

Abstract
Experiments were done to study the dynamic structural motions that determine protein hydrogen exchange (HX) behavior. The replacement of a solvent-exposed lysine residue with glycine (Lys8Gly) in a helix of recombinant cytochrome c does not perturb the native structure, but it entropically potentiates main-chain flexibility and thus can promote local distortional motions and large-scale unfolding. The mutation accelerates amide hydrogen exchange of the mutated residue by about 50-fold, neighboring residues in the same helix by less, and residues elsewhere in the protein not at all, except for Leu98, which registers the change in global stability. The pattern of HX changes shows that the coupled structural distortions that dominate exchange can be several residues in extent, but they expose to exchange only one amide NH at a time. This ‘local fluctuation’ mode of hydrogen exchange may be generally recognized by disparate near-neighbor rates and a low dependence on destabilants (denaturant, temperature, pressure). In contrast, concerted unfolding reactions expose multiple neighboring amide NHs with very similar computed protection factors, and they show marked destabilant sensitivity. In both modes, ionic hydrogen exchange catalysts attack from the bulk solvent without diffusing through the protein matrix.
Keywords: Cytochrome c, hydrogen exchange, local fluctuation, glycine mutations
One wants to understand the structural and dynamic determinants of protein hydrogen exchange (HX). This problem has become important because HX methods are now able to provide direct information on protein structure, structure change, interactions, dynamics, and folding, resolved to the level of individual amino acids (Woodward 1994; Scholtz and Robertson 1995; Wand and Englander 1996).
Exchange rates for hydrogens that are freely exposed to solvent in unstructured polypeptides are quantitatively predictable (Molday et al. 1972; Bai et al. 1993; Connelly et al. 1993). Rates are much slower for hydrogens that are protected by H-bonded structure, whether they are buried or at the solvent-exposed surface. The exchange of amide hydrogens is catalyzed, above pH 4, by direct attack and H-bonding of solvent hydroxide ion to the exchangeable hydrogen (Berger and Linderstrøm-Lang 1957; Eigen 1964; Englander and Kallenbach 1983). To allow for hydrogen bonding to hydroxide ion, a sizable separation of any protecting H-bond is necessary (Milne et al. 1998), requiring some transient distortion of local structure. Many dynamical models for the structural events that allow protected hydrogens to exchange have been considered. Extended cooperative unfolding reactions, both global and subglobal, have now been documented in particular cases. The suggestion that more local transient distortions determine the exchange of many other hydrogens (Bai et al. 1995; Milne et al. 1998) has become controversial (Hilser and Freire 1996; Wooll et al. 2000).
We explored a mutational approach. A surface lysine residue in recombinant cytochrome c (Cyt c) was changed to glycine. Glycine substitutions make available parts of Ramachandran phi-psi space that are sterically excluded for nonglycine residues. This can be expected to promote the population of non-native unfolded states and also smaller distortional modes that enter that space.
As anticipated, the glycine mutation imposes a large change on protein stability even though the mutation has no effect whatsoever on the native protein structure, and it produces a distinct pattern of HX effects and noneffects on neighboring residues. The results allow local fluctuations to be distinguished from larger unfoldings, define their structural range, and can help to illuminate structural details of the underlying motions.
Results
Structure
The recombinant pseudowild-type cytochrome c (pWT Cyt c; oxidized form) used here duplicates mammalian equine Cyt c except that the two normally occurring peripheral histidines (His26 and His33) are replaced by asparagines and the N-terminus is not acetylated. The structure of pWT Cyt c is essentially identical to the highly studied mammalian protein, with minor NMR chemical shift changes in the immediate vicinity of the altered side chains.
The Lys8Gly mutation (K8G) studied here is placed at a surface exposed site. It does not perturb the structure. Changes in NMR chemical shift are less than 0.06 ppm except for Gln12 NH (0.09 ppm), which H-bonds to residue 8. The expected change in CαH chemical shift for the Lys to Gly change (0.4 ppm) (Wishart and Case 2001) is not seen, perhaps indicating a compensating change.
Stability
Protein stability was determined by guanidinium chloride (GdmCl) melting experiments over the range of pH used in the HX experiments. Figure 1 ▶ shows a broad maximum in stability around neutral pH with the Lys8Gly mutant less stable by 1.38 ± 0.12 kcal/mole at all pH values, in the range of other similar variants (Serrano et al. 1992; Srivastava and Sauer 2000).
Figure 1.

Unfolding free energy from GdmCl melting (oxidized Cyt c, 0.5 M KCl, 20°C). The Lys8Gly mutation is destabilizing by 1.4 kcal/mole across the pH range used in the HX experiments. (Stability increases by 0.45 kcal/mole in D2O where HX NMR measurements were made.)
Independently, it is often possible to measure global stability directly under native conditions from HX measurements, using equation 4, if NHs can be identified that exchange only from the transient globally unfolded state (Bai et al. 1994; Huyghues-Despointes et al. 2001). For Cyt c, the Leu 98 NH has this character (Bai et al. 1995), but it exchanges too slowly to measure in the parent pWT protein in the absence of added denaturant (∼100 years at pH 7, 20°C). A faster HX rate for Leu98 in the K8G variant could be seen at relatively high pH (pH* 8.4; months) in the range expected for the 1.4 kcal/mole destabilization.
Hydrogen exchange
The various amide hydrogens in Cyt c exchange at very different rates. Hydrogens that are either too fast or too slow can often be brought into the measurable time window at lower or higher pH. We measured HX at pH* 5.3, 6.1, 7.5, 7.9, and 8.4, which made it possible to compare 50 amide protons in the pWT and Lys8Gly proteins. Exchange rates increased linearly with OH− ion concentration, with measured slope (logkex versus pH) of 1.0 ± 0.3 total outside range, indicating exchange through an EX2 mechanism (equation 3).
The Lys8 NH in pWT Cyt c is protected from exchange by a factor of 104. Figure 2 ▶ shows HX rates through the pWT (Lys8) and mutant (Gly8) proteins, represented by the ΔG°ex parameter, computed from the measured rates using equations 1, 3, and 4. The exchange of almost all of the hydrogens could be measured at more than one solution pH*, allowing for increased accuracy by averaging ΔG°ex measurements. (Note that this parameter may not properly reflect the free energy level of local fluctuational distortions, although it does appear to correctly evaluate unfolding reactions; see Discussion.) The HX rate ratio, kK8G/kpWT, in Figure 3A ▶, shows the effect of the Lys8Gly mutation on the residues that could be measured in both proteins. Figure 3B ▶ illustrates the most pertinent HX data.
Figure 2.

ΔG°ex values calculated for the determining opening reactions from HX rates (equations 1, 3, 4; 0.5 M KCl, 20°C), using kint calibrated for unstructured molecules, usually averaged from experiments at more than one pH. (Use of the ΔG°ex parameter is convenient but may not accurately represent the free energy of the structural distortion; see Discussion).
Figure 3.


HX rate changes. (A) The HX rate ratio, kK8G/kpWT, for residues that could be measured in both pWT and Lys8Gly proteins. The ratio is relatively independent of kint except for residue 8 itself, where a factor of 2 enters (not applied in the graph shown). (B) HX data for residues near Lys8 (closed symbols) or Gly 8 (open symbols), measured at pH* 6 or corrected to pH* 6 when measured at another pH* (residue 8 at pH* 5.3; residue 10 at pH* 8.4).
The Lys8 to Gly8 mutation increases the HX rate of its own amide proton by 84-fold. This points to a 42-fold increase in the fraction of time open (exchange competent) when the twofold change in intrinsic exchange rate (kint) between Lys and Gly is considered (Bai et al. 1993; largely an inductive effect). Computed ΔΔG°ex is then 2.2 kcal/mole (equation 4). Lys7 is accelerated by 2.7-fold, and Ile9 and Phe10 by 19 and 15-fold, respectively. Other residues in the helix are accelerated by less than twofold in HX rate. Exchange elsewhere in the protein is wholly unaffected except for the global marker Leu98.
Discussion
Questions in protein HX
Over the 50 years since Linderstrøm-Lang and his colleagues first studied protein hydrogen exchange, many models for the dynamic structural processes that determine HX rate have been considered. These may be separated into cooperative unfolding models and noncooperative local fluctuation models (Miller and Dill 1995).
Cooperative unfolding reactions, both global and subglobal, have been well documented. Huyghes-Despointes et al. (2001) catalog over a dozen proteins plus various destabilizing mutants in which the exchange rate of the very slowest hydrogens, when computed as in equation 4 (including a proline isomer effect; Bai et al. 1994), correctly measures the free energy of the global unfolding equilibrium. More limited subglobal unfoldings have been demonstrated in a number of proteins (Bai et al. 1995; Chamberlain et al. 1996; Chamberlain and Marqusee 1997; Hiller et al. 1997; Bhuyan and Udgaonkar 1998; Fuentes and Wand 1998; Llinas et al. 1999; Chu et al. 2002; Hoang et al. 2002). All of these unfolding reactions have a recognizable signature. They cause multiple neighboring hydrogens to exchange with a common ΔG°ex (EX2 exchange, equation 3) or kop (EX1 exchange, equation 5) and a common dependence on destabilizing conditions that promote cooperative unfoldings.
Most hydrogens in Cyt c and in other proteins do not exhibit these signature characteristics. Neighboring hydrogens often exchange at very different rates that are essentially unaffected by denaturant (Hilton et al. 1981; Wagner and Wuthrich 1982; Kim and Woodward 1993; Mayo and Baldwin 1993; Bai et al. 1995). These characteristics suggest more local structural fluctuations that break protecting H-bonds noncooperatively and expose hydrogens to exchange more or less individually (Bai et al. 1995).
Present knowledge leaves open a number of fundamental questions. How local and noncooperative are these supposed fluctations? Do HX catalysts attack from the solvent or by diffusing through the protein? What is the validity of the ΔG°ex calculation? Can HX rates be predicted in general based on a presumption of extended unfolding reactions (Hilser and Freire 1996; Wooll et al. 2000)?
Local fluctuations versus concerted unfolding
Residues in the Cyt c N-terminal helix (residue 2 to 15) exchange with a range of rates (Fig. 2 ▶) and are insensitive to added denaturant (Bai et al. 1995), suggesting HX by way of local fluctuations. Residue 8 is in the middle of the helical length and on the solvent-exposed surface. The Lys8 to Gly8 mutation has no effect on Cyt c structure whatsoever, but it imposes a pattern of HX effects and noneffects locally and remotely. The rates and the pattern of changes observed allow some important conclusions to be drawn.
The results show that residue 8 does not exchange through an extended unfolding reaction. When the Gly8 NH is transiently made exchange competent, residues 7, 9, and 10 cannot simultaneously become exchange competent more than 0.02, 0.07, and 0.001 of the time, respectively, or else they would exchange much faster than is observed. This follows from the fact that the neighbors are much slower than Gly8, by factors of 50, 15, and 1000. A similar calculation holds for the parent Lys8 protein (<0.3, 0.16, and 0.05 of the time), and also for residue 9, because it is much faster than Phe10 and a little faster than Lys7. Thus residues 8 and 9 exchange by way of independent opening reactions that expose only the individual amide NH to exchange, rather than a more extensive concerted unfolding (under strongly native conditions). The same logic holds if it is conceived that exchange is due to motions of the acceptor amides (residues 4 and 5), which in any case are unlikely to be affected by the Lys8Gly change.
In the other direction, is it possible that the structural distortions that expose a neighbor to exchange represent an unfolding reaction that includes residue 8? The neighbors exchange more slowly, which allows for a common unfolding that includes residue 8 but does not govern its exchange. The Lys8 to Gly8 substitution sharply promotes its own opening, and it decreases the stability against unfolding reactions. If an unfolding reaction that governs the HX of any other residue includes also residue 8, then the Lys to Gly mutation would promote the unfolding. A roughly 10-fold acceleration in HX rate of the neighbor might be expected, corresponding to the measured global unfolding destabilization of 1.4 kcal/mol observed for the Lys8 to Gly8 substitution. In fact, the Gly8 mutation accelerates residues 7 and 11 to 15 by much less (1.1–2.7-fold). This logic precludes a governing segmental unfolding for residue 7 or 11–15 that includes residue 8. An unfolding-dependent exchange of the slowest exchangers, Ile9 (but unfolding only to the left) and Phe10, cannot be excluded on this basis. It can be noted that Ile9 and Phe10 show no denaturant dependence (Bai et al. 1995), but Phe10 does have an unusually large ΔH°ex (40 kcal/mole; Milne et al. 1999).
In the same way, the absence of Gly8-induced changes in HX rate elsewhere in the protein precludes rate-determining unfoldings that would reach out to residue 8, whether those remote hydrogens are faster or slower than residue 8. The point is illustrated by the one exception, the remote global unfolding marker Leu98, which is accelerated by the Lys8Gly mutation because residue 8 necessarily participates in the transient global unfolding.
These observations are consistently against cooperative HX rate-determining segmental unfolding reactions in the N-terminal helix, and point instead to separate more limited openings. One realizes that cooperative unfolding reactions that involve the entire N-terminal helix do exist, but only at a lower level. The N-terminal helix participates in a very low level unfolding reaction that marks the reversible global unfolding (ΔG°ex = 12.8 kcal/mole at pH 7 and 30°C; Bai et al. 1994). This unfolding rises to dominate HX rates only under conditions that selectively promote large unfoldings relative to more local openings (e.g., added denaturant; Bai et al. 1995). More stable conditions suppress unfolding and allow HX pathways dependent on small fluctuations to dominate.
Earlier observations of disparate HX rates for neighboring residues and near-zero denaturant dependence were taken to suggest that the exchange of many hydrogens is determined by local structural fluctuations (Bai et al. 1995; Milne et al. 1998; Huyghues-Despointes et al. 2001). The present results confirm that conclusion for the residues in the Cyt c N-terminal helix. This demonstration strengthens the likelihood that the exchange of many other hydrogens, in Cyt c and other proteins, identifiable by these same characteristics, proceeds by way of very local fluctuational motions.
How local are local fluctuations?
The present results indicate that the local fluctuations accessed in the N-terminal helix are not as local as might be thought. They involve distortional motions that extend over several residues. Residues upstream (Lys7) and downstream from residue 8, at least through Val11 and possibly further, are detectably accelerated by the Lys8Gly substitution. This indicates that the structural distortions that allow each of these residues to exchange reach to Gly8. The coupled distortions must include newly available Gly8 phi-psi rotations that enter previously inaccessible Ramachandran space.
Thus, local fluctuations are not strictly localized to the individual exchanging residue, although in each case only the individual NH is exposed to exchange. The need for coupled multiresidue displacements in the rigid helical context can be expected, especially so, given that a sizable distortion is necessary to separate the H-bond donor NH and its acceptor CO to allow attack by hydroxide ion catalyst (probably by 5 Å or so; Milne et al 1998, 1999). An interesting corollary is that distortions necessary to achieve exchange in β-structures, loops, and turns may have quite different requirements. This remains to be seen. Also, amides that are buried are likely to require additional distortions to access solvent, over and above those necessary for the exchange of surface residues. This latter consideration probably explains the general correlation of HX rate with helical surface burial, an example of which can be seen in the exchange profile through the N-terminal helix in Figure 2 ▶. A periodic pattern of protection due to depth of burial is superimposed on the large protection due to the fact of H-bonding (for a more complete display, see Fig. 5B in Milne 1998).
Other more detailed information is potentially contained in HX patterns. For example, the motion that exposes the Gly8-NH to exchange does not make the Gln12-NH exchange competent, even though its H-bond acceptor is Gly8-CO.
Diffusional penetration versus external exposure
When amide hydrogens are deprotected by H-bond breakage, do HX catalysts (OH− and H+) attack directly from the outside solvent or do they approach from inside after diffusing through the protein?
A variety of penetration models for protein HX were formulated in the 1970s when it was still conceived that well-protected hydrogens must be those that are well buried. Small distortional motions of various kinds were pictured to allow the internal diffusion of water molecules to access buried hydrogens (Rosenberg and Enberg 1969; Bryan 1970; Nakanishi et al. 1973; Lumry and Rosenberg 1975; Richards 1979; Tsuboi and Nakanishi 1979; Woodward and Hilton 1979; reviewed in Englander and Kallenbach 1983). Discussions favoring penetration mechanisms have often been phrased as a conflict between two limited alternatives (the two-state model), global unfolding and penetration (“exchange from the native state”). Indications of exchange that does not depend on global unfolding were then taken to support a penetration model.
One now knows that most slowly exchanging protein hydrogens are very near the aqueous surface. All of the helical H-bonded groups considered here are less than 2 Å from solvent water except for Phe10 (3 Å surface to surface, which is the chemically pertinent metric, not center of mass to center of mass). Lysines 7, 8, and 13 are on a solvent-exposed surface but they are slowed by factors over 104. In such cases penetration and internal diffusion have no meaning; there is no atom between the amide and solvent. The same is true for many other slowly exchanging protein hydrogens and even for small organic molecules (Haslam and Eyring 1967; Rose and Stuehr 1968; Luo and Baldwin 1997). For the more buried amides, the sizable stereochemical distortions necessary to produce HX competent H-bond separation must potentiate access to the nearby solvent. The exchange pattern seen for the exposed and buried hydrogens in the Cyt c N-terminal helix (Fig. 2 ▶) suggests some extra protection for the buried hydrogens rather than a whole new penetration-dependent pathway.
Deeply buried hydrogens seem better candidates for penetration-dependent exchange. However, the most deeply buried amide hydrogens in Cyt c, a typical globular protein (104 amino acids), are only removed from solvent by up to 4 Å (Milne et al. 1998). These are often “marker” protons that exchange by way of sizable unfolding reactions (Bai et al. 1995), and clearly bring buried amides out to the solvent.
Another discriminating indicator is the temperature dependence of exchange. HX is catalyzed by ionic species and not by simple contact with neutral water molecules. The buried amide NHs of Ile9 and Val11 exhibit ΔH°ex under 15 kcal/mole (after accounting for the temperature dependence of OH− concentration at constant pH; Milne et al. 1999). These low values are characteristic (Barksdale and Rosenberg 1982), and have often been attributed to a water penetration mechanism. However, one now knows that they are far smaller than one can expect for an ion penetration mechanism. The enthalpy for desolvation of hydroxide ion exceeds 100 kcal/mole (Marcus 1994). The enthalpy for breaking buried H-bonds to allow the chemical exchange event would increase this value. The alternative, that the catalyzing ion penetrates without desolvating, would require the massive ion–water complex to diffuse within the protein. For buried hydrogens such as Phe10-NH (2.9 Å from solvent), where H-bond breakage may not immediately expose the NH to solvent, a partial surface insertion/desolvation of the catalyzing OH− ion can be considered, which might explain the large ΔH°ex of 40 kcal/mole (Milne et al. 1999).
It is also pertinent that OH− ions and H3O+ ions must encounter the amide group over 102 and 1010 times on average, respectively, for one successful proton transfer event.
In summary, the facts of near surface location and small temperature dependence make catalyst ion attack from the nearby solvent seem much more likely than deep penetration, internal diffusion, and attack from the inside.
Stability calculation from HX
The glycine mutation used here, at the aqueous surface of Cyt c, destabilizes the protein. The measured destabilization largely reflects the glycine-dependent entropic stabilization of the U state and the loss of this conformational entropy when the glycine is held rigidly in the native helix. Other local contributions due to differential interactions between Lys8 and Gly8 with nearby protein regions seem unlikely, given the absence of changes in NMR chemical shift or HX rate, and the presence of 0.5 M KCl.
A discrepancy then arises. The apparent promotion of the residue 8 opening, 2.2 kcal/mole in free energy, is greater than the change in global destabilization (1.4 kcal/mole). The source of this discrepancy may be found in equations 1–4, which equate ΔG°ex with Kopkch and not just Kop as usually interpreted. A change in effective kch by fourfold between the Lys8 and Gly8 residues seems indicated, suggesting some partial blocking in the exchange competent condition, perhaps present in both molecules. It seems likely that the additional glycine-dependent flexibility allows more extensive solvent exposure, leading to the exaggerated ΔΔG°ex.
A number of studies have demonstrated the accuracy of the residue-dependent kint values in unfolded polypeptides (Roder et al. 1985; Robertson and Baldwin 1991; Bai et al. 1993; Buck et al. 1994; Parker et al. 1997), but their pertinence to local fluctuations of the native state, with less than totally free exposure, has been unclear. The present results raise the possibility that kint and the ΔG°ex values calculated from them may not be reliable for fluctuations in which the main chain is still constrained and hydrogens are still partially protected when exchange occurs. Some caution in drawing conclusions from such numbers seems warranted.
Summary: HX mechanism
The present results confirm a determining role for local fluctuational distortions in the exchange of amide hydrogens in one of the major helices of Cyt c. The fluctuational distortions appear to extend over several residues but expose to exchange only one residue NH at a time.
For local fluctuational HX, the literature suggests protection factors in the range of 105 in helices, less in Ω loops, and possibly more in beta sheets. Important determining factors may include the rigidity of helical structure, the flexibility of loops, and the fluidity of beta structures (Salemme 1981), and also the degree of burial. Unfolding-dependent exchange is mainly determined by the free energy of the unfolding in an EX2 case or by the activation free energy in an EX1 case. In the H-bond broken condition, residual blocking effects (Bai et al. 1995; Krantz et al. 2002) may come into play in either case, but are much more likely for constrained local fluctuations.
Pathways involving cooperative unfolding can dominate HX in less stable structure, or under conditions where unfolding reactions are favored, or when hydrogens are so firmly buried that competing local fluctuation pathways are particularly slow. Unfolding and local fluctuation pathways may be generally distinguished by the relative HX rates of sequence neighbors and by their very different sensitivity to destabilants. It is most interesting that unfolding reactions tend to involve entire cooperative units of native secondary structure (Rumbley et al. 2001; Englander et al. 2002).
We suggest that all known protein HX behavior can be accounted for by local fluctuation pathways and unfolding pathways, global and subglobal, all of which must break protecting H-bonds and expose hydrogens to attack by HX catalysts directly from the bulk solvent.
Materials and methods
Materials
D2O (99%) and guanidinium chloride (GdmCl) were from Isotec and ICN. All chemicals were analytical grade. Buffers used for equilibrium unfolding studies, in H2O unless otherwise noted and in D2O for NMR HX experiments, were 0.1 M phosphate at pH 5.0 to 8.4, with 0.5 M KCl to avoid electrostatic effects. Reported pH of D2O solutions (pH*) are uncorrected pH meter readings. Protein concentration was measured spectrophotometrically using an extinction coefficient of 106 mM−1 cm−1 at 409 nm. GdmCl concentration was determined from refractive index measurements (Nozaki 1972). All experiments were at 20°C.
The pWT Cyt c and the Lys8Gly mutant were expressed in a high yield Escherichia coli system and purified as described elsewhere (Rumbley et al. 2002).
Stability
GdmCl-induced unfolding was measured by circular dichroism (222 nm) and fluorescence simultaneously with an Aviv 202 CD spectrometer equipped with a thermoelectric cell holder, computer controlled syringe pump, and automatic recording. Fluorescence excitation was at 280 nm; emission was selected with a 320 nm cutoff filter.
The stability of pWT and Lys8Gly Cyt c at zero GdmCl was obtained by multiplying the melting midpoint (Cm) by the m value (4.5 kcal mol−1 M−1) determined previously from global unfolding measured by the native state HX experiment as a function of GdmCl concentration (Bai et al. 1995). This procedure was used because the usual linear extrapolation of Cyt c melting data (Pace 1975) underestimates the m value, and therefore the equilibrium stability, due to a significant population (∼18%) of partially folded intermediates in the melting transition zone (Mayne and Englander 2000).
HX by NMR
Hydrogen-deuterium exchange as a function of time was monitored by the time-dependent decrease in amide crosspeak volumes in two-dimensional homonuclear correlated 1H NMR spectra (pulsed field gradient COSY with two to four transients per increment) using a Varian 500 MHz Inova spectrometer. Typical acquisition used 1024 complex data points, 512 time increments, a spectral width of 20 ppm in both dimensions, and low power presaturation to suppress the residual HOD signal. Data analysis used the Felix package on a Silicon Graphics workstation. Appropriately scaled baseline footprints were subtracted from crosspeak volumes normalized to the Tyr97 cross peak. Data analysis and display were performed automatically with home-written software.
Exchange was initiated by moving protein from H2O into D2O exchange buffer by centrifugal gel filtration (Jeng and Englander 1991). Dead time of the experiment was less than 25 min. For more detail, see Bai et al. (1995), Xu et al. (1998), and Milne et al. (1999).
HX data analysis
The exchange rate of a freely exposed amide hydrogen is determined by a proton transfer reaction (Eigen 1964) in which the amide proton is transferred across an H-bond to a hydroxide ion (above pH ∼4; Berger and Linderstrøm-Lang 1957). The chemical exchange rate (kch) is therefore determined by pH and also by an intrinsic second-order rate constant (kint) that depends on temperature, isotope effects, and nearest-neighbor inductive and blocking effects calibrated in structureless models (equation 1) (Molday et al. 1972; Bai et al. 1993; Connelly et al. 1993).
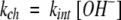 |
1 |
For OH−-ion catalyst to attack and H-bond to a labile hydrogen, preexisting structural H-bonds must first be separated (Linderstrøm-Lang 1958; Eigen 1964; Hvidt and Nielsen 1966; Englander and Kallenbach 1983). In the steady state, the overall exchange rate (kex) is given by equation 2, where kop and kcl are opening and reclosing rates of the protecting H-bond. (The word “open” in the present context is taken to mean HX competent.)
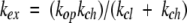 |
2 |
When reclosing is faster than chemical exchange (the EX2 bimolecular exchange condition), equation 2 reduces to equation 3. Measured exchange rate (kex) together with the known kch (equation 1 ) then provides Kop, and hence, Δ G°ex (equations 3, 4). When chemical exchange is faster than reclosing (the EX1 monomolecular exchange limit), the exchange rate is given by equation 5, allowing the measurement of structural opening rates (Hvidt and Nielsen 1966).
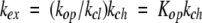 |
3 |
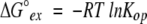 |
4 |
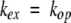 |
5 |
Acknowledgments
This work was supported by the NIH and the Mathers Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HX, hydrogen exchange
Cyt c, cytochrome c
pWT, pseudowild type
GdmCl, guanidinium chloride
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0225803.
References
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S. W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Genet. 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. Protein stability parameters measured by hydrogen exchange. Proteins Struct. Funct. Genet. 20 4–14. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barksdale, A.D. and Rosenberg, A. 1982. Acquisition and interpretation of hydrogen exchange data from peptides, polymers, and proteins. In Methods of biochemical analysis (ed. D. Glick), p. 113. John Wiley & Sons, New York, NY. [DOI] [PubMed]
- Berger, A. and Linderstrøm-Lang, K. 1957. Deuterium exchange of poly-D,L-alanine in aqueous solution. Arch. Biochem. Biophys. 69 106–118. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A.K. and Udgaonkar, J.B. 1998. Two structural subdomains of barstar detected by rapid mixing NMR measurement of amide hydrogen exchange. Proteins Struct. Funct. Genet. 30 295–308. [PubMed] [Google Scholar]
- Bryan, W.D. 1970. The mechanism of hydrogen exchange in proteins. Recent Prog. Surf. Sci. 3 101–120. [Google Scholar]
- Buck, M., Radford, S.E., and Dobson, C.M. 1994. Amide hydrogen exchange in a highly denatured state. Hen egg-white lysozyme in urea. J. Mol. Biol. 237 247–254. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K. and Marqusee, S. 1997. Touring the landscapes: Partially folded proteins examined by hydrogen exchange. Structure 5 859–863. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3 782–787. [DOI] [PubMed] [Google Scholar]
- Chu, R., Pei, W., Takei, J., and Bai, Y. 2002. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry 41 7998–8003. [DOI] [PubMed] [Google Scholar]
- Connelly, G.P., Bai, Y., Jeng, M.F., and Englander, S.W. 1993. Isotope effects in peptide group hydrogen exchange. Proteins Struct. Funct. Genet. 17 87–92. [DOI] [PubMed] [Google Scholar]
- Eigen, M. 1964. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Angew. Chem. Int. Ed. Eng. 3 1–19. [Google Scholar]
- Englander, S.W. and Kallenbach, N.R. 1983. Hydrogen exchange and structural dynamics of proteins and nucleic-acids. Q. Rev. Biophys. 16 521–655. [DOI] [PubMed] [Google Scholar]
- Englander, S.W., Mayne, L., and Rumbley, J. 2002. Submolecular cooperativity produces multi-state protein unfolding and refolding. Biophys. Chem. (in press). [DOI] [PubMed]
- Fuentes, E.J. and Wand, A.J. 1998. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry 37 9877–9883. [DOI] [PubMed] [Google Scholar]
- Haslam, J.L. and Eyring, E.M. 1967. Deuterium oxide solvent isotope effects on N-H. . .O, O-H. . .N, and N-H. . .N intramolecular hydrogen bonds. J. Phys. Chem. 71 4470–4475. [Google Scholar]
- Hiller, R., Zhou, Z.H., Adams, M.W., and Englander, S.W. 1997. Stability and dynamics in a hyperthermophilic protein with melting temperature close to 200°C. Proc. Natl. Acad. Sci. 94 11329–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser, V.J. and Freire, E. 1996. Structure-based calculation of the equilibrium folding pathway of proteins: Correlation with hydrogen-exchange protection factors. J. Mol. Biol. 262 756–772. [DOI] [PubMed] [Google Scholar]
- Hilton, B.D., Trudeau, K., and Woodward, C.K. 1981. Hydrogen exchange rates in pancreatic trypsin inhibitor are not correlated to thermal stability in urea. Biochemistry 20 4697–4703. [DOI] [PubMed] [Google Scholar]
- Hoang, L., Bédard, S., Krishna, M.M.G., Lin, Y., and Englander, S.W. 2002. Cytochrome c folding pathway: Kinetic native state hydrogen exchange. Proc. Natl. Acad. Sci. 99 12173–12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghues-Despointes, B.M., Pace, C.N., Englander, S.W., and Scholtz, J.M. 2001. Measuring the conformational stability of a protein by hydrogen exchange. Methods Mol. Biol. 168 69–92. [DOI] [PubMed] [Google Scholar]
- Hvidt, A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Jeng, M.-F. and Englander, S.W. 1991. Stable submolecular folding units in a non-compact form of cytochrome c. J. Mol. Biol. 221 1045–1061. [DOI] [PubMed] [Google Scholar]
- Kim, K.S. and Woodward, C. 1993. Protein internal flexibility and global stability: Effect of urea on hydrogen exchange rates of bovine pancreatic trypsin inhibitor. Biochemistry 32 9609–9613. [DOI] [PubMed] [Google Scholar]
- Krantz, B.A., Mayne, L., Rumbley, J.N., Englander, S.W., and Sosnick, T.R. 2002. Fast and slow intermediate accumulation and the initial barrier mechanism in protein folding. J. Mol. Biol. (in press). [DOI] [PubMed]
- Linderstrøm-Lang, K.U. 1958. Deuterium exchange and protein structure. In: Symposium on protein structure (ed. A. Neuberger). London: Methuen.
- Llinas, M., Gillespie, B., Dahlquist F.W., and Marqusee, S. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6 1072–1078. [DOI] [PubMed] [Google Scholar]
- Lumry, R. and Rosenberg, A. 1975. The mobile defect hypothesis of protein function. Col. Int. C.N.R.S. L’Eau Syst. Biol. 246 55–63. [Google Scholar]
- Luo, P. and Baldwin, R.L. 1997. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 36 8413–8421. [DOI] [PubMed] [Google Scholar]
- Marcus, Y. 1994. A simple empirical model describing the thermodynamics of hydration of ions of widely varying charges, sizes, and shapes. Biophys. Chem. 51 111–127. [Google Scholar]
- Mayne, L. and Englander, S.W. 2000. Two-state vs. multistate protein unfolding studied by optical melting and hydrogen exchange. Protein Sci. 9 1873–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo, S.L. and Baldwin, R.L. 1993. Guanidinium chloride induction of partial unfolding in amide proton exchange in RNase A. Science 262 873–876. [DOI] [PubMed] [Google Scholar]
- Miller, D.W. and Dill, K.A. 1995. A statistical mechanical model for hydrogen exchange in globular proteins. Protein Sci. 4 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, J.S., Mayne, L., Roder, H., Wand, A.J., and Englander, S.W. 1998. Determinants of protein hydrogen exchange studied in equine cytochrome c. Protein Sci. 7 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, J.S., Xu, Y., Mayne, L.C., and Englander, S.W. 1999. Experimental study of the protein folding landscape: Unfolding reactions in cytochrome c. J. Mol. Biol. 290 811–822. [DOI] [PubMed] [Google Scholar]
- Molday, R.S., Englander, S.W., and Kallen, R.G. 1972. Primary structure effects on peptide group hydrogen exchange. Biochemistry 11 150–158. [DOI] [PubMed] [Google Scholar]
- Nakanishi, M., Tsuboi, M., and Ikegami, A. 1973. Fluctuation of the lysozyme structure. II. Effects of temperature and binding of inhibitors. J. Mol. Biol. 75 673–682. [DOI] [PubMed] [Google Scholar]
- Nozaki, Y. 1972. The preparation of guanidinium chloride. Methods Enzymol. 26 43–50. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1975. The stability of globular proteins. CRC Crit. Rev. Biochem. 3 1–43. [DOI] [PubMed] [Google Scholar]
- Parker, M.J., Dempsey, C.E., Lorch, M., and Clarke, A.R. 1997. Acquisition of native beta-strand topology during the rapid collapse phase of protein folding. Biochemistry 36 13396–13405. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1979. Packing defects, cavities, volume fluctuations, and access to the interior of proteins, including some general comments on surface area and protein structure. Carlsberg Res. Commun. 44 47–63. [Google Scholar]
- Robertson, A.D. and Baldwin, R.L. 1991. Hydrogen exchange in thermally denatured ribonuclease A. Biochemistry 30 9907–9914. [DOI] [PubMed] [Google Scholar]
- Roder, H., Wagner, G., and Wuthrich, K. 1985. Individual amide proton exchange rates in thermally unfolded basic pancreatic trypsin inhibitor. Biochemistry 24 7407–7411. [DOI] [PubMed] [Google Scholar]
- Rose, M.C. and Stuehr, J. 1968. Kinetics of proton transfer reactions in aqueous solutions: Rates of internally hydrogen bonded systems. J. Am. Chem. Soc. 90 7205–7209. [Google Scholar]
- Rosenberg, A. and Enberg, J. 1969. Studies of hydrogen exchange in proteins. II. The reversible thermal unfolding of chymotrypsinogen A as studied by exchange kinetics. J Biol. Chem. 244 6153–6159. [PubMed] [Google Scholar]
- Rumbley, J.N., Hoang, L., Mayne, L., and Englander, S.W. 2001. An amino acid code for protein folding. Proc. Natl. Acad. Sci. 98 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbley, J.N., Hoang, L., and Englander, S.W. 2002. Recombinant equine cytochrome c in E. coli: High level expression, characterization, and folding and assembly mutants. Biochemistry (in press). [DOI] [PubMed]
- Salemme, F.R. 1981. Conformational and geometrical properties of beta-sheets in proteins. 3. Isotropically stressed configurations. J. Mol. Biol. 146 143–156. [DOI] [PubMed] [Google Scholar]
- Scholtz, J.M. and Robertson, A.D. 1995. Hydrogen exchange techniques. Methods Mol. Biol. 40 291–311. [DOI] [PubMed] [Google Scholar]
- Serrano, L., Neira, J.L., Sancho, J., and Fersht, A.R. 1992. Effect of alanine versus glycine in alpha-helices on protein stability. Nature 356 453–455. [DOI] [PubMed] [Google Scholar]
- Srivastava, A.K., and Sauer, R.T. 2000. Evidence for partial secondary structure formation in the transition state for arc repressor refolding and dimerization. Biochemistry 39 8308–8314. [DOI] [PubMed] [Google Scholar]
- Tsuboi, M. and Nakanishi, M. 1979. Overall and localized fluctuation in the structure of a protein molecule. Adv. Biophys. 12 101–130. [PubMed] [Google Scholar]
- Wagner, G. and Wuthrich, K. 1982. Amide proton exchange and surface conformation of BPTI in solution: Studies with 2D NMR. J. Mol. Biol. 160 343–361. [DOI] [PubMed] [Google Scholar]
- Wand, A.J. and Englander, S.W. 1996. Protein complexes studied by NMR spectroscopy. Curr. Opin. Biotechnol. 7 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. and Case, D.A. 2001. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 338 3–34. [DOI] [PubMed] [Google Scholar]
- Woodward, C.K. 1994. Hydrogen exchange rates and protein folding. Curr. Opin. Struct. Biol. 4 112–116. [DOI] [PubMed] [Google Scholar]
- Woodward, C.K. and Hilton, B.D. 1979. Hydrogen exchange kinetics and internal motions in proteins and nucleic acids. Annu. Rev. Biophys. Bioeng. 8 99–127. [DOI] [PubMed] [Google Scholar]
- Wooll, J.O., Wrabl, J.O., and Hilser, V.J. 2000. Ensemble modulation as an origin of denaturant-independent hydrogen exchange in proteins. J. Mol. Biol. 301 247–256. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Mayne, L., and Englander, S.W. 1998. Evidence for an unfolding and refolding pathway in cytochrome c. Nat. Struct. Biol. 5 774–778. [DOI] [PubMed] [Google Scholar]