

Abstract
Binary patterning of polar and nonpolar amino acids has been used as the key design feature for constructing large combinatorial libraries of de novo proteins. Each position in a binary patterned sequence is designed explicitly to be either polar or nonpolar; however, the precise identities of these amino acids are varied extensively. The combinatorial underpinnings of the “binary code” strategy preclude explicit design of particular side chains at specified positions. Therefore, packing interactions cannot be specified a priori. To assess whether the binary code strategy can nonetheless produce well-folded de novo proteins, we constructed a second-generation library based upon a new structural scaffold designed to fold into 102-residue four-helix bundles. Characterization of five proteins chosen arbitrarily from this new library revealed that (1) all are α-helical and quite stable; (2) four of the five contain an abundance of tertiary interactions indicative of well-ordered structures; and (3) one protein forms a well-folded structure with native-like features. The proteins from this new 102-residue library are substantially more stable and dramatically more native-like than those from an earlier binary patterned library of 74-residue sequences. These findings demonstrate that chain length is a crucial determinant of structural order in libraries of de novo four-helix bundles. Moreover, these results show that the binary code strategy—if applied to an appropriately designed structural scaffold—can generate large collections of stably folded and/or native-like proteins.
Keywords: Protein design, binary patterning, de novo proteins, native-like protein structure, combinatorial library, four-helix bundle
The binary code strategy for protein design is based on the premise that appropriate patterning of polar and nonpolar residues can drive a polypeptide chain to fold into segments of amphiphilic secondary structure that anneal together to form a desired tertiary structure (Kamtekar et al. 1993; Xiong et al. 1995; West et al. 1999). A designed binary pattern specifies the order of polar and nonpolar residues. However, within a library of binary patterned sequences, the identity of the side chain at each polar and nonpolar site is varied combinatorially. The combinatorial mixes of polar and nonpolar amino acids are dictated by the organization of the genetic code: six polar residues (Lys, His, Glu, Gln, Asp, and Asn) are encoded by the degenerate codon VAN, and five nonpolar residues (Met, Leu, Ile, Val, and Phe) are encoded by the degenerate codon NTN (V = A, G, or C; N = A, G, C, or T).
Initial application of the binary code strategy focused on designing a library of 74-residue sequences targeted to fold into four-helix bundles (Kamtekar et al. 1993). Purification and characterization of >50 proteins from the initial collection demonstrated that virtually all sequences indeed folded into α-helical structures (Kamtekar et al. 1993; Roy and Hecht 2000).
Because of its combinatorial nature, the binary code strategy does not allow explicit design of specific interresidue interactions. Because unique packing cannot be designed a priori, it is reasonable to question whether “native-like” structures can nonetheless be isolated from binary code libraries a posteriori. Searches for well-folded proteins in the original 74-residue library showed that several proteins possessed some native-like characteristics (Roy et al. 1997a, 1997b; Rosenbaum et al. 1999; Roy and Hecht 2000). However, most proteins in the initial collection formed fluctuating structures, reminiscent of molten globule intermediates.
Why did most sequences from the original collection fail to form native-like structures? One might postulate that fluctuating “molten” structures are exactly what should be expected from a combinatorial strategy that precludes explicit design of specific sequences with predetermined side-chain interactions. However, the alternative result—native-like structures—might have been predicted by numerous studies demonstrating that a well-folded structure can be specified by many different amino acid sequences (Dill 1985; Chothia and Lesk 1987; Bowie et al. 1990; Matthews 1993; Bromberg and Dill 1994; Axe et al. 1996; Gassner et al. 1996; Munson et al. 1996; Riddle et al. 1997). Comparisons of evolutionarily related sequences, theoretical studies using simplified models, and extensive mutagenesis experiments have led to the realization that protein structures are robust, and explicit design of “jigsaw puzzle” packing may not be necessary. For example, Matthews and coworkers replaced up to 10 residues in the core of T4 lysozyme with methionines and found—in contrast to the predictions of the jigsaw puzzle model—that the multiply substituted proteins were active and cooperatively folded (Gassner et al. 1996).
These and other findings (Dill 1985; Chothia and Lesk 1987; Bowie et al. 1990; Lau and Dill 1990; Behe et al. 1991; Matthews 1993; Bromberg and Dill 1994; Axe et al. 1996; Gassner et al. 1996; Munson et al. 1996; Riddle et al. 1997) led us to question whether the tendency of the original binary code proteins to form fluctional structures might not be a failure of the binary code strategy per se but rather a shortcoming of the designed structural scaffold used in its initial implementation. In particular, we questioned whether the α-helices specified by our original scaffold might simply be too short. We reasoned that in the context of the binary code strategy, which cannot specify side chain packing a priori, it might be advantageous to use a scaffold that encodes longer α-helices, and hence, larger interhelical interfaces. Here, we describe the design of a second-generation scaffold, in which the four α-helices are 50% longer. Characterization of several proteins arbitrarily chosen from the new library demonstrates that they are all significantly more stable, and most of them (four out of five) are substantially more native-like than proteins from the original binary code library.
Results
Design and construction of the second-generation binary patterned library
The de novo proteins in the original binary code library were designed to be 74 residues long (Kamtekar et al. 1993). Natural four-helix bundles, however, are invariably longer than 100 residues, with individual α-helices typically longer than 20 residues (Y. Wei and M.H. Hecht, unpubl.). To assess whether a longer structural scaffold would favor well-folded structures over molten globules, we constructed a second-generation library of 102-residue sequences. This new library was not constructed “from scratch.” Instead, to stringently test whether the redesigned features are sufficient to convert a fluctional protein into a well-ordered structure, we chose a typical molten globule-like protein (sequence #86) from the original 74-residue library (Kamtekar et al. 1993) as the starting point for designing the second-generation elongated library.
The new library of sequences was constructed in two steps. In the first step, two minor modifications were incorporated: (1) a tyrosine was inserted after the initiator methionine to facilitate concentration determinations and also to prevent methionine removal in vivo (Bachmair et al. 1986), and (2) a glycine dipeptide was inserted into the central interhelical turn in place of a proline. Proline was undesirable because cis/trans isomerism could lead to multiple (rather than unique) conformations. The modified sequence with these two changes (called n86) served as the template for constructing the second-generation binary code library.
In the second step, sequence n86 was elongated by inserting combinatorially diverse sequences between α-helices 1 and 2, and between α-helices 3 and 4. Each of the four α-helices was elongated from 14 residues to 20 residues (not including N- and C-caps) in accordance with the binary code patterning. The overall design of the structural scaffold for the second-generation library is summarized in Figure 1 ▶. Construction of the second-generation library of synthetic genes using combinatorial mixes of DNA codons encoding polar, nonpolar, N-cap, C-cap, and turn residues is described in Materials and Methods.
Figure 1.

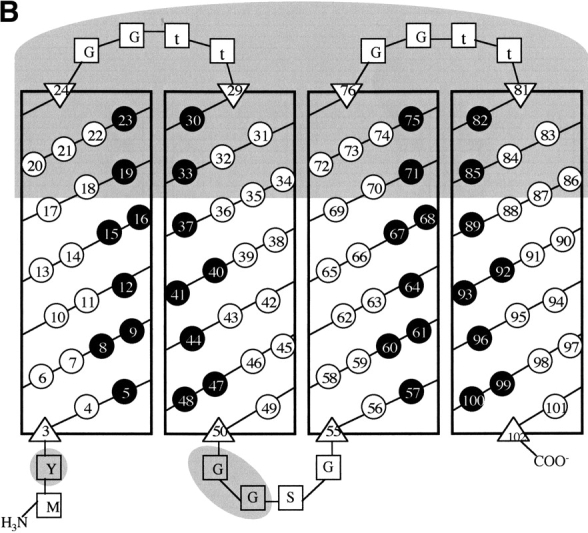
(A) Elongation of the structural scaffold for a combinatorial library of four-helix bundles. Cylinders represent α-helices. Sequences in the original library were 74 residues long; those in the second-generation library are 102 residues long. (B) Helix net diagram showing the design of a 102-residue structural scaffold for the second-generation library of binary patterned four-helix bundles. Each of the four α-helices is shown as a vertical rectangle, with binary patterned polar (○) and nonpolar (•) residues depicted as white and black circles, respectively. Vertical stripes of nonpolar residues indicate the hydrophobic faces of each α-helix. N-cap and C-cap residues are depicted as triangles, and turn residues as squares. Within the interhelical turns, uppercase letters indicate invariant residues, while a lowercase “t” indicates combinatorially diverse turn residues. Features distinguishing this redesigned 102-residue scaffold from the original 74-residue scaffold are shaded in gray.
The newly constructed library of synthetic genes was transformed into Escherichia coli. Clones were screened for correctly sized genes by PCR, and verified by DNA sequence analysis. From the resulting library of correct sequences, five proteins were arbitrarily chosen for biophysical characterization. The sequences of these proteins (and the parental protein from the original library) are shown in Figure 2 ▶. The proteins were purified using published methods (Kamtekar et al. 1993; Johnson and Hecht 1994; Roy and Hecht 2000), and their identities were confirmed by electrospray mass spectrometry (Table 1).
Figure 2.

Amino acid sequences (single-letter code) of de novo proteins. Polar and nonpolar residues in the α-helices are shown in red and green, respectively. (Top) Sequence 86 is from the original binary code library (Kamtekar et al. 1993). Sequence n86 differs from 86 by having a tyrosine inserted after the N-terminal methionine and a glycine dipeptide in place of a proline in the central turn. (Bottom) The newly designed scaffold for the elongated second-generation library. Combinatorial turn residues are shown as t. Combinatorial mixes of amino acids used at polar, nonpolar, N-cap, C-cap, and turn residues are described in the Materials and methods section. Below the redesigned scaffold are five sequences from the new library. The “S” prefix indicates sequences from the second-generation library.
Table 1.
Properties of de novo proteins
| Protein | Mass (Obs) | Mass (calc) | Elution time (min) | CD (222 nm) | Gnd-midpoint | ΔG (kcal/mole) |
| n86 | 8625.9 | 8627.7 | nd | −21,950 | 1.7 M | 3.0 |
| S-23 | 11856.0 | 11855.2 | 23.35 | −25,890 | 3.7 M | 6.6 |
| S-213 | 11857.0 | 11856.3 | 23.35 | −29,690 | 3.4 M | 8.4 |
| S-285 | 12063.2 | 12062.5 | 23.79 | −32,870 | 3.6 M | 9.5 |
| S-824 | 11928.8 | 11928.4 | 23.77 | −29,710 | 3.2 M | 7.7 |
| S-836 | 11927.7 | 11927.4 | 23.76 | −31,550 | 3.0 M | 5.4 |
| cyt C | 12384 | 22.60 |
Mass (obs) was measured using electrospray mass spectrometry. Mass (calc) was calculated from the sequences. Elution times were measured using size-exclusion chromatography performed at the same concentrations as NMR experiments. CD (222 nm) indicates mean residue ellipticity at 222 nm at 20°C. Gnd midpoint is the concentration of guanidine hydrochloride at which 50% of the protein sample is denatured. ΔG indicates the free energy of unfolding in the absence of denaturant, and is derived by extrapolation of the guanidine denaturation data.
The second-generation proteins are monomeric, α-helical, and stable
Size-exclusion chromatography demonstrated that all five proteins are monomeric in solution (Table 1), even at the relatively high concentrations used for NMR spectroscopy. The proteins all formed α-helical structures, with circular dichroism (CD) spectra showing the typical α-helical signatures including a maximum at 190 nm, and minima at 208 and 222 nm (not shown). The magnitudes of the minima at 222 nm (Table 1) are as expected for four-helix bundles.
The stabilities of the second-generation proteins are compared to the parental protein, n86, in Figure 3 ▶. All five of the newly designed 102-residue proteins are substantially more stable than the parental n86 protein. The free energies stabilizing the folded state relative to the unfolded state are approximately two- to threefold more favorable for the elongated proteins relative to the parental n86 protein (Table 1).
Figure 3.

Stability of de novo proteins. The parental protein, n86, is compared to the second-generation proteins, S-23, S-213, S-285, S-824, and S-836. All five of the second-generation proteins are considerably more stable than n86. Denaturation was monitored by measuring the CD signal at 222 nm as a function of the concentration of guanidine hydrochloride.
Homonuclear NMR spectroscopy
Although increased stability is an important goal in protein design, high stability does not necessarily indicate that a protein forms a uniquely folded structure. Indeed, several very stable de novo proteins have been shown to be molten globules (Betz et al. 1993). To assess whether our newly designed binary code proteins are fluctional or well ordered, we examined their NMR spectra. Two-dimensional NOESY spectra are particularly useful for assessing conformational specificity. In such spectra, two main features are important: (1) the dispersion of chemical shifts, and (2) the number of well-resolved NOE cross peaks. Good dispersion indicates that different parts of the molecule occupy distinctly different chemical environments, as would be found in a well-ordered structure, but not a fluctuating molten globule. An abundance of well-resolved NOE cross peaks indicates the existence of tertiary interactions that persist over time. Such interactions are characteristic of well-folded structures, but not molten globules.
Figure 4 ▶ compares the NOESY spectrum of the parental n86 protein with the spectra of the five second-generation proteins. The spectrum of protein n86 shows poor chemical shift dispersion, only a few distinguishable interresidue NH-to-NH contacts, and only a limited number of side-chain interactions. Thus, protein n86 does not form a well-folded structure. The spectrum of one second-generation protein, S-23, although better than that of n86, also does not display the dispersion, resolution, or abundance of NOE cross-peaks that would be expected for a well-folded structure.
Figure 4.


Comparison of the NOESY spectra of the parental protein n86 with the second-generation “S” proteins. (Top) Cross-peaks between side chain protons and amide NH or aromatic CH protons. (Bottom) Cross-peaks between amide NH and/or aromatic CH protons. The spectra of n86 and S-23 show poor chemical shift dispersion, and few distinguishable peaks. In contrast, the spectra of the four second-generation proteins, S-213, S-285, S-824, and S-836, contain numerous sharp NOE cross-peaks, indicating that these 102-residue proteins fold into structures that maintain specific interresidue interactions.
In contrast, the spectra of the four second-generation proteins, S-213, S-285, S-824, and S-836, are well dispersed and contain many well-resolved NOE cross-peaks. These cross-peaks indicate that these four second-generation proteins fold into structures containing many tertiary contacts.
15N,1H-HSQC NMR spectroscopy
Additional information about the native-like versus molten globule-like properties of a protein can be obtained from heteronuclear NMR spectroscopy. Figure 5 ▶ compares the 15N,1H-HSQC spectra of the second-generation proteins with the spectrum of the parental n86 protein. As expected for proteins with different sequences, the HSQC spectra of the five second-generation proteins range in quality. Consistent with the homonuclear results described above, the HSQC spectrum of protein S-23 is only slightly better than that of the parental protein, n86. However, the other four second-generation proteins (S-213, S-285, S-824, and S-836) yield 15N,1H-HSQ spectra that are substantially better than n86: All four display peak dispersion (in both dimensions) that is superior to n86, and is comparable to the dispersion seen for many natural α-helical proteins. Moreover, in contrast to n86, which contains only a few well-resolved cross-peaks, the spectra of these four second-generation proteins contain numerous well-resolved cross-peaks. Based on these spectra, it is clear that the structures of these arbitrarily selected second-generation proteins are substantially more ordered than that of the parental protein, n86.
Figure 5.

15N,1H HSQC NMR spectra of uniformly 15N-labeled proteins. The parental protein n86 is compared to the five second-generation proteins. Well-resolved peaks and good chemical shift dispersion in both dimensions are indicators of well-folded protein structures.
The HSQC spectrum of S-824 is particularly good, suggesting that this protein probably forms a well-ordered structure comparable to those of native proteins. Structure determination of this protein is currently underway.
13C,1H-HSQC NMR spectroscopy
Although the 15N,1H-HSQC spectra described above (and shown in Fig. 5 ▶) suggest that the second-generation proteins form ordered structures, these spectra report primarily on the environment of the backbone. To distinguish well-folded structures from those with extensive local mobility, it is also necessary to analyze the environments of the side chains—especially those in the hydrophobic core. As described recently by Walsh et al. (2001a, b), the degree of side-chain order can be assessed by natural abundance 13C,1H-HSQC NMR spectroscopy.
We measured natural abundance 13C,1H-HSQC NMR spectra for the five second-generation proteins. The methyl 13C,1H correlations for these proteins are shown in Figure 6 ▶. In these spectra, the γ and δ methyl resonances of isoleucine side chains appear in the 13C dimension (F1 in Fig. 6 ▶) at ∼11 and ∼15 ppm, respectively. Methyl groups from the side chains of Val, Met, Leu, and Thr occur between 18 and 24 ppm.
Figure 6.

Natural abundance 13C,1H-HSQC NMR spectra for the five second-generation proteins. γ and δ methyl resonances of isoleucine side chains appear in the 13C dimension at ∼11 ppm and ∼15 ppm, respectively. Methyl groups from the side chains of Val, Met, Leu, and Thr are clustered between 18 and 24 ppm. Spectra for proteins S-213, S-285, S-824, and S-836 display well-resolved peaks and good dispersion, which indicate well-ordered side chains.
The 13C,1H-HSQC spectrum of S-23 shows that the side chains of this protein are not well ordered. This is not surprising because the homonuclear NOESY spectrum of S-23 (Fig. 4 ▶) showed poor dispersion and few NOEs. However, the 13C,1H-HSQC spectra of the four other second-generation proteins display well-resolved peaks and good dispersion. Although we have not yet assigned the peaks in these spectra, it is clear from the isoleucine regions, that all (or almost all) of the isoleucine methyl resonances are well resolved. For example, for protein S-824, there are five isoleuecines in the sequence (see Fig. 2 ▶), and five resonances can be seen in both the γ methyl and the δ methyl regions. More detailed studies of the structures and dynamics of these proteins are in progress.
Differential scanning calorimetry
The NMR experiments described above were performed to assess the structural properties of the de novo proteins. An orthogonal method for distinguishing between molten globules and native-like structures is differential scanning calorimetry (DSC), which measures their thermodynamic properties. The thermal denaturation of molten globules typically occurs over a broad temperature range with a relatively low ΔH. In contrast, native structures denature cooperatively with relatively sharp transitions and larger enthalpies.
Figure 7 ▶ compares the thermal denaturation of protein n86 with the second-generation protein S-824. The elongated protein is significantly more thermally stable. Moreover, it denatures over a relatively narrow temperature range, and with a larger ΔH. Because of the high thermal stability of S-824, the denaturation was not complete even at 110°C. Therefore, calculation of a precise ΔH was not possible. Nonetheless, by assuming the peak is symmetric, we estimate a ΔH between 110 and 130 kcal/mole.
Figure 7.

Differential scanning calorimetry comparing thermal denaturation of the parental protein n86 with the second-generation protein S-824. Protein S-824 is substantially more stable and denatures with a much larger enthalpy.
The cooperativity of protein denaturation depends on both the quality and the quantity of interactions: Either enhanced structural order or increased chain length (or both) can be responsible for an increase in ΔH. Therefore, when interpreting our thermodynamic results, it is important to compare the measured ΔH for protein S-824 to the native and molten globule states of a reference protein of similar size and structure. Cytochrome b562 is a natural four-helix bundle containing 106 residues (compared to 102 in protein S-824). The ΔH reported for the denaturation of native cytochrome b562 is 104 kcal/mole (Robinson et al. 1998). This is similar—or slightly lower—than the ∼120 kcal/mole we estimate for S-824. The ΔH of S-824, however, is two- to threefold greater than the ΔH (46 kcal/mole) reported for the denaturation of the molten globule form of apocytochrome b562 (Feng et al. 1994).
The enthalpy of denaturation for protein S-824 is also approximatly threefold greater than that measured for the parental sequence n86. This difference is considerably larger than would be expected solely from the difference in size (102 residues versus 75 residues, that is, ∼33%). Thus, the calorimetrically measured thermodynamic properties of S-824 (Fig. 7 ▶) are consistent with the spectroscopically measured structural properties (Figs. 4 ▶–6 ▶) in demonstrating that this second-generation protein is significantly more stable and more native-like than the parental protein, n86.
Discussion
Earlier work on binary patterned libraries of polar (○) and nonpolar (•) amino acids demonstrated that designs based on the α-helical periodicity ○•○○••○○•○○••○ specify proteins that are soluble and α-helical (Kamtekar et al. 1993; Roy et al. 1997a, 1997b; Rosenbaum et al. 1999; Roy and Hecht 2000). Among these α-helical collections, de novo proteins that bind cofactors and catalyze reactions occur quite frequently (Moffet et al. 2000, 2001; Moffet and Hecht 2001). More recent work showed that libraries based on the β-strand pattern ○•○•○•○ yield proteins that form β-sheet structures. By varying either the details of the β-sheet design, or the conditions of the experiment (or both) we have produced collections of binary patterned β-sheet proteins that form either monomeric structures, amyloid-like fibrils, self-assembled monolayers, or template assembled biomaterials (West et al. 1999; Xu et al. 2001; Brown et al. 2002; Wang and Hecht 2002).
Although these earlier studies demonstrated that binary patterning can be used to guide the design of various protein structures and functions, a nagging question remained: can the binary code strategy also produce libraries of sequences that recapitulate the structural and thermodynamic properties of well-folded native proteins?
In assessing the potential of the binary code strategy to produce well-folded structures, we considered two opposing hypotheses: (1) Well-ordered structures may be difficult to achieve and therefore must either be designed explicitly or selected evolutionarily. If this hypothesis were correct, then binary patterned libraries would rarely yield well-folded proteins. (2) Alternatively, for a given structural scaffold, well-ordered structures may be achievable with many different combinations of side chains. This hypothesis is supported by statistical studies demonstrating that in natural proteins apolar side chains have little or no inherent preference for specific packing interactions, and “can pack together efficiently in a large number of ways” (Behe et al. 1991). This would lead to the expectation that well-folded structures would occur relatively frequently in binary patterned libraries.
Which of these two hypotheses is actually correct depends, at least in part, on the structural scaffold used in the design. Some scaffolds will rarely, if ever, yield well-folded structures. Because our earlier α-helical library (Kamtekar et al. 1993) was based on a scaffold considerably shorter than found in natural four-helix bundles (74 versus >100 residues), it became apparent that a realistic test of the binary code strategy required the design of a second-generation library using a longer structural scaffold.
The current work describes the construction of this second-generation library. Five proteins from the new library were purified and characterized. The structural and thermodynamic methods used to analyze these proteins demonstrate that (1) all five are α-helical and quite stable; (2) four of the five adopt structures that are reasonably well ordered; and (3) at least one of these proteins, S-824, is very well ordered and appears to fold into a structure that is native-like or near native-like.
The designation of a protein structure as “native-like” or “near native-like” is not entirely black and white. In the early days of protein design (Regan and DeGrado 1988; Hecht et al. 1990), a structure capable of producing the data shown in Figures 3 ▶–7 ▶ would have been considered a successful native-like de novo protein. However, during the past decade, as protein designers have gained experience, and as methods for probing structural rigidity have become more sophisticated, the goal of producing native-like de novo proteins has become somewhat of a moving target. For example, DeGrado and coworkers (1999) reported the NMR structures of a three-helix bundle, which according to several criteria appeared native-like (Walsh et al. 1999). However, more recent analyses of the response of this protein to mutations, as well as NMR studies of side chain dynamics, has demonstrated that the protein is both more malleable and more dynamic than those natural proteins that have been examined at the same level of detail (Walsh et al. 2001a, 2001b). Thus, a detailed understanding of the native-like properties of the second-generation binary code proteins will require further studies of their structures and dynamics. Such studies are underway.
Despite our incomplete knowledge of the detailed structures and dynamics of these proteins, it is clear from the data presented above that proteins from the new library are substantially more stable and dramatically more ordered than the parental proteins from which they were derived. The five proteins characterized in this study were chosen arbitrarily: Neither genetic selections in vivo nor high throughput screens in vitro were required to select these sequences from the overall library. Hence, they presumably represent a fairly unbiased sampling of the second-generation library. Therefore, our observation that well-ordered structures occur in such a small sampling suggests that the first of the two hypotheses enumerated above is false: Structures that are reasonably well ordered are not difficult to achieve; they need not be selected evolutionarily or designed residue by residue.
Although four of the five second-generation proteins appear well ordered, one protein (S-23) is not. Because the identities of individual amino acids clearly play a role in side chain packing, some combinations of amino acids will not be compatible with well-folded structures. Therefore, even for a well-designed scaffold, some binary patterned sequences will not fold into well-ordered structures: in the current library, sequence S-23 is closer to a molten globule.
Although S-23 is the least native-like of the five second-generation proteins (Figs. 4 ▶–6 ▶), it has the highest midpoint for guanidine denaturation (Fig. 3 ▶). Thus, as noted previously (Betz et al. 1993), enhanced stability alone does not prove a structure is more ordered or more native-like.
Although the reason that S-23 is an outlier is not yet known, we consider two possibilities: (1) S-23 is the only sequence with no tryptophans. Trp was shown recently to induce native-like structure in otherwise flexible chains (Klein-Seetharaman et al. 2002), and the absence of Trp in S-23 may tip the balance towards the molten globule state. (2) S-23 contains acidic side chains at positions 27, 28, 79, and 80. It is the only sequence with negative charges at all four of these sites. These positions are designed to be in the interhelical turns at the “top” of the four-helix bundle (Fig. 1 ▶). They would be in close proximity in the designed structure, and the local concentration of uncompensated negative charges may disfavor an ordered structure.
Despite the occasional molten globule, our results indicate that the second of the two opposing hypotheses enumerated above is closer to the truth: for a given structural scaffold, well-folded structures can be specified by many different amino acid sequences. Consequently, when binary patterning is applied to an appropriately designed structural scaffold, the resulting libraries contain a relatively large number of well-ordered and/or native-like structures.
Materials and methods
Construction of the second-generation library
Sequence 86 (74 residues) was converted to sequence n86 using PCR overlap extension to (1) insert a Tyr after the initiator Met, and (2) insert a glycine dipeptide in place of a proline in the central turn.
Sequence n86 was then used as the template for constructing a new combinatorial library of 102-residue sequences. The newly inserted regions of sequence are shaded gray in Figure 1 ▶. The inserted regions were designed as follows:
Combinatorial polar residues (Lys, His, Glu, Gln, Asp, Asn) were encoded at positions 18, 20, 21, 22, 31, 32, 34, 35, 70, 72, 73, 74, 83, 84, 86, and 87 by the degenerate DNA codon VAM. (V = A, G, or C; M = A or C; N = A, G, C, or T; S = G or C).
Combinatorial nonpolar residues (Met, Leu, Ile, Val, Phe) were encoded at positions 19, 30, 33, 71, 82 and 85 by the degenerate codon NTS.
The nonpolar NTS codon does not encode Trp. To facilitate occasional incorporation of this structure inducing hydrophobic residue (Klein-Seetharaman et al. 2002) into the cores of our proteins, we used an alternative codon, TKG (K = G or T) to encode an equimolar mixture of Trp and Leu at positions 23 and 75.
N-cap positions (29 and 81) in the newly inserted regions were encoded by AVC. This degenerate codon encodes a mixture of Asn, Ser, and Thr; residues that are favored at N-caps in natural proteins (Aurora and Rose 1998).
At the new C-cap positions (24 and 76), we used our standard polar codon VAM. Five of the six residues encoded by VAM occur frequently as C-caps in natural proteins (Aurora and Rose 1998).
In all three interhelical turns, glycine was incorporated at the position following the C-cap of the preceding helix. This is consistent with the strong preference for Gly at this position in natural proteins (Aurora and Rose 1998).
Combinatorial turn residues (“t” at positions 27, 28, 79, and 80) were encoded by the standard polar codon, VAM.
The new regions of sequence (gray in Fig. 1 ▶) were incorporated using two sequential steps of full plasmid PCR to insert long stretches of combinatorially diverse sequences first between α-helices 1 and 2, and then between α-helices 3 and 4. The steps involved in this construction are shown schematically in Figure 8 ▶.
Figure 8.

Construction of the second-generation library of binary patterned sequences. α-Helices were elongated by insertion of combinatorially diverse sequences into the gene encoding sequence n86. In step 1, PCR primers were annealed to DNA encoding the C-terminal section of α-helix 1 and the N-terminal section of α-helix 2. New sequence was inserted (shaded regions) by PCR amplification of the entire plasmid. Following PCR, plasmids were recircularized by blunt end ligation. The combinatorially diverse product of step 1 was then used as the PCR template for step 2, and PCR primers were annealed to DNA encoding the end of α-helix 3 and the beginning of α-helix 4. Again, new sequence was inserted (shaded regions) by PCR amplification of the entire plasmid, and plasmids were recircularized by blunt-end ligation.
Colony PCR was used to screen clones for those containing genes with the correct length. Sequences with insertions or deletions were discarded. Clones that had acquired “bonus” substitution mutations were corrected by site-directed (PCR) mutagenesis. Proteins were expressed and purified using methods similar to those described previously (Kamtekar et al. 1993; Johnson and Hecht 1994; Roy and Hecht 2000).
Size-exclusion chromatography
Elution times were measured using a Superdex 75 HR/10/30 gel filtration column (Pharmacia). Size-exclusion chromatography was performed at the same concentrations as NMR experiments in 50 mM sodium acetate-acetic acid buffer, pH 4.0.
Circular dichroism spectroscopy
CD measurements were performed at 20°C in 10 mM NaPO4 (pH 6.8), 40 mM NaF using an Aviv model 62 DS spectrapolarimeter.
NMR spectroscopy
Spectra were acquired at 25°C and 600 MHz on a Varian Unity/INOVA spectrometer, using WATERGATE solvent suppression (Piotto et al. 1992). Data processing was done using NMRPipe (Delaglio et al. 1995), while NMRView (Johnson and Blevins 1994) was used for visualization and data analysis. Protein concentration was ca. 1 mM in a buffer containing 50 mM sodium acetate/acetic acid at pH 4.0. (Several experiments run at pH = 6.8 yielded similar results.)
NOESY Spectra were acquired in 95%H2O/5%D2O with a 150-msec mixing time using gradients for suppression of radiation damping during t1 and for artifact suppression (Sklenar 1995). Data acquisition parameters for the NOESY spectra were as follows: The spectral window was 8 kHz with the carrier positioned at the water resonance (4.769 ppm). 4K* and 480* data points were collected in t2 and t1, respectively (t2[max] = 511 ms, t1[max] = 60 msec) with 16 scans collected for each FID. Data processing included digital filtering for residual solvent signal suppression, apodization with combined shifted cosine and Gaussian functions, zero filling in both dimensions, reconstruction of two missing complex data points in t1 by backwards linear prediction (LP), and automated polynomial baseline correction in both dimensions. The final size of the frequency domain data was 8K × 2K data points.
1H,15N HSQC spectra were recorded on uniformly 15N-labeled proteins, prepared from cultures grown in minimal medium with 15N-labeled ammonium chloride as the sole nitrogen source. HSQC spectra were recorded using sensitivity enhanced gradient selected HSQC technique (Kay et al. 1992). The spectra were acquired with an 8 kHz spectral window for 1H, positioning the carrier on the water resonance (4.769 ppm), and 2 kHz frequency range for 15N (centered at 118.56 ppm). 1024* and 128* data points were collected in t2 and t1, respectively (t2[max] = 128 msec, t1[max] = 64 msec), averaging eight transients for each FID. Data processing included digital filtering for suppression of the residual solvent signal, combined shifted cosine and Gaussian apodization, and zero filling in both dimensions. Final data size was 2K × 2K data points.
Natural abundance 13C,1H-HSQC spectra were run at 599.5 MHz (1H) and 150.8 MHz (13C) using the pulse sequence in ProteinPack provided with the Vnmr software (Varian, Inc.). The carrier was positioned at 4.769 ppm (water resonance) for 1H and 35.00 ppm for 13C. Spectral windows were 8 KHz for 1H and 12 kHz for 13C. In t2 1024* points were collected (t2[max] = 126 msec) averaging 64 scans for each FID, while 256* data points were acquired in t1 (t1[max] = 21 msec). Data processing included digital filtering for removal of the residual solvent signal, combined shifted cosine and Gaussian apodization, and zero filling prior to Fourier transform. Final data size was 2K × 2K data points.
Differential scanning calorimetry
Scans were performed on a MicroCal. MC-2 calorimeter. Samples were in 50 mM NaAc buffer, pH 4.1. Calorimetric analysis of protein S-836 (not shown) demonstrated that it denatures at even higher temperatures than protein S-824. Proteins S-23, S-213, and S-285 denature at higher guanidine concentrations than either S-824 or S-836, and no attempts were made to measure their calorimetric denaturations.
Acknowledgments
We thank Luciano Mueller (Bristol-Myers Squibb) for recording some of the HSQC spectra and George McLendon for use of the DSC instrument. This work was supported by NIH Grant RO1 GM62869.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.0228003.
References
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe, D.D., Foster, N.W., and Fersht, A.R. 1996. Active barnase variants with completely random hydrophobic cores. Proc. Natl. Acad. Sci. 93 5590–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmair, A., Finley, D., and Varshavsky, A. 1986. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234 179–186 [DOI] [PubMed] [Google Scholar]
- Behe, M.J., Lattman, E.E., and Rose, G.D. 1991. The protein-folding problem: The native fold determines packing, but does packing determine the native fold? Proc. Natl. Acad. Sci. 88 4195–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz, S.F., Raleigh, D.P., and DeGrado, W.F. 1993. De-novo protein design from molten globules to native-like states. Curr. Opin. Struct. Biol. 3 601–610. [Google Scholar]
- Bowie, J.U., Reidhaar-Olson, J.F., Lim, W.A., and Sauer, R.T. 1990. Deciphering the message in protein sequences: Tolerance to amino acid substitutions. Science 247 1306–1310. [DOI] [PubMed] [Google Scholar]
- Bromberg, S. and Dill, K.A. 1994. Side-chain entropy and packing in proteins. Protein Sci. 3 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C.L., Aksay, I.A., Saville, D.A., and Hecht, M.H. 2002. Template-directed assembly of a de novo designed protein. J. Am. Chem. Soc. 124 6846–6848. [DOI] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1987. The evolution of protein structures. Cold Spring Harbor Symp. Quant. Biol. 52 399–405. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe—A multidimensional spectral processiong system based on unix pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. 1985. Theory for the folding and stability of globular proteins. Biochemistry 24 1501–1509. [DOI] [PubMed] [Google Scholar]
- Feng, Y., Sligar, S.G., and Wand, A.J. 1994. Solution structure of apocytochrome b562. Nat. Struct. Biol. 1 30–35. [DOI] [PubMed] [Google Scholar]
- Gassner, N.C., Baase, W.A., and Matthews, B.W. 1996. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc. Natl. Acad. Sci. 93 12155–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht, M.H., Richardson, J.S., Richardson, D.C., and Ogden, R.C. 1990. De novo design, expression, and characterization of Felix: A four-helix bundle protein of native-like sequence. Science 249 884–891. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRview—A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Johnson, B.H. and Hecht, M.H. 1994. Recombinant proteins can be released from E. coli cells by repeated cycles of freezing and thawing. Biotechnology 12 1357–1360. [DOI] [PubMed] [Google Scholar]
- Kamtekar, S., Schiffer, J.M., Xiong, H., Babik, J.M., and Hecht, M.H. 1993. Protein design by binary patterning of polar and non-polar amino acids. Science 262 1680–1685. [DOI] [PubMed] [Google Scholar]
- Kay, L., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663–10665. [Google Scholar]
- Klein-Seetharaman, J., Oikawa, M., Grimshaw, S.B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L.J., Dobson, C.M., and Schwalbe, H. 2002. Long-range interactions within a nonnative protein. Science. 295 1719–1722. [DOI] [PubMed] [Google Scholar]
- Lau, K.F. and Dill, K.A. 1990. Theory for protein mutability and biogenesis. Proc. Natl. Acad. Sci. 87 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1993. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62 139–160. [DOI] [PubMed] [Google Scholar]
- Moffet, D.A. and Hecht, M.H. 2001. De novo proteins from combinatorial libraries. Chem. Rev. 101 3191–3203. [DOI] [PubMed] [Google Scholar]
- Moffet, D.A., Certain, L.K., Smith, A.J., Kessel, A.J., Beckwith, K.A., and Hecht, M.H. 2000. Peroxidase activity in heme proteins derived from a designed combinatorial library. J. Am. Chem. Soc. 122 7612–7613. [Google Scholar]
- Moffet, D.A., Case, M.A., House, J.C., Vogel, K., Williams, R.D., Spiro, T.G., McLendon, G.L., and Hecht, M.H. 2001. Carbon monoxide binding by de novo heme proteins derived from designed combinatorial libraries. J. Am. Chem. Soc. 123 2109–2115. [DOI] [PubMed] [Google Scholar]
- Munson, M., Balasubramanian, S., Fleming, K.G., Nagi, A.D., O’Brien, R., Sturtevant, J.M., and Regan, L. 1996. What makes a protein a protein? Hydrophobic core designs that specify stability and structural properties. Protein Sci. 5 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto, M., Sauder, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Regan, L. and DeGrado, W.F. 1988. Characterization of a helical protein designed from first principles. Science 241 976–978. [DOI] [PubMed] [Google Scholar]
- Riddle, D.S., Santiago, J.V., Bray-Hall, S.T., Doshi, N., Grantcharova, V.P., Yi, Q., and Baker, D. 1997. Functional rapidly folding proteins from simplified amino acid sequences. Nat. Struct. Biol. 4 805–809. [DOI] [PubMed] [Google Scholar]
- Robinson, C.R., Liu, Y., O’Brien, R., Sligar, S.G., and Sturtevant, J.M. 1998. A differential scanning calorimetric study of the thermal unfolding of apo- and holo-cytochrome b562. Protein Sci. 7 961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum, D.M., Roy, S., and Hecht, M.H. 1999. Screening combinatorial libraries of de novo proteins by hydrogen-deuterium exchange and electrospray mass spectrometry. J. Am. Chem. Soc. 121 9509–9513. [Google Scholar]
- Roy, S. and Hecht, M.H. 2000. Cooperative thermal denaturation of proteins designed by binary patterning of polar and nonpolar amino acids. Biochemistry 39 4603–4607. [DOI] [PubMed] [Google Scholar]
- Roy, S., Helmer, K.J., and Hecht, M.H. 1997a. Detecting native-like properties in combinatorial libraries of de novo proteins. Fold. Design. 2 89–92. [DOI] [PubMed] [Google Scholar]
- Roy, S., Ratnaswamy, G., Boice, J.A., Fairman, R., McLendon, G., and Hecht, M.H. 1997b. A protein designed by binary patterning of polar and nonpolar amino acids displays native-like properties. J. Am. Chem. Soc. 119 5302–5306. [Google Scholar]
- Sklenar, V. 1995. Suppression of radiation damping in multidimensional NMR experiments using magnetic field gradients. J. Mag. Reson. Ser. A. 114 132–135. [Google Scholar]
- Walsh, S.T., Cheng, H., Bryson, J.W., Roder, H., and DeGrado, W.F. 1999. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. 96 5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, S.T., Lee, A.L., DeGrado, W.F., and Wand, A.J. 2001a. Dynamics of a de novo designed three-helix bundle protein studied by 15N, 13C, and 2H NMR relaxation methods. Biochemistry 40 9560–9569. [DOI] [PubMed] [Google Scholar]
- Walsh, S.T., Sukharev, V.I., Betz, S.F., Vekshin, N.L., and DeGrado, W.F. 2001b. Hydrophobic core malleability of a de novo designed three-helix bundle protein. J. Mol. Biol. 305 361–373. [DOI] [PubMed] [Google Scholar]
- Wang, W. and Hecht, M.H. 2002. Rationally designed mutations convert de novo amyloid-like fibrils into monomeric beta-sheet proteins. Proc. Natl. Acad. Sci. 99 2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, M.W., Wang, W., Patterson, J., Mancias, J.D., Beasley, J.R., and Hecht, M.H. 1999. De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. 96 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, H., Buckwalter, B.L., Shieh, H.M., and Hecht, M.H. 1995. Periodicity of polar and non-polar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Natl. Acad. Sci. 92 6349–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, G., Wang, W., Groves, J.T., and Hecht, M.H. 2001. Self-assembled monolayers from a designed combinatorial library of de novo beta-sheet proteins. Proc. Natl. Acad. Sci. 98 3652–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]