as previously reported) to develop a series of vectors for rapid screening for expression in Escherichia coli. A high level of recombinant protein expression is a requirement for purification of protein for structural determination and other purposes. In other applications, successful complementation of a missing enzyme activity in E. coli, as well as directed evolution studies and metabolic engineering, often require a much lower level of protein expression. In this report we describe the construction of a number of new pHOST vectors that can be screened for both low- and high-level expression. We isolated a mutant vector for MBP fusions that exhibited a more optimal level of expression for complementation of aerobic respiration in hemA− E. coli, our functional assay for the alternative oxidase. We then demonstrated the use of our system to rapidly screen for both optimal functional expression and optimal overexpression of the alternative oxidase as well as two other members of a family of membrane-bound diiron carboxylate proteins, the plastid terminal oxidase and 5-demethoxyquinone hydroxylase.
Keywords: Di-iron proteins, membrane proteins, protein expression, alternative oxidase, clk-1, Coq7, IMMUTANS, ubiquinone biosynthesis
The technical advances that have led to the sequencing of entire genomes have now necessitated the development of technology to analyze the enormous numbers of genes encoded in these genomes. One particularly challenging aspect is the determination of protein structures in a high-throughput fashion. Several initiatives having this goal are underway, and typically a pool of genes predicted to encode nonmembrane proteins are expressed in E. coli using a single affinity tag (generally a hexahistidine N-terminal fusion driven by a T7 promoter), and whatever expresses as a soluble protein is then carried through to purification for subsequent crystallization or NMR structure determination. Preliminary data on nonmembrane proteins in a Methanobacterium thermoautotrophicum pilot project from one such initiative shows a 50% attrition rate from the cloned gene to the expression of soluble protein, from a sample size of more than 200 genes (Christendat et al. 2000). If these data are representative, about half the genes will require further screening of promoters and fusions to obtain soluble protein. Not surprisingly, two pilot studies on high-throughput production of nonmembrane proteins have shown that screening several different N-terminal fusions increases the chance of having the expressed protein appear in the soluble fraction (Hammarström et al. 2002; Shih et al. 2002).
Other current uses for expression screening are in the optimization of “functional” expression for the directed evolution of enzymes or the metabolic engineering of bacteria. In both cases, the goal is to attain a homogeneous and viable population of cells that express the active enzyme to a controlled level. In addition, with directed evolution experiments, there is a preference for expression levels at the low range of the available enzyme assay (Matsumura et al. 2001). Screening for expression of a functional enzyme can also be used to verify that a given promoter/protein fusion is capable of producing correctly folded protein. As many expression vectors are high-copy number plasmids utilizing promoters yielding “all or nothing” expression (Keasling 1999), it can often be difficult to attain the control necessary to achieve an active enzyme, or a given level of enzyme activity.
Predicted membrane proteins account for a large fraction of the genes in sequenced genomes, and a disproportionately large fraction of proteins with unsolved structures. Because of the difficulties associated with the expression, purification, and crystallization of membrane proteins, the few studies providing statistics on protein expression have pointedly excluded these genes from their target lists (Christendat et al. 2000; Braun et al. 2002; Hammarström et al. 2002). Interfacial membrane proteins bind monotopically to the interfacial region of the membrane without transversing the bilayer. Their affinity for the membrane varies, as some bind predominantly through electrostatic interactions, whereas others require detergent for solubilization. The number of interfacial membrane proteins is unknown because they cannot be readily identified by hydropathy analysis or sequence motif. In this study, we use three related interfacial membrane proteins: the alternative oxidase (AOX) (Berthold et al. 2000), a plastid terminal (quinol) oxidase (PTOX) (Carol et al. 1999; Wu et al. 1999), and an enzyme involved in the hydroxylation of a ubiquinone biosynthetic intermediate (Coq7) (Stenmark et al. 2001).
Here we demonstrate the successful application of the univector plasmid recombination system (Liu et al. 1998, 2000) for screening for both overexpression and functional expression of AOX, PTOX, and Coq7. Using this system, we have cloned the genes into a single “universal” vector, and then have recombined each gene into several “pHOST” expression vectors for expression screening. We report the construction of several new pHOST vectors, as well as the isolation of a mutant maltose-binding protein (MBP) fusion vector that allows IPTG-inducible low-level expression. In addition to rapidly evaluating a number of expression vectors for overexpression, by using the appropriate E. coli mutants we have been able to assess whether a given construct yields an active enzyme and thus merits further work. The use of this system should be of benefit both to laboratories involved in large-scale expression projects as well as to those laboratories optimizing expression levels for a single protein of interest.
Results
Cloning of genes and construction of pHOST expression vectors
The chief advantage of the univector plasmid recombination system is that the gene needs to be cloned only once, into a “universal” entry vector “pUNI”, from which it can be easily shuttled into a variety of “pHOST” expression vectors using Cre-recombinase (Fig. 1 ▶). The gene is cloned such that it is positioned in-frame behind the loxP site in pUNI. The loxP site in the pHOST expression vectors is located such that following recombination the coding sequence derived from pUNI follows the promoter, N-terminal fusion, and the loxP sequence itself in a continuous reading frame. We have cloned the Arabidopsis thaliana AOX and PTOX genes into pUNI using routine methods, to create pUNI–AtAO and pUNI–AtIm, respectively. For the three Coq7 genes we tested the method of in vivo recombination (Jones and Howard 1991; Yao et al. 1992; Bubeck et al. 1993). This method proved to be both rapid and robust. Between 10 and 30 colonies of pUNI containing the inserted gene were obtained by cotransformation of the two linear DNA fragments (the gene and the vector) directly from the PCR reaction mixtures into chemically competent E. coli cells. The number of colonies is low, because of the inherently low level of recombinase activity in E. coli. However, nearly every colony contained the desired product. When background colonies resulting from undigested plasmid template were rigorously excluded, and a single product was found in the PCR reaction, the success rate approached 100%. This was the case for Coq7 from Pseudomonas aeruginosa, Thiobacillus ferrooxidans, and Schizosaccharomyces pombe. Each of these genes was cloned into pUNI10, forming pUNI–PaCOQ, pUNI–TfCOQ, and pUNI–SpCOQ, respectively.
Figure 1.
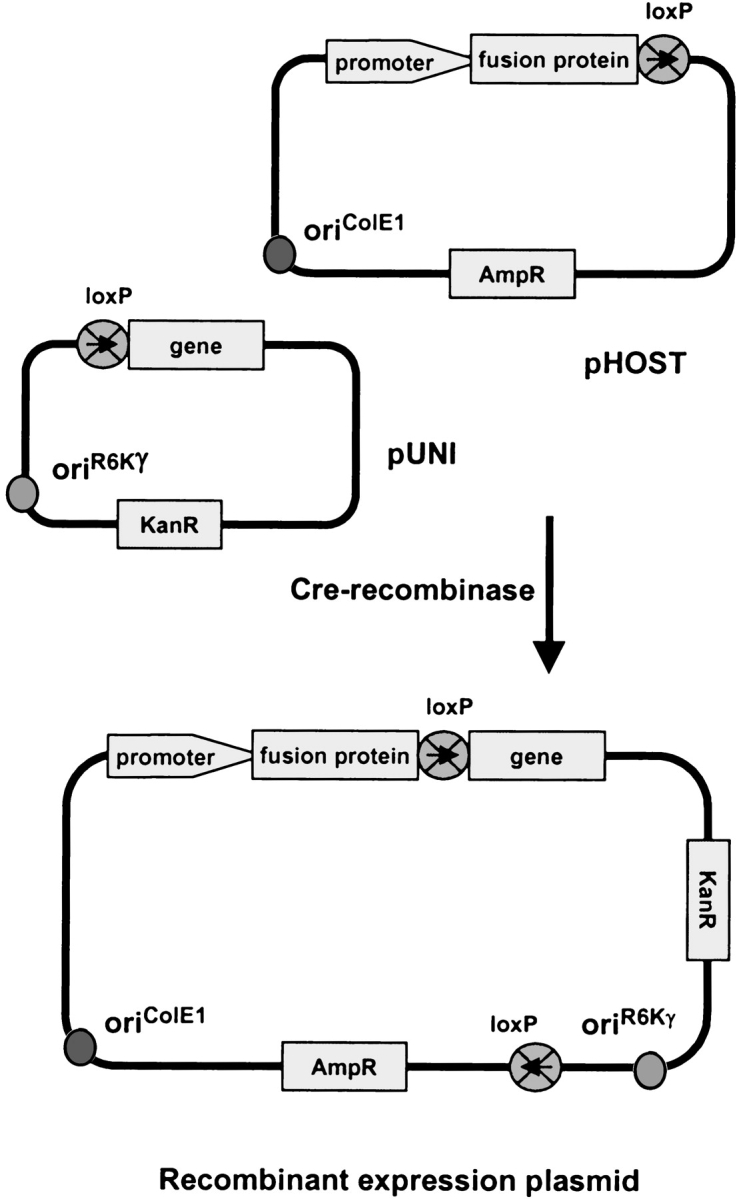
A diagram of the recombination between the pUNI entry vector and the pHOST to form a recombinant expression plasmid. The pUNI plasmid has an origin of replication from plasmid R6Kγ that requires use of a pir-containing strain of E. coli such as BW23474. The pUNI and pHOST plasmids are mixed with Cre-recombinase, incubated, and the mixture is transformed into a strain of E. coli that lacks pir. Selection for recombinants on kanamycin precludes replication of the unrecombined pHOST vector, and the use of E. coli TOP10 or DH5α guarantees that no unrecombined pUNI plasmids will form viable colonies. Recombination occurs in a directional manner at the loxP site, such that the gene follows the promoter and fusion protein in the recombinant expression plasmid. This figure is modifed from Liu et al. 2000.
Bacterial host vectors (pHOSTs designated “pHB-”) for the expression of genes from pUNI were constructed by inserting the loxP site into the expression vectors pMALc2X, pET43a, pQE–82L, and pBAD–GFPcycle3 as described in Materials and Methods, to yield pHB–MALc2X, pHB–PET43, pHB–QE82, and pHB–GFP, respectively (Table 1). Exchanging MalE and NusA in pMALc2X and pET43 gave a T7-promoter driven MBP fusion (pHB–MALT7) and a tac-promoter driven NusA fusion (pHB–NUSc2). The region surrounding the loxP site in the newly constructed plasmids is shown in Figure 2 ▶. The pUNI vectors containing AOX, PTOX, and Coq7 were recombined with the pHOST vectors shown in Table 1. Successful recombination was verified by restriction digest of the isolated plasmids.
Table 1.
pHOST expression vectors used in this study
| pHOST | Base vectora | Promoter | Fusion | Protease siteb | Sourcea |
| pHB3-His6 | pET15b, (1) | T7 | 6-His | Tb | (7) |
| pHB-QE82 | pQE82L, (2) | T5 | 6-His | — | This work |
| pBAD/thioE | pBAD/thio, (3) | BAD | His-patch thioredoxin | En | (3) |
| pHB-GFPc3 | pBADcyc3, (4) | BAD | GFP | — | This work |
| pHB2-GST | pGEX-2T, (5) | tac | GST | Tb | (7) |
| pHB-MALc2X | pMALc2X, (6) | tac | MBP | Xa | This work |
| pHB-MALc2M | pMALc2X, (6) | mutant tac | mutant MBP | Xa | This work |
| pHB-MALcM | pMALc2, (6) | mutant tac | mutant MBP | Xa | This work |
| pHB-MAL-T7 | pET43a, (1) | T7 | MBP | Xa | This work |
| pHB-PET43 | pET43a, (1) | T7 | NusA, 6-His, S-tag | Tb, En | This work |
| pHB-NUSc2 | pMALc2X, (6) | tac | NusA, 6-His, S-tag | Tb, En | This work |
a Numbers in parenthesis refer to the source of the vector. (1) Novagen (Madison, WI); (2) Qiagen (Chatsworth, CA); (3) Invitrogen (Carlsbad, CA); (4) Maxygen (Redwood City, CA); (5) Amersham Pharmacia (Uppsala, Sweden); (6) New England Biolabs (Beverly, MA); (7) Stephen Elledge (Baylor College of Medicine, Houston, TX).
b Tb, thrombin; En, enterokinase; Xa, factor Xa.
Figure 2.

Linker regions of newly constructed pHOST vectors showing location of loxP recombination site. Unique restriction sites are shown underlined with a solid line, and nonunique sites, with a dashed line. The linker region for pHB-MALcM is identical to pHB-MALc2X, with the exception that the AvaI site is not unique.
Complementation of aerobic respiration in E. coli
The AOX expression vectors shown in Table 2 were introduced into hemA− E. coli to test their ability to restore aerobic respiration. The complementation assay involves a strain of E. coli that is deficient in heme biosynthesis and is therefore unable to synthesize the terminal components of the aerobic respiratory chain, cytochrome bo oxidase and cytochrome bd oxidase. The presence of a functioning alternative oxidase allows respiratory electron flow through NADH dehydrogenase, the ubiquinol pool, and the alternative oxidase. The results of the complementation assay are shown in Table 2. Each N-terminal fusion yielded AOX activity either in the induced or uninduced state, with the exception of the pET15a-derived pHB3–His6.
Table 2.
Functional expression of A. thaliana AOX1a
| Expression plasmid | Uninduced | Induceda |
| pUNI-AtAO | ||
| Recombined with pHOST | ||
| pHB2-GST | +++ | o |
| pHB3-His6 | o | o |
| pHB-QE82 | o | ++ |
| pHB-GFPc3 | o | ++ |
| pBAD/thioE | +++ | ++++ |
| pHB-NUSc2 | +++ | +++inh |
| pHB-MALc2X | +++inh | oinh |
| pHB-MALc2M | o | ++++ |
| pHB-MALcM | o | ++++ |
| Nonrecombined | ||
| pAtAOmKX | ++++ | ++++ |
| pAtAOc2 | +++inh | oinh |
| pAtAOc2MED | + | +++++ |
| pAtAOcM | + | +++++ |
| No AOX gene | ||
| pUC118 | o | o |
| pBAD/thioE | o | o |
| pHB-MALc2X | o | o |
| pHB-MALcM | o | o |
The relative colony size of hemA−E. coli 64 h after plating is shown. “o” denotes background size of colony, which was visible only as a refractive difference at the surface of the agar. “+” through “+++++” denotes increasing size of visible colonies. An “inh” superscript indicates an inhomogeneity in colony size, with the median size reported.
a Induced with 0.1 mM IPTG or arabinose, as required by the promoter.
Using the same complementation assay, the expression hosts pHB2–GST, pHB–QE82, pHB–GFPc3, pHB–MALc2X, and pHB–MALc2M recombined with pUNI–AtIm were tested for functional expression of PTOX. In no case was there colony formation, indicating an absence of ubiquinol oxidase activity. However, because PTOX is a plastoquinol oxidase, it is possible that the ubiquinol found endogenously in the E. coli membrane does not support catalytic activity. We also tested PTOX constructs with 0.5 mM benzoquinone present in the medium, but were unable to demonstrate activity.
The assay for functional Coq7 also involves the restoration of aerobic growth (Stenmark et al. 2001). Here, a strain of E. coli is used that is lacking the enzyme catalyzing the penultimate step in ubiquinone biosynthesis, the hydroxylation of 5-demethoxyquinone. Expression of Coq7 restores ubiquinone biosynthesis, and this can be monitored as aerobic growth of colonies (Stenmark et al. 2001). The results of the complementation are shown in Table 3. As a control, the ability of the unrecombined pHOST vectors pBAD/thioE and pHB–MALc2M to restore aerobic respiration was also tested, and as expected these empty vectors did not complement. Remarkably, every pHOST vector was able to support activity, with the exception of pHB2–GST under conditions of IPTG induction. Both bacterial Coq7s were demonstrated to be active, whereas the S. pombe Coq7 was not.
Table 3.
Functional expression of Coq7 using ubiF-E. coli
| Genea | pHOST | Uninduced | Induced |
| Pa Coq7 | pHB2-GST | +++++ | o |
| pHB3-His6 | +++++ | +++++ | |
| pHB-QE82 | +++++ | +++++ | |
| pBAD/thioE | +++++ | +++++ | |
| pHB-MALc2M | +++++ | +++++ | |
| Tf Coq7 | pBAD/thioE | +++++ | ++ |
| Sp Coq7 | pBAD/thioE | o | o |
| Noneb | pBAD/thioE | o | o |
| pHB-MALc2M | o | o |
The data for the pHB-MALc2M and pBAD/thioE constructs of P. aeruginosa and T. ferrooxidans were reported previously (Stenmark et al. 2001). “o” denotes background colony size (∼0.03 mm2). “+” through “+++++” denotes increasing size of colonies observed after overnight incubation at 37°C, with the maximum size ∼80-fold greater than background.
a Pa, Pseudomonas aeruginosa; Tf, Thiobacillus ferrooxidans; Sp, Schizosaccharomyces pombe.
b Denotes unrecombined pHOST plasmids.
Isolation of a mutant vector for a lower level of IPTG-inducible expression
Study of the A. thaliana AOX expressed in E. coli has been hindered by the availability of only a single expression plasmid (the original pAOX [Kumar and Söll 1992] or its variant pAtAOmKX [Berthold 1998]). This plasmid can produce reasonable levels of AOX activity as assayed in isolated membranes, but only when grown under certain conditions from a strain of heme-deficient E. coli. Although the base vector, pcDNAII, contains lacZ preceded by an IPTG-inducible promoter, AOX is not in-frame (Kumar and Söll 1992), and there is no increase in expression with addition of IPTG (pAtAOmKX in Table 2; unpubl. immunoblot data). Apparently, expression is driven by a cryptic promoter and an unidentified ribosome binding site as well. The expression level from this plasmid in hemA− E. coli is limited by the activity of the enzyme, and thus it is difficult (or impossible) to obtain sufficient levels of low-activity (or inactive) mutant AOX protein for study.
It was our interest in identifying a plasmid capable of inducible AOX expression in a respiratory-competent strain of E. coli that led us to characterize the basis for a large colony that arose spontaneously on an IPTG plate during the functional assay of AOX. Under IPTG-induced conditions, an MBP–AOX fusion expression plasmid pAtAOc2 normally produces a heterogeneous array of colony size, with the majority being background size (i.e., no complementation) (Table 2). Isolation of the plasmid (designated pAtAOc2MED) from the spontaneously arising large colony and transformation of the plasmid into fresh hemA− E. coli yielded colonies of uniformly large size (Table 2). We demonstrated that the large phenotype remained after the exchange of fresh AOX coding region into the mutant vector (pAtAOcM). We further narrowed the source of the phenotype to a 1.2-kb ApaI–BglII fragment containing the 3′ coding region of lacI and the 5′ UTR and 5′ coding region of MBP by constructing two vectors, pHB–MALcM and pHB–MALc2M. These two vectors each contained the ApaI–BglII fragment derived from pAtAOc2MED, but the rest of each vector was derived from either pAtAOc2MED or pMALc2X (for details of the construction, see Materials and Methods). Each vector, when recombined with pUNI–AtAO, gave identical-size large colonies in the functional assay. Sequencing of the ApaI–BglII region revealed mutations in two areas. A change from T to C was found at position −95 in the regulatory region of MBP, which falls within the TATA box of the tac promoter. In addition, there were mutations in the third codon of MBP, ACT to ATC, resulting in a change from Ile to Thr. It was not investigated whether the mutations in both the promoter and the coding region were required for the low-expression phenotype, but it is plausible that both regions contribute (Goldstein and Doi 1995; Wallis et al. 1995). These mutations caused a decrease in both the uninduced and induced level of expression, as inferred from the smaller colony size in plates lacking IPTG, and a larger colony size in the presence of IPTG (Table 2). A preliminary assessment of the difference in IPTG-induced protein expression between pAtAOcM and the original expression plasmid was made by quantifying AOX protein on immunoblots of isolated membranes. We estimate a four- to sevenfold decrease in the AOX protein level expressed from pAtAOcM compared to pAtAOc2 (data not shown).
Overexpression of membrane-bound di-iron proteins
The pUNI vectors containing the AOX, Im and Coq7 genes were recombined into the pHOST vectors listed in Tables 4 and 5, and the resultant plasmids were isolated and verified as recombinants by restriction digest. The plasmids were then transformed into E. coli C43 (DE3) for expression. This strain of E. coli has been shown to be appropriate for the expression of membrane and soluble proteins that express poorly or not at all in E. coli BL21 (DE3) (Miroux and Walker 1996). A test of the MBP/AOX fusion showed that similar expression levels were obtained in E. coli strains BL21 (DE3), C41 (DE3), and C43 (DE3). For consistency, we chose to do all our expression testing in the latter strain.
Table 4.
Overexpression of A. thaliana AOX and PTOX
| Expressed level in: | |||
| Gene | pHOST | Membrane | Inclusion bodies |
| AOX | pHB2-GST | o | +++ |
| pHB3-His6 | o | ++ | |
| pHB-QE82 | o | o | |
| pBAD/thioE | o | +++ | |
| pHB-MALc2X | +++++ | ++++ | |
| pHB-MAL-T7 | o | o | |
| pHB-PET43 | o | o | |
| pHB-NUSc2 | ++ | ++ | |
| PTOX | pHB2-GST | + | +++ |
| pHB-QE82 | o | o | |
| pBAD/thioE | o | ++ | |
| pHB-MALc2X | ++++ | ++++ | |
Relative expression levels as observed on Coomassie-stained denaturing polyacrylamide gels. “o” denotes no obvious expression, and “+” through “++++,” increasing protein level.
Table 5.
Overexpression of Coq7
| 37°C, 2-h Induction | 30°C, 4-h Induction | ||||
| Genea | pHOST | Membrane | Inclusion bodies | Membrane | Inclusion bodies |
| Pa Coq7 | pHB2-GST | o | ++++ | o | + |
| pHB3-His6 | o | o | ndb | nd | |
| pHB-QE82 | o | o | ndb | nd | |
| pBAD/thioE | o | ++ | o | o | |
| pHB-MALc2X | ++ | ++++ | ++++ | ++++ | |
| Tf Coq7 | pHB2-GST | nd | nd | o | ++ |
| pBAD/thioE | nd | nd | o | o | |
| pHB-MALc2X | nd | nd | ++++ | ++++ | |
Relative expression levels as observed on Coomassie-stained denaturing polyacrylamide gels of isolated membranes and inclusion bodies. “o” denotes no obvious expression, and “+” through “++++,” increasing protein level.
a Pa, Pseudomonas aeruginosa; Tf, Thiobacillus ferrooxidans.
b nd, not determined.
E. coli C43 (DE3) freshly transformed with the indicated expression plasmids were cultured and harvested as described in Materials and Methods. To assay the expression level, the cells were broken by sonication, and the inclusion body fraction separated from a fraction containing both soluble and membrane-bound proteins. For some expressions, the soluble and membrane protein sample was further fractionated to obtain a membrane pellet. Samples were loaded on a denaturing SDS-urea gel on the basis of equal cell density, and expression level was assessed by visual inspection. Results are shown in Tables 4 and 5. A number of pHOST expression vectors were able to produce inclusion bodies of the fusion proteins, but only pHB–MALc2X recombinants gave a high level of membrane-bound protein. This was true for each of the three diiron proteins expressed.
For high-level expression of AOX, PTOX, and Coq7 as MBP fusions, membranes were isolated from a large-scale culture, and loaded on a denaturing SDS-urea gel on an equal protein basis. A Coomassie-stained gel of isolated E. coli membranes containing the overexpressed di-iron carboxylate proteins is shown in Figure 3 ▶. The level of expression, as derived from a densitometric scan, is 20–25% of the total membrane protein for AOX and PTOX, and 50% of the total membrane protein for Coq7.
Figure 3.
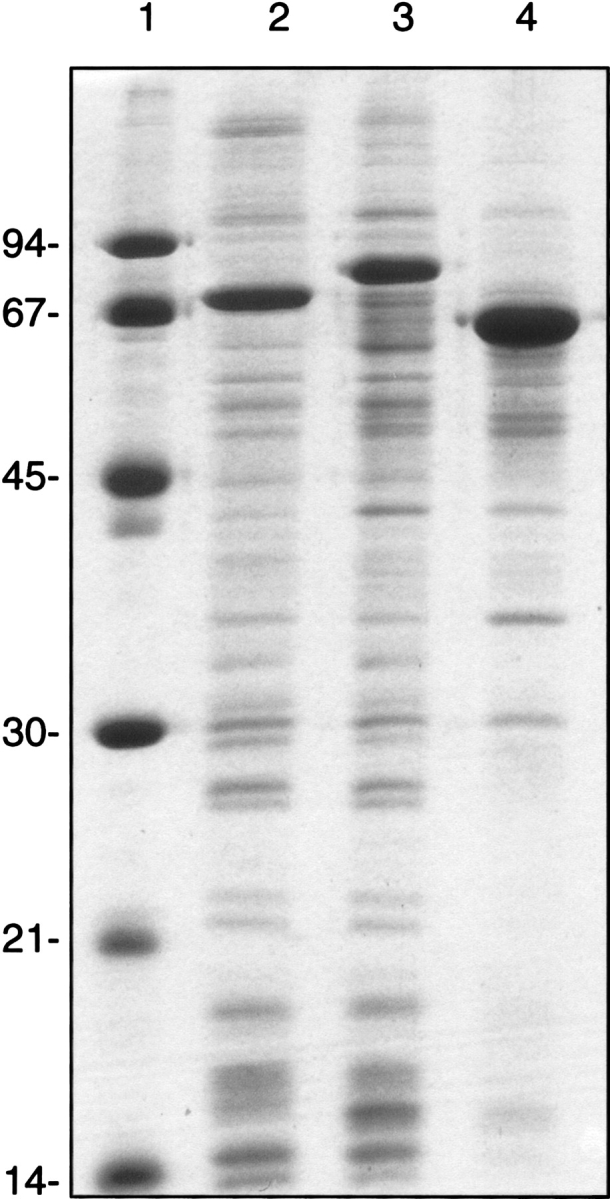
SDS-polyacrylamide gel electrophoresis of di-iron proteins expressed from pHB-MALc2X recombinants. Lane 1, molecular weight standards. Lane 2, membrane fraction from E. coli C43 (DE3) expressing the MBP-AtAOX fusion. Lane 3, membrane fraction from E. coli C43 (DE3) expressing the MBP-PTOX fusion. Lane 4, membrane fraction from E. coli JF496 expressing the MBP-PaCoq7 fusion. Two micrograms of protein loaded per lane. The expected mass of each fusion is MBP-AtAOX, 78.5 kD; MBP-PTOX, 85.7 kD, and MBP-PaCoq7, 68.8 kD.
Discussion
To rapidly screen a variety of vectors for optimal overexpression and functional expression, we chose to use a plasmid recombination system. Several systems are currently commercially available. We selected the univector system, because we were able to purify the Cre recombinase produced as a GST fusion (Liu et al. 2000), resulting in a very minimal cost per recombination reaction. The univector system, however, limits one to recombination at only the 5′ end of the gene of interest. Because we were interested in investigating the effect of promoters and N-terminal fusions, this was adequate for our needs. The low cost and ease of use made it worthwhile for us to construct a series of expression vectors compatible with this system (Table 1).
Cloning by in vivo recombination
There are several methods in general use that can be applied for cloning genes into the pUNI vector. One can add appropriately placed restriction sites in the PCR primers, but cloning restricted fragments of PCR products is known to be inefficient, owing to the losses incurred during product cleanup and the difficulty in completely removing the DNA polymerase prior to digest. The use of TA cloning with topo-isomerase is a popular method, but this limits one to commercially available versions of pUNI and can become expensive. We have found cloning by in vivo recombination to be an efficient and inexpensive alternative. This method requires the two primers to be synthesized with a 15–18 base extension that is complementary to the area of insertion in the target vector. In a separate reaction, two primers complementary to pUNI–AtIm were used to create a linearized plasmid fragment. The method uses a recA− strain of E. coli, and we have found that the recA− pir-116 E. coli BW23474, the strain essential for pUNI replication, works well in this regard. One factor that promotes this high efficiency is the small size of the pUNI plasmid (2220 bp), which aids both its synthesis by PCR and its transformation efficiency. Another factor that is likely to influence the success of this method is the lack of a promoter upstream from the cloning region in pUNI10, which guarantees that there is no selection against the plasmid due to leaky protein expression. In this work we have used in vivo recombination for cloning of the three Coq7 genes, using genomic DNA as a template. Subsequent to this study, we have used this method with success for cloning genes into pUNI from digested plasmid templates, cDNA libraries, as well as directly from bacterial cells added to the PCR reaction (unpubl. results).
Functional expression of AOX and Coq7
The ability to express a gene simultaneously from a series of vectors allows one to readily make comparisons between the vectors. We were fortunate in the case of both AOX and Coq7, in that complementation of the appropriate mutant E. coli resulted in the restoration of aerobic growth, giving us a simple assay for function of these two genes. By assaying various expression constructs for functional expression, we could assess whether any given N-terminal fusion was deleterious to activity. The presence of enzyme activity likewise suggests that the protein is properly folded. AOX and Coq7 tolerate each of the N-terminal fusions tested (Tables 2, 3), and for PTOX it was not possible to determine. The absence of complementation of hemA− E. coli by PTOX is consistent with its role as a plastoquinol oxidase (Josse et al. 2000).
The most striking difference between functional expression of AOX and Coq7 is the level of expression that gives optimal complementation. Uninduced expression of P. aeruginosa Coq7 from all vectors gave a wild-type level of colony growth (i.e., comparable to the growth of E. coli DH5α; Table 3), whereas an uninduced level of AOX expression could support aerobic respiration in hemA− E. coli from only about half of the pHOSTs tested (Table 2). This is consistent with the roles of each of these enzymes in the cell: In contrast to the alternative oxidase, each Coq7 enzyme needs to turn over fewer times in that it produces a stable product, ubiquinone, which itself catalyzes aerobic respiration. That an exceedingly low level of Coq7 expression is sufficient to complement ubiF− E. coli is underscored by the success of leaky (uninduced) expression from a T7 promoter in E. coli JF496, a strain that does not contain the T7 RNA polymerase (Table 3). Likewise, given the apparent low level of expression necessary for complementation, it is particularly striking that the S. pombe Coq7 did not complement at all (Table 3). This result is consistent with the accumulating evidence that in yeast Coq7 the enzyme is a component of a multimeric complex, and may be unstable in the absence of the other subunits (Hsu et al. 2000).
Low-expression variant of pMALc2X
The isolation of a mutant MBP expression vector was a fortuitous outcome of the complementation assay. Because the alternative oxidase does not pump protons, it is a poor substitute for the E. coli cytochrome bo oxidase, the oxidase endogenously expressed in E. coli under normal oxygen tensions. Cells respiring with only AOX can take 2–3 d to show visible colonies, compared to overnight for wild type. The mutant MBP vector allowed a more suitable level of AOX expression, which resulted in the earlier appearance of colonies.
The construction of a set of AOX-expressing vectors with univector and nonrecombined variants allowed a close examination for differences between them. The functional assay for AOX activity magnifies expression differences, because an increased AOX expression improves the energetics of the cell (up to a point), which then allows a further increase in protein expression. Slight differences in expression as observed by colony size can be seen between the univector and nonrecombined variants of the low expression plasmids, with the nonrecombined versions having an increase in both the induced and uninduced expression (Table 2). This is the first observation of a difference between the univector system and corresponding nonrecombined plasmids, and likely represents a small actual difference in expression level.
Screening for maximal membrane-bound protein expression
The screen for overexpression showed that for each of the three diiron proteins, the maltose binding protein was the optimal fusion, with no other fusion yielding an appreciable level of expression in a native (membrane-bound) form (Tables 4, 5). Expression of AOX as a nonrecombinant MBP fusion (from pAtAOc2) produced the same result (Table 4). Our success with MBP is consistent with previous studies comparing the effect of N-terminal fusions upon solubility, in that MBP has been found to be one of the more effective fusion partners (although most of the proteins have not been functionally characterized) (Kapust and Waugh 1999; Braun et al. 2002; Hammarström et al. 2002; Shih et al. 2002). There was also some expression of membrane-bound AOX as a NusA fusion (Table 4).
Several vectors did produce fusion protein of the correct size as inclusion bodies; for AOX these included GST, 6His, His-patch thioredoxin, and NusA as N-terminal fusions (Table 4). Interestingly, the success of the MBP plasmid may have had as much to do with the promoter as the nature of the N-terminal fusion, as the T7 version of this plasmid (Table 4) showed no expressed protein either in the membrane or as inclusion bodies. The NusA fusion followed the same pattern. NusA had been identified through a computer search as a particularly soluble protein and developed as a fusion protein on this premise (Davis et al. 1999). In our tests, it is the only fusion besides MBP that gave expression of a membrane-bound form of AOX. However, this was true when NusA replaced MBP in the tac-driven expression plasmid, but not of the original plasmid (pET43) in which a T7 promoter was present (Table 4). For expression of AOX it is clear that the promoter plays a significant role in the expression of protein in its native, membrane-bound form. In contrast, in a previous study of the T7 and tac promoters driving a MBP fusion of the soluble penicillin-binding protein, the fusion was equally well-expressed under either promoter, at a level of 33% of the total cell protein (Pryor and Leiting 1997).
The overexpression of Coq7 showed the varied interplay of expression condition and vector. For the GST fusion, lowering the induction temperature from 37 to 30°C and harvesting at 4 h after induction (rather than 2 h), resulted in a lower level of expressed protein (here only as inclusion bodies). In contrast, the same shift in conditions gave a sizable increase in the level of membrane-bound protein for the MBP fusion (Table 5).
Future prospects for protein expression
One problem with most of the currently available expression vectors is what is called “all or none” expression, in that for neither IPTG- nor arabinose-inducible promoters is it possible to truly titrate the level of expression (Keasling 1999). When an intermediate level of expression is found on the scale of a culture, it has been shown to consist of a heterogeneous cell population, in which some cells are maximally induced (“on”) and others are completely uninduced (“off”). In this study we have shown that at some level of expression (either “on” or “off,” depending on the vector) AOX is active, and therefore membrane-bound and correctly folded, for every promoter/fusion combination, with the exception of the T7/His-tagged pHB3–His6. This suggests that it would be possible to overexpress AOX with many different N-terminal fusions. However, we observed a visibly overexpressed protein band on a Coomassie-stained gel for only the tac/MBP or the tac/NusA promoter/fusion combinations. The disparity between these results is likely to be a function of the promoter strength or a combination of transcription and translation rates. This conclusion is supported by our observation that when the tac and T7 promoters drove expression of either the MBP or NusA fusions, the tac promoter gave a high level of expression, whereas the T7 promoter yielded none. The conclusion is further supported by the isolation of a mutant vector (pAtAOc2MED) giving an optimized level of functional expression. In this vector, the mutations were found in positions with the potential of modulating both transcription and translation. Our results imply that by modulation of the promoter strength and/or the translation rate, it would be possible to express AOX at high level with the N-terminal fusion of choice. Indeed, expression of a His-tagged trypanosomal AOX, at a level sufficient for purification, has been reported both from pQE32 (Chaudhuri et al. 1998) and also from pET15b (Fukai et al. 1999), which is the T7 promoter/fusion combination that did not work in our hands. (With a fusion as small as the hexahistidine tag, it is likely that the AOX coding sequence also serves to modulate expression.) These results suggest that the ability to select a given N-terminal fusion and obtain with it an optimized level of expression is a function of the availability of the vector of interest at the correct promoter strength. An array of vectors with a gradation of promoter strengths would be generally useful for the many applications—complementation assays, metabolic pathway engineering, directed evolution studies, as well as optimized overexpression—that require controlled or modulated levels of protein expression.
For the emerging structural and functional proteomics programs, technologies for gene-independent cloning into multiple vectors, as well as expression screening using a range of different expression conditions, will clearly be essential components to optimize success rates of protein expression. These experiments should preferably be done in a format allowing experiments to be done in parallel and with a large degree of automation. Although such developments are underway in several laboratories, including our own, it is still very uncertain which strategies will be the most favorable. The recombination cloning strategies used in the present study, as well as related recombination systems, have the potential to be an important component of such strategies. We have shown in the present study that good expression can be obtained for a family of membrane-bound diiron carboxylate proteins, and that the presence of the loxP recombination sequence within the open reading frame does not constitute a major problem for expression of these proteins. We have also shown that the method of in vivo recombination provides a rapid means for cloning genes into the pUNI entry vector. From there, the gene can be efficiently transferred to a variety of expression vectors, and as this method is not dependent upon restriction enzymes, it is gene independent. This system should be applicable to any number of proteins (soluble or membrane bound) for screening for overexpression, and with an appropriate complementation assay, for the screening of functional expression.
Materials and methods
Cloning genes into pUNI10
The univector entry vector, pUNI10 (Liu et al. 1998), was modified by ligation of two complementary 15-mers (aattcccgctagcgc) and (tcgagcgctagcggg) into the EcoRI and XhoI digested vector (Kang and Inouye 1993) to insert an NheI site (underlined) for cloning of the alternative oxidase. This vector was named pUNI–10n. The A. thaliana alternative oxidase gene AOX1a had been previously amplified using pAOX (from M. Kumar and D. Söll, Yale University) as a template and the primers (cggaattcagcatgcatatggctagcacgatt) and (caggatccgtcgactcaatgatacccaat), and cloned into the EcoRI-BamHI sites of pMALc2 (New England BioLabs) to yield pAtAOc2. In this vector, the coding region of the alternative oxidase (omitting the signal sequence) is fused to the C-terminus of MBP. The 885-bp NheI–BamH fragment of pAtAOc2 containing the alternative oxidase was ligated into the similarly digested pUNI–10n, giving pUNI–AtAO. The gene for the A. thaliana PTOX (Im) was subcloned from a plasmid template (from Steven Rodermel, Iowa State University), using the primers (tactggaattcatatggcaacgattttgcaagacg) and (acgatggatccttattaacttgtaatggatttctt) to omit the transit peptide and add restriction sites. It was initially blunt ligated into the suicide vector pGATA (Trudel et al. 1996), and then transferred by restriction digest to pUNI10 with EcoRI and BamHI, to yield pUNI–AtIm.
Coq7 from P. aeruginosa and T. ferrooxidans were cloned into pUNI10 using the method of in vivo recombination (Yao et al. 1992) as described previously (Stenmark et al. 2001). The cloning of Coq7 from S. pombe genomic DNA (a gift from C. Andréasson and P.O. Ljungdahl, Karolinska Institute) utilized the same method with the primers (gttatctggaattcatatgttgtcacgtagacag) and (gcggccgcggatccttattaaaatcgtttacacatcc).
pUNI vectors were propagated in E. coli BW23474 using 40 μg/mL kanamycin in LB medium. All genes cloned from PCR products were sequenced for verification.
E. coli BW23474, and plasmids pUNI10, pHB2–GST, pHB3–His6, and pQL123 were a gift from S. J. Elledge (Baylor College of Medicine). E. coli JF496 was obtained from CGSC (Yale University), and E. coli C43 (DE3) from J. E. Walker (Medical Research Council Laboratory). E. coli TOP10 was from Invitrogen. The sources for the vectors used for pHOST construction are noted in Table 1.
Construction of pHOST vectors
pHB–MALc2X
pAtAOc2 was submitted to an NdeI partial digest followed by full digestion with HindIII, and the 6.6-kb vector fragment was ligated to the 54-bp NdeI–HindIII fragment of pTAG–Flag containing the loxP site. This plasmid, in which the AOX gene was replaced by loxP in-frame with the MBP coding sequence, was named pHB–MALc2. pHB–MALc2X was created by digesting pMALc2X with EcoRI and HindIII, and inserting the similarly digested loxP fragment from pHB–MALc2. The linker region of these and other new pHOST vectors are shown in Figure 2 ▶.
pHB–QE82
pHB–MALc2 was digested with SphI and HindIII to yield a 41-bp loxP fragment, which was ligated to the vector fragment of the SphI–HindIII digested pQE82L to produce pHB–QE82.
pHB–PET43
pHB–MALc2X was digested with EcoRI and HindIII to produce a 52-bp loxP-containing fragment, which was ligated to the vector fragment of EcoRI–HindIII digested pET43a to give pHB–PET43.
pHB–NUSc2
The 1.7-kb NdeI–HindIII fragment of pET43a containing NusA was ligated to the 5.4-kb NdeI–HindIII fragment of pMALc2X to give pNUS–c2, with NusA under a tac promoter. pHB–MALc2X was digested with EcoRI and HindIII, and the 52-bp loxP fragment was ligated to the vector fragment of similarly digested pNUS–c2 to yield pHB–NUSc2.
pHB–MALT7
The 1.2-kb NdeI–HindIII fragment of pMALc2X containing MalE was ligated to the 5.5-kb NdeI–HindIII vector fragment of pET43a to give pMAL–T7, with the MBP under a T7 promoter. pHB–MALc2X was digested with EcoRI and HindIII, and the 52-bp loxP fragment was ligated to the EcoRI–HindIII digested pMAL–T7 to give pHB–MALT7.
pHB–GFP
pBAD–GFPcycle3 (Crameri et al. 1996) was digested with SacI and religated to remove the stop codon following the coding sequence of GFP, producing pGFPc3mS. pHB–GFP was constructed from a three-piece ligation of the 1.8-kb AflII–KpnI fragment and the 3.6-kb BamHI–AflII fragment of pGFPc3mS together with the 72-bp KpnI–BamHI fragment of pUNI10 containing loxP.
Recombination of pUNI and pHOST vectors
A GST fusion of Cre-recombinase was expressed and purified as described (Liu et al. 2000). The recombination method followed that described by Elledge and coworkers, with slight modifications (Liu et al. 2000). A pUNI vector (50 ng) was mixed with a pHOST vector (50 ng) in a 10-μL reaction volume containing 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 30 mM NaCl, and 0.1 mg/mL BSA, and 1–2 μL of crude Cre-recombinase. The reaction was incubated for 20 min at 37°C, followed by 5 min at 65°C, and then 5 μL was transformed into 50 μL CaCl2-competent E. coli TOP10 using a 30-sec heat shock at 42°C. SOC medium (500 μL) was added immediately, and the cells were incubated 45 min at 37°C, and plated on LB agar containing 40 μg/mL kanamycin. Recombinant plasmids (usually 100% under this selection) were verified by restriction digest of the isolated plasmid DNA. Recombination simulations for determination of restriction patterns and size of fusion proteins, as well as pHOST construction, were done with Clone Manager (SciEd Software) with the Cre-recombinase recognition sequence entered as a restriction site.
Complementation assays
To determine whether AOX or PTOX plasmids produced active enzyme, the heme-deficient E. coli strain SASX41BD containing the indicated plasmid was plated on medium that promoted aerobic growth. First, a preculture from a single colony was grown in M63+SG (Berthold 1998) with 50 μg/mL ALA and 60 μg/mL ampicillin to an A600 of 0.3–1.0 and diluted 1–3 × 105 into the same medium exclusive of ALA. Fifty microliters of the dilution were spread on GA agar (Berthold 1998) for a desired colony density of 60–150 per plate. The diluted cells were also spread on GA media containing 50 μg/mL ALA to verify the dilution. The plates were incubated at 37°C. The relative colony size on GA plates were noted at 48 and 64 h. Three AOX expression plasmids served as size standards to ensure that valid comparisons could be made between experiments.
For the complementation assay to test Coq7 activity, plasmids containing Coq7 were transformed into the ubiF− E. coli strain JF496. Cells were grown as described previously (Stenmark et al. 2001).
Isolation of a mutant pMALc2 vector for a lower level of expression
The A. thaliana AOX was subcloned from pAOX (Kumar and Söll 1992) into pMALc2 to yield pAtAOc2. pAtAOc2MED was isolated from a spontaneously arising large colony under IPTG induction during an assay of functional expression from pAtAOc2. Replacing the AOX coding region in pAtAOc2MED with that from the original pAtAOc2 (a 929-bp EcoRI–HindIII fragment) gave pAtAOcM. A pHOST expression vector, pHB–MALcM was constructed from this plasmid by replacing AOX with the loxP site. Upon recombination with pUNI–AtAO, this vector represented the univector variant of the original plasmid pAtAOcM. pHB–MALc2M was constructed by moving the 1.2-kb ApaI–BglII fragment of pAtAOc2MED into pHB–MALc2X to create pHB–MALc2M. (The two pHOST vectors, pHB–MALcM, and pHB–MALc2M, have slight differences that correspond to the differences between New England BioLab’s original pMALc2 vector and the newer version, pMALc2X.).
Overexpression
To assay for overexpression of AOX, PTOX, and Coq7, a freshly transformed colony of plasmid in E. coli C43 (DE3) (Miroux and Walker 1996) was picked and grown overnight in LB medium with the appropriate antibiotic (60 μg/mL ampicillin; or for the pHOST recombinants based on pHB–MAL–T7 or pHB–PET43, 25 μg/mL carbenicillin). LB media with antibiotic was then inoculated with overnight culture and grown at 37°C to an OD600 of approximately 0.3. For expression experiments at 30°C, the temperature was lowered at this point. At an OD600 of approximately 0.8, an aliquot was removed as the uninduced control, and the culture was induced with IPTG or arabinose. Aliquots of the culture were harvested at the indicated times after induction. The cells were pelleted (5000 × g, 10 min) and resuspended in Buffer A (100 mM sodium phosphate, 10 mM Tris-Cl, 0.1 mM MgCl2, pH 7.5) containing Complete™ protease inhibitor cocktail (Roche Diagnostics) and stored at −80°C until use. The cell suspension was adjusted to an OD600 of 20, lysozyme added, and a 500-μL aliquot was sonicated with a microtip probe until reaching a constant clarity using 10-sec pulses intermittent with 1 min on wet ice. The sample was centrifuged at 18,000 × g for 10 min to pellet inclusion bodies and cell debris. The pellet was resuspended to the original volume with 0.5 mL Buffer A. Three volumes of the resuspended pellet (inclusion body fraction) or of the supernatant (soluble and membrane proteins) were each mixed with one volume of 4× sample buffer (0.15 M Tris, pH 6.8, 3.3 M urea, 0.4 M dithiothreitol, 10% glycerol, 8% sodium dodecyl sulfate, 0.8 mg/mL bromphenol blue), and incubated at 25°C for 10 min. The samples were loaded (5 μL per lane) on a 2.5 M urea 12% polyacrylamide gel (Berthold and Siedow 1993). The gels were stained with Coomassie blue using the modified Fairbanks method (Wong et al. 2000) and inspected visually for protein expression. When isolated membranes were needed, the supernatant from the low speed spin was ultracentrifuged (200,000 × g, 2 h) to separate membranes from soluble proteins. The membrane pellet was resuspended in the original volume using Buffer A, and incubated with sample buffer as above.
For large-scale membrane preparations, E. coli was grown in LB containing 60 μg/mL ampicillin and 40 μM FeSO4. Cultures were incubated at 37°C with the temperature lowered to either 18 or 25°C prior to induction with IPTG. Cells were harvested following overnight incubation. Membranes were prepared from a protocol similar to that noted above with adjustment of volumes, with the exception that cells were broken using one pass through a French Press at 18,000 psi.
Acknowledgments
We thank Stephen Elledge for introducing us to the univector recombination system and providing us with plasmids and E. coli strains. We thank Dieter Söll for pAOX, Steven Rodermel for the PTOX clone, and Tobias Cornvik for technical assistance. We would like to acknowledge the Swedish Research Council, the Wallenberg Consortium North, and the Göran Gustafsson Foundation for Research in Natural Science and Medicine for financial support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AOX, alternative oxidase
Coq7, 5-demethoxyquinone hydroxylase
IPTG, isopropylthio-β-d-galactoside
MBP, maltose binding protein
pHOST, a generic term in the univector system for an expression vector containing an in-frame loxP site
pUNI, the generic term in the univector system for the entry vector (into which a coding sequence is inserted)
PTOX, plastid terminal oxidase
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0223703.
References
- Berthold, D.A. 1998. Isolation of mutants of the Arabidopsis thaliana alternative oxidase (ubiquinol:oxygen oxidoreductase) resistant to salicylhydroxamic acid. Biochim. Biophys. Acta 1364 73–83. [DOI] [PubMed] [Google Scholar]
- Berthold, D.A. and Siedow, J.N. 1993. Partial purification of the cyanide-resistant alternative oxidase of skunk cabbage (Symplocarpus foetidus) mitochondria. Plant Physiol. 101 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold, D.A., Andersson, M.E., and Nordlund, P. 2000. New insight into the structure and function of the alternative oxidase. Biochim. Biophys. Acta 1460 241–254. [DOI] [PubMed] [Google Scholar]
- Braun, P., Hu, Y., Shen, B., Halleck, A., Koundinya, M., Harlow, E., and LaBaer, J. 2002. Proteome-scale purification of human proteins from bacteria. Proc. Natl. Acad. Sci. 99 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck, P., Winkler, M., and Bautsch, W. 1993. Rapid cloning by homologous recombination in vivo. Nucleic Acids Res. 21 3601–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carol, P., Stevenson, D., Bisanz, C., Breitenbach, J., Sandmann, G., Mache, R., Coupland, G., and Kuntz, M. 1999. Mutations in the Arabidopsis gene IMMUTANS cause a variegated phenotype by inactivating a chloroplast terminal oxidase associated with phytoene desaturation. Plant Cell 11 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri, M., Ajayi, W., and Hill, G.C. 1998. Biochemical and molecular properties of the Trypanosoma brucei alternative oxidase. Mol. Biochem. Parasit. 95 53–68. [DOI] [PubMed] [Google Scholar]
- Christendat, D., Yee, A., Dharamsi, A., Kluger, Y., Savchenko, A., Cort, J.R., Booth, V., Mackereth, C.D., Saridakis, V., Ekiel, I., et al. 2000. Structural proteomics of an archaeon. Nat. Struct. Biol. 7 903–908. [DOI] [PubMed] [Google Scholar]
- Crameri, A., Whitehorn, E.A., Tate, E., and Stemmer, W.P.C. 1996. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol. 14 315–319. [DOI] [PubMed] [Google Scholar]
- Davis, G.D., Elisee, C., Newham, D.M., and Harrison, R.G. 1999. New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 65 382–388. [PubMed] [Google Scholar]
- Fukai, Y., Amino, H., Hirawake, H., Yabu, Y., Ohta, N., Minagawa, N., Sakajo, S., Yoshimoto, A., Nagai, K., Takamiya, S., et al. 1999. Functional expression of the ascofuranone-sensitive Trypanosoma brucei brucei alternative oxidase in the cytoplasmic membrane of Escherichia coli. Comp. Biochem. Physiol. C 124 141–148. [DOI] [PubMed] [Google Scholar]
- Goldstein, M.A. and Doi, R.H. 1995. Prokaryotic promoters in biotechnology. Biotechnol. Annu. Rev. 1 105–128. [DOI] [PubMed] [Google Scholar]
- Hammarström, M., Hellgren, N., van den Berg, S., Berglund, H., and Härd, T. 2002. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 11 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, A.Y., Do, T.Q., Lee, P.T., and Clarke, C.F. 2000. Genetic evidence for a multi-subunit complex in the O-methyltransferase steps of coenzyme Q biosynthesis. Biochim. Biophys. Acta 1484 287–297. [DOI] [PubMed] [Google Scholar]
- Jones, D.H. and Howard, B.H. 1991. A rapid method for recombination and site-specific mutagenesis by placing homologous ends on DNA using polymerase chain reaction. BioTechniques 10 62–66. [PubMed] [Google Scholar]
- Josse, E.-M., Simkin, A.J., Gaffé, J., Labouré, A.-M., Kuntz, M., and Carol, P. 2000. A plastid terminal oxidase associated with carotenoid desaturation during chromoplast differentiation. Plant Physiol. 123 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, C. and Inouye, M. 1993. One-step insertion of oligonucleotide linkers or adaptors to DNA using unphosphorylated oligonucleotides. BioTechniques 15 659–668. [PubMed] [Google Scholar]
- Kapust, R.B. and Waugh, D.S. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasling, J.D. 1999. Gene-expression tools for the metabolic engineering of bacteria. Trends Biotechnol. 17 452–460. [DOI] [PubMed] [Google Scholar]
- Kumar, A.M. and Söll, D. 1992. Arabidopsis alternative oxidase sustains Escherichia coli respiration. Proc. Natl. Acad. Sci. 89 10842–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q., Li, M.Z., Leibham, D., Cortez, D., and Elledge, S.J. 1998. The univector plasmid-fusion system, a method for rapid construction of recombinant DNA without restriction enzymes. Curr. Biol. 8 1300–1309. [DOI] [PubMed] [Google Scholar]
- Liu, Q., Li, M.Z., Liu, D., and Elledge, S.J. 2000. Rapid construction of recombinant DNA by the univector plasmid-fusion system. Methods Enzymol. 328 530–549. [DOI] [PubMed] [Google Scholar]
- Matsumura, I., Olsen, M.J., and Ellington, A.D. 2001. Optimization of heterologous gene expression for in vitro evolution. BioTechniques 30 474–476. [DOI] [PubMed] [Google Scholar]
- Miroux, B. and Walker, J.E. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260 289–298. [DOI] [PubMed] [Google Scholar]
- Pryor, K.D. and Leiting, B. 1997. High-level expression of a soluble protein in Escherichia coli using a His6-tag and maltose-binding-protein double-affinity fusion system. Protein Expr. Purif. 10 309–319. [DOI] [PubMed] [Google Scholar]
- Shih, Y.-P., Kung, W.-M., Chen, J.-C., Yeh, C.-H., Wang, A.H.-J., and Wang, T.-F. 2002. High-throughput screening of soluble recombinant proteins. Protein Sci. 11 1714–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, P., Grünler, J., Mattsson, J., Sindelar, P.J., Nordlund, P., and Berthold, D.A. 2001. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J. Biol. Chem. 276 33297–33300. [DOI] [PubMed] [Google Scholar]
- Trudel, P., Provost, S., Massie, B., Chartrand, P., and Wall, L. 1996. pGATA: A positive selection vector based on the toxicity of the transcription factor GATA-1 to bacteria. BioTechniques 20 684–693. [DOI] [PubMed] [Google Scholar]
- Wallis, O.C., Sami, A.J., and Wallis, M. 1995. The effect of changes in nucleotide sequence coding the N-terminus on expression levels of ovine growth hormone variants in Escherichia coli. Biochim. Biophys. Acta 1261 360–368. [DOI] [PubMed] [Google Scholar]
- Wong, C., Sridhara, S., Bardwell, J.C.A., and Jakob, U. 2000. Heating greatly speeds Coomassie blue staining and destaining. BioTechniques 28 426–432. [DOI] [PubMed] [Google Scholar]
- Wu, D., Wright, D.A., Wetzel, C., Voytas, D.F., and Rodermel, S. 1999. The IMMUTANS variegation locus of Arabidopsis defines a mitochondrial alternative oxidase homolog that functions during early chloroplast biogenesis. Plant Cell 11 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, Z., Jones, D.H., and Grose, C. 1992. Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl. 1 205–207. [DOI] [PubMed] [Google Scholar]