

Abstract
Long insertions into a loop of a folded host protein are expected to have destabilizing effects because of the entropic cost associated with loop closure unless the inserted sequence adopts a folded structure with amino- and carboxy-termini in close proximity. A loop entropy reduction screen based on this concept was used in an attempt to retrieve folded sequences from random sequence libraries. A library of long random sequences was inserted into a loop of the SH2 domain, displayed on the surface of M13 phage, and the inserted sequences that did not disrupt SH2 function were retrieved by panning using beads coated with a phosphotyrosine containing SH2 peptide ligand. Two sequences of a library of 2 × 108 sequences were isolated after multiple rounds of panning, and were found to have recovery levels similar to the wild-type SH2 domain and to be relatively intolerant to further mutation in PCR mutagenesis experiments. Surprisingly, although these inserted sequences exhibited little nonrandom structure, they do not significantly destabilize the host SH2 domain. Additional insertion variants recovered at lower levels in the panning experiments were also found to have a minimal effect on the stability and peptide-binding function of the SH2 domain. The additional level of selection present in the panning experiments is likely to involve in vivo folding and assembly, as there was a rough correlation between recovery levels in the phage-panning experiments and protein solubility. The finding that loop insertions of 60–80 amino acids have minimal effects on SH2 domain stability suggests that the free energy cost of inserting long loops may be considerably less than polymer theory estimates based on the entropic cost of loop closure, and, hence, that loop insertion may have provided an evolutionary route to multidomain protein structures.
Keywords: Loopentropy, random sequences, phage display
Whereas the majority of multidomain proteins are formed via end-to-end linkages of domains, 28% of domains are discontinuous, suggesting that they may have evolved through the insertion of one domain into a loop of another domain (Jones et al. 1998). Specific examples include dsbA and the Escherichia coli DNA polymerase I (Russell 1994). Evolutionary benefits to forming multidomain proteins via loop insertions as opposed to end-to-end linkages would include stronger coupling between domains and increased rigidity, as the domains are linked via two connections as opposed to one, promoting allosteric interactions between the two domains.
For loop insertion to be a viable evolutionary route to new proteins, it is necessary that the parent domain retains stability and function after the insertion event. Recent experiments have shown that insertions of folded domains into surface loops are generally accepted with a minimal effect on the parent domain’s activity. For example, insertions of either dihydrofolate reductase (DHFR) or β-lactamase into four surface loops in phosphoglycerate kinase (PGK) were shown to have only a small effect on PGK’s activity (Collinet et al. 2000), and the maltodextrin-binding protein retained activity upon insertion of β-lactamase into two of the three surface loops examined (Betton et al. 1997).
From an evolutionary point of view, however, it may be more relevant to examine how insertions of unstructured sequences affect the stability of parent domains, because the insertion of incomplete domains or relatively unstructured segments is a more likely event than the insertion of intact domains. Theoretical studies have examined this question using a simple polymer model in which the inserted residues are treated as an increase in loop length, resulting in an increase in the entropic cost of loop closure (Chan and Dill 1988). In this model, the change in configurational entropy of loop closure and the corresponding change in the free energy of folding are a function of the probability that the two ends of the loop will be close in space and is given by
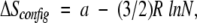 |
1 |
in which a is related to the distance between the loop ends required for closure to occur (between −2 and −8 cal mole−1K−1) and N is the number of residues in the loop.
Experimental studies on the effect of loop length on protein stability have matched well with theory. Insertions of up to 10 glycine residues in the 4-helix bundle protein Rop resulted in an average free-energy loss of 0.26 kcal mole−1 per glycine residue (Nagi and Regan 1997). Additionally, insertion studies on CI2 (Ladurner and Fersht 1997) and α-spectrin SH3 (Viguera and Serrano 1997) indicate a 0.1 kcal mole−1 loss in free energy per residue added to an existing loop. These studies also found that the insertion of the first few residues contributed more to the overall loss in free energy than later insertions as expected, given the logarithmic dependence of the loop entropy on loop length.
Both Equation 1 and extrapolation of the experimental results suggest that the insertion of 80–100 unstructured amino acids, the size of a small domain, would result in a destabilization of the parent domain on the order of 4–6 kcal mole−1. Because this decrease in free energy is of the same order as the free energy of folding of many small, single-domain proteins, such insertions could disrupt the folding and activity of the parent domain. Given this observation, is it plausible that domain insertion is an evolutionary mechanism for the creation of novel folds and multidomain proteins?
Here, a library of 2 × 108 random sequences is inserted into a loop of a host protein, the lck SH2 domain, in a loop entropy reduction screen to identify folded proteins. The basic premise of the screen is that insertion of long disordered sequences into a loop of a host protein will substantially destabilize the host because of the entropic cost of closing a loop in a disordered chain. If the inserted sequence folds spontaneously into a stable structure with its amino- and carboxy-termini close in space, however, this entropic cost is diminished. The host protein function can, therefore, be used to select folded inserted sequences without relying on specific properties of the inserted sequence. We find, however, that the insertion of long sequences into the loop of an existing domain is not as inherently disruptive as thought previously. Many sequences are compatible with in vitro folding and binding activity, but only a very small fraction of long inserts appear to be compatible with in vivo folding and function. The data suggest that the primary requirement of the inserted sequence for retention of function in vivo is that it cannot negatively impact the solubility of the created chimeric protein.
Results
The lck SH2 domain was used as a host protein for the loop entropy reduction screen because it was determined previously that the selection method was able to discriminate between a folded SH3 domain and an unfolded SH3 domain inserted in a surface loop of SH2 (see Fig. 1 ▶ for insertion point; Minard et al. 2001). In the design of the random sequences to be inserted into the SH2 domain, several factors were considered. First, to ensure the presence of full-length sequences, it was imperative that the random sequences lack stop codons. Second, to avert possible complications of the loop entropy screen by the presence of disulfide bonds, cysteine residues were avoided. Third, with the exception of cysteine residues, the amino acid percentages of the random sequences were designed to be similar to those found in naturally occurring proteins. Fourth, in order to avoid any unintentional patterning in the sequences resulting from codon positioning, the library was constructed from multiple randomized codons. To accommodate these concerns, three randomized codons, RNN, NNG, and NHY (R = A, G; H = A, C, T; Y = C, T; and N = A, C, T, G), were chosen. The selected codons lacked nonsuppressible stop codons and, when equally represented in the library, result in amino acid percentages similar to those found in small, single-domain proteins in the PDB (Target %, Table 1).
Figure 1.

Structure of the lck SH2 domain (Eck et al. 1993). The surface loop in SH2 used for insertion is indicated with an arrow. Images were made using the Raster 3D (Bacon and Anderson 1988; Merrit and Murphy 1994) and Molscript programs (Kraulis 1991).
Table 1.
Design and target amino acid percentages for random sequence library
| Amino acid | RNN (%) | NHY (%) | NNG (%) | Combined (%) | Target (%) | Difference (%) |
| Ala | 12.5 | 8.25 | 6.3 | 9 | 8.5 | 0.5 |
| Arg | 6.3 | 0 | 12.5 | 6.3 | 4.6 | 1.7 |
| Asn | 6.3 | 8.3 | 0 | 4.8 | 4.7 | 0.1 |
| Asp | 6.3 | 8.3 | 0 | 4.8 | 6.0 | −1.2 |
| Cys | 0 | 0 | 0 | 0 | 1.5 | −1.5 |
| Gln | 0 | 0 | 12.5 | 4.2 | 6.0 | −1.8 |
| Glu | 6.3 | 0 | 6.3 | 4.2 | 3.9 | −0.3 |
| Gly | 12.5 | 0 | 6.3 | 6.3 | 7.8 | −1.5 |
| His | 0 | 8.3 | 0 | 2.8 | 2.3 | 0.5 |
| Ile | 9.4 | 8.3 | 0 | 5.9 | 5.7 | 0.2 |
| Leu | 0 | 8.3 | 12.5 | 6.9 | 8.1 | −1.2 |
| Lys | 6.3 | 0 | 6.3 | 4.2 | 5.7 | −1.5 |
| Met | 3.1 | 0 | 6.3 | 3.2 | 2.1 | 1.1 |
| Phe | 0 | 8.3 | 0 | 2.8 | 4.0 | −1.2 |
| Pro | 0 | 8.3 | 6.3 | 4.8 | 4.7 | 0.1 |
| Ser | 6.3 | 8.3 | 6.3 | 6.9 | 6.1 | 0.8 |
| Thr | 12.5 | 8.3 | 6.25 | 9.0 | 5.8 | 3.2 |
| Trp | 0 | 0 | 6.3 | 2.1 | 1.5 | 0.6 |
| Tyr | 0 | 8.3 | 0 | 2.8 | 3.9 | −1.1 |
| Val | 12.5 | 8.3 | 6.3 | 9 | 6.9 | 2.1 |
| Nonpolar (AILMFWV) | 37.6 | 14.2 | 37.5 | 38.8 | 36.7 | 2.1 |
| Acidic (DE) | 12.5 | 8.3 | 6.3 | 9 | 9.9 | −0.9 |
| Basic (HKR) | 12.5 | 8.3 | 18.8 | 13.2 | 12.6 | 0.6 |
| Polar (DEHKNQR) | 31.3 | 24.8 | 37.5 | 31.2 | 33.1 | −1.9 |
Long sequences of the randomized codons were constructed and inserted into the SH2 domain to yield a library with a complexity of 2 × 108 (see Materials and Methods). Sequencing of library members showed that ∼50% of the inserted sequences were full length. Out-of-frame sequences typically contained a nonsuppressible stop codon, effectively removing them from the library, reducing the actual complexity of the library to ∼1 × 108. For sequences lacking insertions/deletions, the actual amino acid percentages were found to be similar to the design scheme, confirming that the design of the random sequences was maintained during library construction.
In an attempt to isolate folded inserted sequences, the library was displayed on the surface of phage and panned against paramagnetic beads coated with the SH2 ligand, a phosphotyrosyl peptide, using previously described methods (Gu et al. 1995; Minard et al. 2001). Ideally, phage displaying folded SH2 host domains with minimally disruptive inserted sequences should be isolated on the basis of their ability to bind the SH2 ligand. Binding of the phage was performed under two conditions: (1) a short, 2-h incubation at room temperature, and (2) a long, overnight incubation at 4°C. To enrich the number of positives, multiple rounds of panning were performed in which the bound phage from the previous round were amplified and used as the starting material for the next round of panning for each condition. The phage recovery levels after each round of panning are shown in Table 2. The stringency of the panning procedure was increased for each successive panning round by increasing the number of wash steps (round 1, 3 washes; round 2, 4 washes; round 3, 6 washes), resulting in lower recovery levels for round 3.
Table 2.
Library recovery levels on phosphotyrosyl peptide coated beads
| Precent of phage recovered on phosphotyrosyl peptide-coated beads | |||
| Round 1 | Round 2 | Round 3 | |
| 2-h incubation | |||
| Library | 3.5 × 10−5 | 8.0 × 10−4 | 2.0 × 10−5 |
| Backgrounda | 3.6 × 10−6 | 5.8 × 10−5 | 2.0 × 10−6 |
| ON incubation | |||
| Library | 1.0 × 10−4 | 2.0 × 10−3 | 2.0 × 10−4 |
| Background | 3.2 × 10−6 | 1.2 × 10−6 | 1.4 × 10−6 |
a Background recovery was taken to be recovery levels of phage displaying the protein L domain.
Sequence properties of the selected phage after each round of panning are summarized in Table 3. Overall, it was found that short inserted sequences out-competed longer sequences. Specifically, the percentage of long sequences, 60–120 amino acids, decreased from 93% to 45% in the 2-h incubation condition and to 33% in the overnight incubation condition by the third round of panning. Consequently, the number of short sequences, primarily the sequence resulting from the ligation of the two end cassettes used for library construction (see Materials and Methods), greatly increased by the third round of panning. It is not surprising that shorter inserted sequences out-compete longer sequences, as the entropic cost of loop closure is reduced.
Table 3.
Sequence charaacteristics of selected library members
| 2-hr incubation | ON incubation | |||||
| Among long sequences | Among long sequences | |||||
| Panning round | % long sequences (60–120 aa) | % inframe | % 283 | % long sequences (60–120 aa) | % inframe | % 283 |
| 0 | 93 | 54 | 0 | 93 | 54 | 0 |
| 1 | 50 | 75 | 0 | 57 | 25 | 0 |
| 2 | 34 | 82 | 41 | 19 | 50 | 33 |
| 3 | 42 | 75 | 42 | 33 | 91 | 64 |
However, the shorter sequences did not completely dominate the selection sequences, and sufficient numbers of long sequences were present for analysis. Among long insertions, sequencing indicated that the percentage of in-frame sequences increased from 54% to 75% in the 2-h incubation condition and to 95% in the overnight incubation condition by the third round of panning, as would be expected if the selection procedure was working correctly. Interestingly, one inserted sequence, 283, was sequenced independently multiple times, and by the third round of panning, represented a significant portion of the long sequences (see Table 4 for sequence information).
Table 4.
Inserted sequences chosen for biophysical characterization
| Seq. no. | Sequence, including linker region resulting from end cassettes |
| 217 | GSGSGSKDLSSFSVINDTKVHMYAGSNHMNRSIKSYDLLMNqLNGSDPLSPTHAKSTqRIAGPKGSGSGS |
| 227 | GSGSGSNDRETLTLIFMARALQLIGSIFPETSTMIVKqKMSTLTGSVVKSMYPWTFGSqINKPKGSGSGS |
| 283 | GSGSGSAPEAMYAPDAVKMVHEFGGSNYTGMTIKMSTSLTVTDEGSSLTTQPHAENTRRDDQNIGSRFAGESHVNNTTKTTKLEGSGSGS |
| 290 | GCGSGSGTGGLTNLEHSSPTNMNSGSAVKTEFFTAHDQTINRATGSGDANGNNLNFGLKAPTAEGSGSGS |
| 333 | GSGSGSDSQGLHLAGTAKRDNSVTGSNTQSQPFTVNRLEMHSHMGSAPLEAAPLGVTMQTIRTTGSILRTLDAWGYSKGAAMLAGSDTLNGSFKDVNKEALVLRGSGSGS |
| 344 | GSGSGSNSTIQSVqTDGPEAHPIIGSTYTTPPTTVNDMKAFPNMGSELQNTAHMKHTPLTPQHNGSGSGS |
| 425 | GSGSGSNIKKLPVLRTPSLVHRFTGSGSWTEDSMTFSQRDHTLIGSVITGPSSQATDGPRNTTTGSGSGS |
Because of the large number of selected library members, a method was devised to identify the most promising sequences for further characterization. All long in-frame inserts identified by sequencing bound phage after each panning round were pooled together, amplified using error-prone PCR (Cadwell and Joyce 1994) to introduce variation, and cloned back into the SH2 host protein. A total of 31 parent sequences were pooled with the exclusion of 283 to avoid it from once again dominating the selection. The resulting library of mutated positives had a complexity of 5 × 107. Sampling of the library members indicated that, on average, each sequence contained 1.5 mutations. The library was then displayed on the surface of phage, subjected to multiple rounds of panning, and retained phage sequenced after each round of panning to identify sequences that dominated the selection procedure. It was found that, by the third and fourth rounds of panning, only two sequences remained, 290 and 425; 290 represented 69% and 80% of the third and fourth round sequences, respectively, whereas 425 represented 31% and 20% of the third and fourth round sequences, respectively (see Table 4 for sequences.). Sequencing indicated that the number of amino acid mutations present in the 290 and 425 inserts decreased significantly by the final round of panning; after rounds 1 and 2, 290 and 425 contained, on average, 1.3 and 1.6 mutations, respectively, whereas after round 4, they contained, on average, 0.4 and 0.5 mutations, respectively. Surprisingly, this result suggests that the 290 and 425 wild-type sequences were already, to some extent, optimal for insertion into the SH2 domain without loss of function.
Because of their significant presence in the selection procedures, 283, 290, and 425 were chosen for further characterization. Two additional sequences identified in the original selection procedure, 333 and 344, were also chosen; 333 because of its length and high hydrophobicity, and 344 because, next to 290 and 425, it was the third most represented sequence in the first and second rounds of panning of the error-prone library. To serve as negative controls, two sequences from the library that had not undergone any selection, 217 and 227, were chosen for characterization.
Phage displaying each of the inserted sequences in the SH2 host domain were made and tested for their ability to independently bind beads coated with phosphotyrosyl peptide. Table 5 displays the binding recovery relative to background binding levels. As expected, both the 290 and the 425 insertions result in very high recovery levels. The remaining inserts, including 283, were recovered at levels similar to background. It is surprising that the 283 insertion, given its dominance in the original selection procedure, is only recovered slightly above background levels. Subtle deviations in phage production for single sequences versus libraries of sequences may result in varying levels of expression on the surface of phage, explaining the observed discrepancy.
Table 5.
Summary of sequence and biophysical characteristics of inserted sequences
| Inserted sequence no. | Insert size (aa) | Insert recovery level (fold over backgrounda) | % Hydrophobic residues (inserted seq) | % Polar residues (inserted seq) | Expression level | Pellet/SN | ΔGunf (kcal/mole) | ΔΔGunf (kcal/mole) | SPR binding |
| none | 0 | 280 | — | — | high | 10/90 | 1.5 | — | ++ |
| 217 | 60 | 9 | 24.1 | 36.1 | high | 80/20 | 0.8 | −0.7 | ++ |
| 227 | 60 | 3 | 36.2 | 25.7 | high | 80/20 | NMb | NM | − |
| 283 | 80 | 5 | 19.2 | 34.5 | high | 80/20 | 1.7 | +0.2 | + |
| 290 | 60 | 3700 | 17.2 | 32.6 | high | 50/50 | 0.9 | −0.6 | + |
| 333 | 100 | 3 | 28.2 | 28.0 | high | 80/20 | 0.7 | −0.8 | + |
| 344 | 60 | 9 | 18.9 | 32.6 | high | 50/50 | — | — | NM |
| 425 | 60 | 770 | 24.1 | 27.4 | high | 50/50 | 1.3 | −0.2 | + |
a Background recovery was taken to be recovery levels of phage displaying the protein L domain.
b Not measured.
The SH2 insertion variants were cloned into an expression vector to allow for overexpression and protein purification (see Materials and Methods). The wild-type SH2 domain, as well as the insertion variants, was expressed at very high levels. All of the fusion proteins displayed varying degrees of insolubility when overexpressed in bacterial cytosol; wild-type SH2, SH2[290], SH2[344], and SH2[425] were equally distributed between the soluble and insoluble fractions, whereas SH2[217], SH2[227], SH2[283], and SH2[333] were found predominantly in the insoluble fraction. Notably, the two sequences with the highest retention levels in the phage-panning experiments were also among the most soluble proteins in the expression studies.
For four of the five insertion variants examined, the circular dichroism spectra, a monitor of the amount of secondary structure present, resembles that of wild-type SH2 with slight variances, indicating either a decrease or increase in structural content (Fig. 2 ▶). The spectra for SH2[217] and SH2[425] are similar to that of wild-type SH2 except for the presence of a second minimum near 220 nm, suggestive of an increase in α helical content. Conversely, the minima for the SH2[283] and SH2[290] spectra are shifted from 208 nm to a lower wavelength, suggestive of an increase in random coil content. The spectrum of SH2[344] in small amounts of denaturant (500 mM guanidine) resembled that of a random coil, indicating the protein is unfolded in near-native conditions (data not shown). Due to aggregation problems in the absence of denaturants, the CD spectra of SH2[227] and SH2[333] could not be measured.
Figure 2.

CD spectra of SH2 insertion variants. Spectra of SH2 (•), SH2[217] (○), SH2[283] (▵), SH2[290] (□), and SH2[425] (crosses) were taken in 50 mM sodium phosphate (pH 7) and 500 uM β-mercaptoethanol. Protein concentrations were 5.5 ± 1.2 μM. At these protein concentrations, the molar ellipticity was not affected by protein concentration, indicating that aggregation was not occurring.
To examine the extent of structure present in the inserted sequences, 283, 290, and 425 were studied in isolation. Expression studies showed that the sequences were highly expressed and present predominantly in the soluble fraction. CD spectra of the purified proteins are shown in Figure 3 ▶. The spectra closely resemble that of a random coil with a primary minimum below 210 nm. A smaller, secondary minimum is present near 220 nm, suggesting a small degree of α helical content. The addition of stabilizing agents, such as sodium sulfate, did not enhance the structural content of the sequences. Also, although the amplitude of the CD signal at 222 nm decreased with the addition of denaturants, no cooperative folding/unfolding transition was observed (data not shown). Therefore, it appears that the sequences in isolation contain little, if any, persistent secondary structure. Additionally, subtraction of the CD spectra of the isolated sequences from the spectra of the SH2 fused sequences closely resembled the wild-type SH2 spectra, indicating that the sequences did not gain significant secondary structure in the fusion protein (data not shown).
Figure 3.

CD spectra of 283, 290, and 425 sequences in isolation. Spectra of 283 (▵), 290 (□), and 425 (crosses) were taken in 100 mM sodium phosphate (pH 7), 50 mM NaCl and 120, 20, and 250 mM guanidine, respectively. Protein concentrations were 20 ± 0.4 μM.
For five of the six sequences examined, insertion did not result in a dramatic reduction in SH2 stability as monitored by guanidine denaturation experiments (Fig. 4 ▶; Table 5; see Materials and Methods). Both the 425 and 283 insertions had little effect on SH2 stability; ΔΔGunf = −0.2 and +0.2 kcal mole−1, respectively, whereas the 217, 290, and 333 inserts decreased the stability of SH2 by roughly half; ΔΔGunf = −0.7, −0.6, −0.8 kcal mole−1, respectively. As expected from the CD spectra, only the insertion of 344 resulted in a complete loss of SH2 stability. It is interesting that the 290 insertion results in a similar decrease in SH2 stability as the 217 and 333 inserts, given its large recovery levels observed in the phage-panning experiments. A possible explanation for this discrepancy may lie in the presence of a serine |Zy cysteine point mutation in the SH2[290] amino-terminal linker sequence (Table 4). During phage production, the linker cysteine residue may be forming a disulfide bond with the cysteine in SH2 at position 217, which is reasonably close in space, whereas during over expression and purification, the interaction may be lost. Preliminary evidence supports the importance of the serine |Zy cysteine mutation; reversion of the cysteine residue back to a serine results in a dramatic reduction of phage-panning recovery levels close to background (data not shown).
Figure 4.

Guanidine titration melts of SH2 insertion variants. Denaturation curves of SH2 (•), SH2[217] (○), SH2[283] (▵), SH2[290] (□), SH2[333] (⋄), SH2[344] (♦), and SH2[425] (crosses) were monitored at 222 nm in 100 mM sodium phosphate (pH 7), 50 mM NaCl, and 500 uM β-mercaptoethanol. A denaturation melt of SH2[227] could not be measured due to extensive aggregation problems. The data were fit as described in Scalley et al (1997). Protein concentrations were 8 ± 0.2 μM for SH2 and 5 ± 0.2 μM for other proteins.
It was interesting that, whereas most insertions into SH2 had a relatively small effect on SH2 stability, all of the SH2 insertion variants, except SH2[290] and SH2[425], had low recovery levels in the phage-panning experiments. To probe this apparent contradiction further, we utilized surface plasmon resonance (SPR) techniques to provide an alternate measure of SH2 binding activity. Varying amounts of purified protein were injected over a streptavidin-coated chip onto which the biotinylated phosphotyrosyl peptide had been immobilized previously (see Materials and Methods). An example of the raw data and subsequent data analysis is shown for wild-type SH2 in Figure 5 ▶, A and B. SH2[227], the aggregation-prone insertion variant, was the only protein that exhibited no binding to the phosphotyrosyl peptide. The binding behavior of SH2[217], SH2[333], and SH2[425] was found to be similar to that of wild-type SH2 with an increased binding affinity for SH2[217] and a slightly decreased binding affinity for SH2[333] and SH2[425]. SH2[283] and SH2[290] are not shown in Figure 5B ▶, because their binding was multiphasic with a slow second-binding phase, precluding similar data analysis. The SPR data supports the conclusion that SH2[217], SH2[283], SH2[290], SH2[333], and SH2[425] are folded and functional. The discrepancy between binding activities measured with either SPR or phage panning may indicate an additional level of selection present in the phage experiments. In particular, insertion variants that are folded and functional in vitro may not be capable of folding and assembling onto the surface of phage, perhaps as a result of a propensity to aggregate.
Figure 5.

(A) Raw data of SH2 binding to the phosphotyrosyl peptide from SPR experiments. Increasing amounts of the SH2 domain were injected over the immobilized phosphotyrosyl peptide chips. The arrows indicate the beginning and end of the injection. The signal from a reference cell with only biotin bound to the chip was subtracted from the raw data. (B) Binding curves of SH2 and SH2 insertion variants. SH2 (•), SH2[217] (○), SH2[333] (⋄), and SH2[425] (crosses). The equilibrium value taken from the raw data, Req, was plotted as a function of protein concentration. The solid line represents the fit of the data to the following equation: Req = Rmax ([SH2]/KD-[SH2]), in which Rmax is the signal observed upon maximum binding and KD is the dissociation constant. The plots were normalized to a Rmax of 100 response units (RU).
Discussion
The results of the experiments described in this work indicate that the loop entropy reduction screen is not effective at isolating folded random sequences. Whereas the 290 and 425 insertions were highly recovered during phage panning, the isolated sequences contained minimal structural features. Interestingly, both of these insertion variants, SH2[290] and SH2[425], were found to be the most soluble in the expression studies. Also, it was found that a number of the SH2 insertion variants that lacked functionality in the phage experiments displayed near wild-type binding activity levels in in vitro SPR-binding assays. Together, these results suggest that the insertion of the unstructured sequences studied here do not greatly affect the in vitro folding and activity of the SH2 domain. The phage-panning experiments, in contrast, indicate that the insertions almost universally impair in vivo folding and assembly. Only two insert sequences of the 2 × 108 screened were recovered at the level of wild-type SH2, and these two sequences were surprisingly intolerant of amino acid replacement in the PCR mutagenesis experiments. It is likely that problems with in vivo folding and assembly are, at least in part, related to a decrease in solubility, as there was a rough correlation between in vivo recovery and in vitro solubility.
How are these results reconciled with the previous study indicating that the loop entropy reduction-phage display screen, utilizing the SH2 domain as a host protein, was capable of discriminating between folded and unfolded inserts (Minard et al. 2001)? The efficacy of the loop entropy reduction screen, by use of a folded and strongly destabilized SH3 domain as models for folded and unfolded inserted sequences, respectively, was only tested for phage-panning experiments, in which the insertion of the destabilized SH3 domain resulted in a dramatic reduction in phage recovery levels. Therefore, given the finding in this work that the phage-panning experiments are more stringent than in vitro folding and activity requirements, the results presented here are consistent with the earlier, proof-of-concept experiments.
It is unexpected that extending a loop by the insertion of long random sequences does not result in a large disruption of folding and activity. CD experiments performed on three of the isolated sequences, 283, 290, and 425, indicated that they contained minimal secondary structure. Both theory and experiment predict that unstructured insertions lead to a large increase in configuration entropy of folding and, thus, a significant loss in the free energy of folding. Using Equation 1, an insertion of 80–100 amino acids would result in a free energy loss on the order of 4–6 kcal mole−1, but the energetic cost cannot exceed the free energy loss upon formation of a complex between two independent chains, which has been estimated to be between 3 and 10 kcal mole−1 (Brady and Sharp 1997). Given this observation, it is striking that only one of the insertions examined here results in a complete loss of stability, whereas the other inserts result in only a 0.2–0.8 kcal mole−1 loss in free energy. Because the effects of the insertions surely depend on the host protein, the generality of this observation is difficult to ascertain. A similar result was seen, however, in at least one other case, Doi and coworkers probed the sensitivity of RNase HI to insertion of random sequences of 120–130 amino acids into a surface loop and found that ∼10% of the insertion variants retained activity, despite an apparent lack of structure in the inserted sequences (Doi et al. 1997).
Why do the inserted sequences have a smaller effect on stability than expected? An explanation may be that the inserted sequences are forming stabilizing interactions with surface residues on the host protein. In this scenario, the putative stabilizing interactions formed with the surface will counteract the entropic cost of loop closure, allowing the host protein to remain folded and functional. Alternatively, the inserted sequences may undergo a partial hydrophobic collapse with little concurrent secondary structure formation. This collapse would restrict the conformational space available to the inserted sequence, thereby reducing the entropic cost of loop closure. Additionally, such behavior could explain how the soluble, isolated sequences, 283, 290, and 425, were able to avoid proteolysis in expression studies.
The results of this study are relevant to the evolution of multidomain protein structures. We have found that insertion of long sequences into the loop of an existing domain may not be as inherently disruptive as thought previously. The primary requirement that the inserted sequence must meet for retention of the host protein’s function appears to be that its insertion does not promote insolubility and aggregation. This requirement is fairly difficult to achieve in the case of insertion into SH2, as only two of 2 × 108 sequences were recovered at high levels and the sequences of the two positives were intolerant of substitutions introduced by PCR mutagenesis. However, given that an inserted sequence does not negatively affect solubility, insertion can provide a neutral environment for the inserted sequence to evolve into a compact and folded structure, allowing for the evolution of a multidomain protein.
Materials and methods
Construction and panning of random sequence library
The random sequence library was constructed by the self-ligation of a highly degenerate cassette: 5′-agatctRNNNHYNNGRNNNN GNHYNHYNNGRNNNHYRNNNNGNNGRNN
NHYNNGNHYRNNggatcc-3′, in which R = A, G; H = A, C, T; N = A, C, T, G; and Y = C, T; and AGATCT and GGATCC are BglII and BamHI restriction sites, respectively. BamHI and BglII restriction sites were chosen because they produce identical 5′-overhangs and the ligation of BamHI and BglII ends destroys both restriction sites. The cassettes were, therefore, ligated in a directional manner by the presence of T4 ligase (10 U/μg DNA, NEB), BamHI (150 U/μg DNA) and BglII (250 U/μg DNA). The self-ligation of the random cassettes was limited by the presence of two end cassettes; a start cassette with the sequence 5′-gtcgacggtagcggctcaggatcc-3′, containing a BamHI site at the 3′ end and introducing a SalI site at the 5′ end (underlined), and a stop cassette with the sequence 5′-agatctgggtcgggaagcggtacc-3′, containing a BglII site at the 5′ end and introducing a KpnI site at the 3′ end (underlined). The end cassettes also introduce a (Gly–Ser)3 repeat to serve as a flexible linker between the random sequences and the SH2 domain, alleviating possible strain incurred due to insertion of a folded domain. Ligation products of the desired length (3–6 randomized cassettes yielding sequences of 180–360 bp) were purified using acrylamide electrophoresis and cloned into the SH2 phagemid vector via the SalI and KpnI restriction sites. The ligation products were plated on agar containing carbenicillin, tetracycline, and 1% glucose, and grown overnight at 37°C. Library phage were prepared as discussed below.
Phage panning was performed as described previously (Minard et al. 2001). The biotinylated-phosphotyrosyl peptide used in the panning experiments was synthesized by SigmaGenosys with the following sequence: (GS)7GEPQ[pY]EE. The bound phage were infected into Xl1blue E. coli strain (Stratagene) and plated on agar containing carbenicillin, tetracycline, and 1% glucose. After overnight growth at 37°C, the cells were harvested and stored as a bacterial stock at −80°C. For amplification, ∼108 cells from the bacterial stock were added to 25 mL of LB containing carbenicillin, tetracycline, and 1% glucose. After 2 h of growth at 30°C, the cells were spun down and resuspended in 25 mL of fresh LB solution, and, to induce phage assembly, 50 μL of M13 helper phage (1011 cfu/mL, NEB). The cultures were grown overnight at 30°C and the phage particles were purified using two successive PEG-NaCl precipitations. The phage particles were resuspended in 10 mM Tris-HCl, 1 mM EDTA, and 100 mM NaCl (STE), and stored at −80°C.
Expression, purification, and circular dichroism experiments
To allow for overexpression, all sequences were cloned into the pet29b expression system (Novagen). The methods described by Gu et al (1995) were used for the expression and purification of the proteins, with the exception that bacterial growth was conducted at 30°C, as opposed to 37°C. The wild-type SH2 sequence was taken to be that used in previously published experiments (Minard et al. 2001). Circular dichroism spectra and equilibrium denaturation experiments were performed as described previously (Scalley et al. 1997).
Surface plasmon resonance-binding experiments
SPR experiments were performed on a Biacore 2000 instrument using methods similar to those described in Panayotou et al (1993). The buffer used for all binding experiments consisted of 20 mM HEPES, 150 mM NaCl, 3.4 mM EDTA, 0.005% Tween 20, and 4 mM DTT. The biotinylated-phosphotyrosyl peptide used in the binding experiments was the same as used in panning experiments. Immobilization of the biotinylated peptide onto streptavidin sensor chips (Biacore) was conducted at a flow rate of 10 μL/min and 25°C. A 5-μL wash with 0.1% SDS was used to remove any nonspecifically bound material. To monitor binding, a 10-min injection of wild-type SH2 and the SH2 insertion variants was conducted at a 10 μL/min flow rate, followed by free flow of buffer to initiate dissociation. Regeneration of the chip surface was carried out using a 5-μL injection of 0.1% SDS. As a control, the proteins were injected simultaneously over a cell of the streptavidin sensor chip to which only biotin had been bound.
Acknowledgments
We thank David Kim, Alex Watters, Karen Butner, and Alan Davidson for helpful conversations pertaining to molecular biology and phage panning techniques. This work was supported in part by a grant from the National Institutes of Health (D.B) and by the PHS NRSA T32 GM07270 from NIGMS (M.S.-K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0232003.
References
- Bacon, D.J. and Anderson, W.F. 1988. A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graph. 6 219–220. [Google Scholar]
- Betton, J.M., Jacob, J.P., Hofnung M., and Broome-Smith, J.K. 1997. Creating a bifunctional protein by insertion of β-lactamase into the maltodextrin-binding protein. Nat. Biotechnol. 15 1276–1279. [DOI] [PubMed] [Google Scholar]
- Brady, G.P. and Sharp, K.A. 1997. Entropy in protein folding and in protein-protein interactions. Curr. Opin. Struct. Biol. 7 215–221. [DOI] [PubMed] [Google Scholar]
- Cadwell, R.C. and Joyce, G.F. 1994. Mutagenic PCR. PCR Methods Appl. 3 S136–S140. [DOI] [PubMed] [Google Scholar]
- Chan, H.S. and Dill, K.A. 1988. Intrachain loops in polymers. J. Chem. Phys. 90 492–509. [Google Scholar]
- Collinet, B., Herve, M., Pecorari, F., Minard, P., Eder, O., and Desmadril, M. 2000. Functionally accepted insertions of proteins within protein domains. J. Biol. Chem. 275 17428–17433. [DOI] [PubMed] [Google Scholar]
- Doi, N., Itaya, M., Yomo, T., Tokura, S., and Yanagawa, H. 1997. Insertion of foreign random sequences of 120 amino acid residues into an active enzyme. FEBS Lett. 402 177–180. [DOI] [PubMed] [Google Scholar]
- Eck, M.J., Shoelson, S.E., and Harrison, S.C. 1993. Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of p56lck. Nature 362 87–91. [DOI] [PubMed] [Google Scholar]
- Gu, H., Yi, Q., Bray, S.T., Riddle, D.S., Shiau, A.K., and Baker, D. 1995. A phage display system for studying the sequence determinants of protein folding. Protein Sci. 4 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S., Stewart, M., Michie, A., Swindells, M.B., Orengo, C., and Thornton, J.M. 1998. Domain assignment for protein structures using a consensus approach: Characterization and analysis. Protein Sci. 7 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT—a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24 946–950. [Google Scholar]
- Ladurner, A.G. and Fersht, A.R. 1997. Glutamine, alanine or glycine repeats inserted into the loop of a protein have minimal effects on stability and folding rates. J. Mol. Biol. 273 330–337. [DOI] [PubMed] [Google Scholar]
- Merrit, E.A. and Murphy, M.E.P. 1994. Raster 3D version 2.0. A program for photorealistic molecular graphics. Acta. Cyrstallog. Sect. D. 50 869–873. [DOI] [PubMed] [Google Scholar]
- Minard, P., Scalley-Kim, M., Watters, A., and Baker, D. 2001. A “loop entropy reduction” phage-display selection for folded amino acid sequences. Protein Sci. 10 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagi, A.D. and Regan, L. 1997. An inverse correlation between loop length and stability in a four- helix-bundle protein. Fold Des. 2 67–75. [DOI] [PubMed] [Google Scholar]
- Panayotou, G., Gish, G., End, P., Truong, O., Gout, I., Dhand, R., Fry, M.J., Hiles, I., Pawson, T., and Waterfield, M.D. 1993. Interactions between SH2 domains and tyrosine-phosphorylated platelet-derived growth factor β-receptor sequences: Analysis of kinetic parameters by a novel biosensor-based approach. Mol. Cell Biol. 13 3567–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, R.B. 1994. Domain insertion. Protein Eng. 7 1407–1410. [DOI] [PubMed] [Google Scholar]
- Scalley, M.L., Yi, Q., Gu, H., McCormack, A., Yates, J.R., and Baker, D. 1997. Kinetics of folding of the IgG binding domain of peptostreptococcal protein L. Biochemistry 36 3373–3382. [DOI] [PubMed] [Google Scholar]
- Viguera, A.R. and Serrano, L. 1997. Loop length, intramolecular diffusion and protein folding. Nat. Struct. Biol. 4 939–946. [DOI] [PubMed] [Google Scholar]