

Abstract
Membrane proteins and water-soluble proteins share a similar core. This similarity suggests that it should be possible to water-solubilize membrane proteins by mutating only their lipid-exposed residues. We have developed computational tools to design water-soluble variants of helical membrane proteins, using the pentameric phospholamban (PLB) as our test case. To water-solublize PLB, the membrane-exposed positions were changed to polar or charged amino acids, while the putative core was left unaltered. We generated water-soluble phospholamban (WSPLB), and compared its properties to its predecessor PLB. In aqueous solution, WSPLB mimics all of the reported properties of PLB including oligomerization state, helical structure, and stabilization upon phosphorylation. We also characterized the truncated mutant WSPLB (21–52) comprising only the former transmembrane segment of PLB. This peptide shows a decreased specificity for forming a pentameric oligomerization state.
Keywords: Protein design, computational methods, phospholamban, water-soluble membrane protein
Approximately 30% of the open reading frames of the genomes of higher eukaryotes code for proteins that span or are associated with cell membranes (Stevens and Arkin 2000). To date, roughly 13,000 X-ray or NMR derived structures of water-soluble proteins have been deposited in the PDB, while only ∼35 structures of membrane proteins are currently known, due to inherent difficulties in membrane protein purification and crystallization. Although membrane-associated proteins are very difficult to characterize, water-soluble proteins are amenable to a wide range of biophysical experimental techniques. The interiors of integral membrane proteins and water-soluble proteins are similar (Rees et al. 1989) in terms of amino acid composition and packing angles, although some of the fine details differ (Bowie 1997; Eilers et al. 2002). The greatest difference between soluble and membrane-spanning proteins is the hydrophobicity of the amino acids on the exterior surface, where the amino acids that contact the lipid bilayer in membrane-spanning proteins are more hydrophobic relative to those seen on the surface of water-soluble proteins. Thus, a membrane-spanning protein might be made water-soluble by mutating its hydrophobic surface residues, if there is no alteration of the core. Such a technique would allow us to bypass the membrane to study membrane protein structures, while addressing fundamental questions about the forces that stabilize the native states of both water and membrane-soluble proteins.
We have developed fully automated methods to introduce water-solubility to membrane helices, using the integral membrane protein phospholamban (PLB) as a model system for several reasons. First, it contains a single, helical membrane-spanning segment, and the entire protein is only 52 amino acids in length (Fujii et al. 1986, 1987). The transmembrane region is proposed to form a structurally simple pentameric (Louis et al. 1982; Jones et al. 1985; Wegener et al. 1986; Watanabe et al. 1991) coiled-coil (Simmerman et al. 1996). Second, a large body of mutagenesis data exists to guide the choice of residues that can be safely mutated without compromising the structural integrity of the protein (Arkin et al. 1994; Simmerman et al. 1996; Kimura et al. 1997). Finally, the biological importance of PLB makes any structural information we might glean from studying it particularly interesting. PLB is an integral membrane protein of cardiac sarcoplasmic reticulum, and is the primary downstream target of a phosphorylation cascade resulting from β-adrenergic stimulation. Its primary function is the regulation of the Ca2+-dependent ATPase SERCA2a (Vorherr et al. 1992; Jones and Field 1993; Toyofuku et al. 1994; Cornea et al. 1997, 2000).
PLB contains a cytosolic (1–25) and transmembrane domain (26–52) (Fig. 1 ▶) (Simmerman et al. 1986), which together are 68%–78% α-helical as determined by circular dichroism (CD). The transmembrane domain is 73%–82% α-helical in nondenaturing micelles composed of octylglucoside or C12E18, while in sodium dodecyl sulfate (SDS) micelles it is 90% α-helical (Simmerman et al. 1989). NMR studies of the N-terminal peptide comprising residues 1–25 showed that it has little to no secondary structure in aqueous solutions (Terzi et al. 1992; Hubbard et al. 1994; Mortishiresmith et al. 1995; Quirk et al. 1996; Li et al. 1998). These residues are not essential for pentamer formation, and the transmembrane segment alone can form oligomers in detergent (Kovacs et al. 1988).
Figure 1.

The sequence of canine wild-type PLB compared to several soluble mutants: WSPLB, ADA-FULL, SIMM-FULL (Li et al. 2001), PLB-COMP-1, and PLB-COMP-2 (Sabine et al. 2000). In human PLB, Asn27 is substituted by Lys. Positions S16 and T17 are phosphorylated by PKA and PKC. The differences between WSPLB and PLB are shown in red, the differences between SIMM-FULL and WSPLB are shown in blue, and the differences between PLB-COMP-1 (and 2) and SIMM-FULL are shown in green.
PLB is phosphorylated on serine 16 and threonine 17 (Fig. 1 ▶) by cAMP-dependent protein kinase (PKA) and Ca2+-dependent protein kinase (PKC) respectively (Simmerman et al. 1986; Wegener et al. 1989) following β-adrenergic stimulation. Phosphorylation increases the degree of association of PLB in SDS micelles and phospholipid bilayers, and also decreases its ability to activate SERCA2a (Wegener et al. 1989; Brittsan et al. 2000; Chu et al. 2000). These observations suggest that it is the monomeric form of PLB that interacts with SERCA2a. The cytoplasmic region of PLB (1–25) is predominately positively charged (4 Arg, 1 Lys, 1 Asp, and 1 Glu) and phosphorylation of S16 and T17 changes the pI from 10 to 6.7 (Jones et al. 1985). Thus, one model proposes that phosphorylation reduces the net positive charge on each monomer, relieving their electrostatic repulsion, and favoring pentamer formation.
All of the residues essential for pentamer formation are in the PLB transmembrane domain (Wegener et al. 1986), determined in two mutagenesis studies using SDS-PAGE to monitor pentamer disruption (Arkin et al. 1994; Simmerman et al. 1996). In both studies, amino acid positions in the transmembrane region were assumed to be in the protein core if their mutation disrupted the formation of PLB pentamers. Based on these data the PLB transmembrane domain was modeled as a left-handed coiled coil, containing L37, L44, L51, I40, and I47 in the apolar core with leucines at the a positions, and isoleucines at the d positions (Simmerman et al. 1996).
Previously, two groups have produced water-soluble versions of PLB. Frank et al. (2000) placed the core residues of PLB into a helix that contained the solvent-exposed residues of the water-soluble five-helix bundle cartilage oligomeric matrix protein (COMP) (PDB accession code 1vdf). This COMP-PLB hybrid (Fig. 1 ▶) exhibited poor solubility in water but, when fused with maltose binding protein, produced a protein that appeared to form pentamers and higher aggreagates based on sedimentation velocity. Subsequently, Li et al. (2001) made two variants of a water-soluble PLB, SIMM-FULL and ADA-FULL (Fig. 1 ▶), achieving better solubility and pentamer formation, but with dynamic properties reminiscent of a molten globule. Although these studies represent significant advances, it was difficult to assess the extent to which the water-soluble constructs had the same structure as native PLB.
Here we describe a fully automated computational approach for water-solubilizing membrane proteins, which should be generally applicable to a variety of α-helical membrane proteins. Based on analysis of mutagenesis data, we computationally generated a model of the PLB homopentamer and redesigned the exterior residues to introduce water-solubility (WSPLB, MW = 6293.4 Daltons). To address the structural similarity between PLB and WSPLB we demonstrated the effect of phosphorylation on the stability of WSPLB oligomers. We also examined the determinants of pentamer versus tetramer formation in WSPLB and established that although full-length WSPLB peptides are uniquely in a monomer–pentamer equilibrium, a more stable heterogeneous mixture of tetramers and pentamers is present when the region (1–20) is removed. Our ability to model and predict the behavior of WSPLB upon either phosphorylation or truncation reflects a similarity of its structure with PLB.
Results
Design
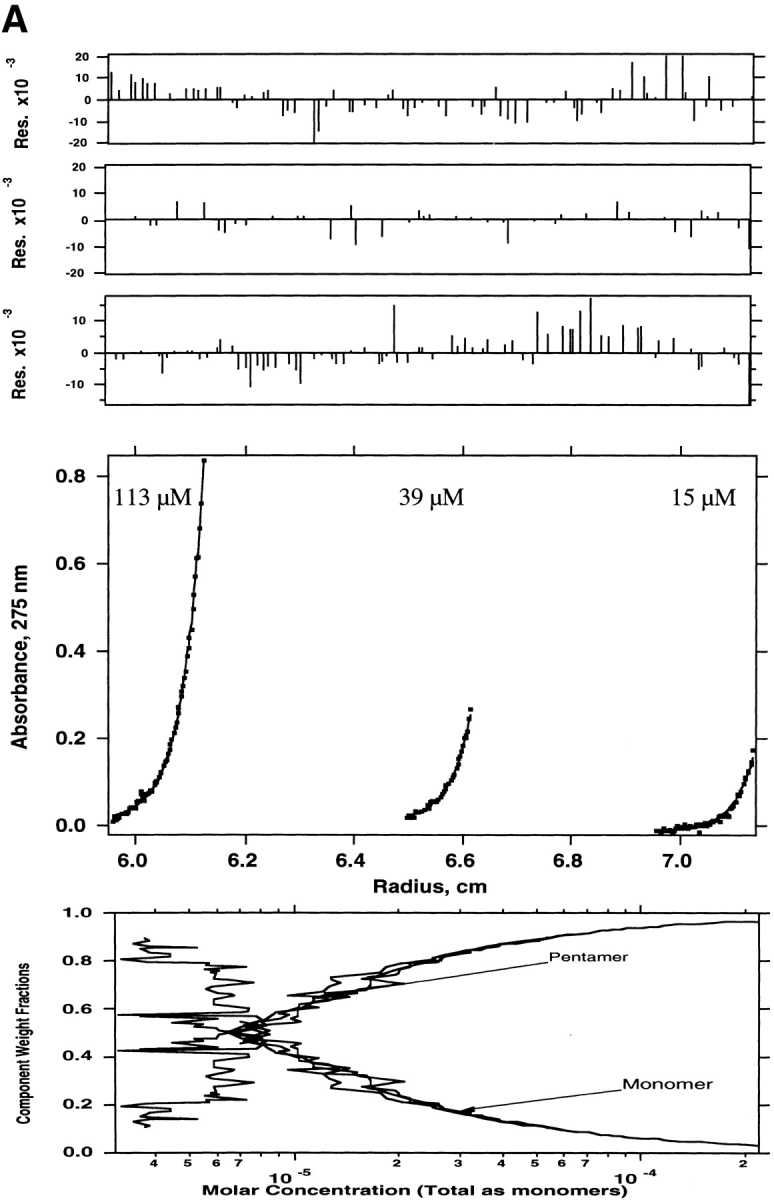
Mutational data from Arkin et al. (1994) and Simmerman et al. (1996) were mapped into a numerical form, defining a perturbation index that ranges from 0 (a mutation that does not disrupt formation of the native pentamer) to 1 (a mutation that completely disrupts pentamer formation). An average perturbation index was calculated for each position in the PLB transmembrane domain and then graphed as a function of sequence position (Dieckmann and DeGrado 1997). The data were analyzed according to a sine wave describing the variation in the perturbation index (P) as a function of the position in the sequence (x):
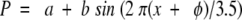 |
1 |
where a, b, and φ are fitting parameters, and the value of 3.5 residues is that of the left-handed coiled coil (Crick 1953) shown for PLB (Arkin et al. 1994; Simmerman et al. 1996). The value of φ is the phase, which defines the face of the helix that projects towards the interior of the structure. In agreement with Simmerman et al. (1996), this identifies the heptad repeat shown in Figure 1 ▶ in which Leu and Ile occupy the a and d positions, respectively. Using this assignment we modeled a C5 symmetric parallel left-handed coiled coil model for residues 31–52 using methods described previously (North et al. 2001). This orientation is similar to that seen in the crystal structure of COMP (Malashkevich et al. 1996), a soluble pentameric coiled coil, and also seen in the original model of Simmerman et al. (1996), and a more recent model of Arkin et al. (Torres et al. 2000).
Based on this model, we decided that solvent-exposed residues F32, F35, I38, L39, L42, L43, I45, C46, I48, V49, and L52 could be mutated without disturbing the structural integrity of the bundle. These residues showed minimal contact with neighboring helices. We began our sequence design by choosing Tyr as a replacement for Phe 32, providing us with a chromophore that could be used for concentration measurements, as well as aiding solubility while minimizing sequence perturbations.
An amino acid based approach—rather than an atom-based approach—was chosen for the design of the solvent exposed residues because of the difficulties in specifying unique orientations for solvent-exposed side chains. Side chains of surface residues tend to be highly mobile (Zhao et al. 2001) and often adopt multiple conformations in solution. The search for a single rotamer/sequence combination for these solvent-exposed residues, therefore, was abandoned for a more general residue-centric approach. The use of amino acid pairwise potential should be analogous to calculating the mean-field energy between residues (Lee and Subbiah 1991).
The solvent-exposed residues were chosen using a computer program that was designed to minimize the residue-based energy function of the entire transmembrane sequence. Following a Monte Carlo/simulated annealing approach, the program was used to optimize the remaining 10 variable amino acids against the background of the fixed (core and nonhelix spanning) residues. The energy function used to score sequences is described in Materials and Methods, and was chosen to optimize both intra- and interhelical interactions, as well as produce a sequence that was hydrophilic enough to be water soluble. This algorithm resulted in the selection of the sequence of WSPLB that was expressed in Escherichia coli. This protein was phosphorylated at S16 with cAMP-dependent protein kinase providing pWSPLB. Finally, we synthesized a peptide corresponding to residues 21–52, denoted WSPLB (21–52).
CD spectroscopy
Circular dichroism (CD) spectroscopy was used to examine the secondary structure of WSPLB. The spectra are concentration dependent (data not shown), as would be expected for a self-associating peptide. However, at concentrations greater than approximately 100 μM, the spectra are essentially independent of concentration and show a double minimum at 208 and 222 nm, the hallmarks of the α-helix (Fig. 2 ▶). At 125 μM, the ellipticity at 222 nm [θ222] is −17,400 deg cm2 dmol−1, which is similar to the range of values (−20,000 to −25,000 deg cm2 dmole−1) observed for the full-length native PLB in DMPC vesicles (Arkin et al. 1995) and C12E8, and octyl glucoside detergents (Simmerman et al. 1989). Also, at similar protein concentrations, the spectrum of the phosphorylated form pWSPLB was the same within experimental error, suggesting that the secondary structures of the proteins are similar. Analysis of the spectra of WSPLB indicate that approximately 25 residues are in an α-helical conformation (Chakrabartty et al. 1991), corresponding to the transmembrane helical region. The similarity of the spectra of WSPLB and native PLB suggest that they may have similar overall structures, with a disordered amino terminal segment followed by an α-helical coiled coil within the C-terminal half of the protein.
Figure 2.

CD spectra of 125 μM WSPLB and WSPLB (21–52). WSPLB spectra taken in 10 mM sodium phosphate pH 7.5, 50 mM NaCl, 1 mM TCEP-HCl. WSPLB (21–52) spectra taken in 15 mM MOPS pH 7.0, 50 mM NaCl, 1 mM EDTA, and 1 mM TCEP-HCl.
To provide additional support for this suggestion, we examined the CD spectrum of the truncated peptide WSPLB (21–52) (Fig. 2 ▶). The helicity calculated for this peptide is approximately 85% (Chakrabartty et al. 1991) ([θ222] = −29,453 deg cm2 dmole−1), again corresponding to about 25 residues in a helix.
Oligomeric state of WSPLB
Size-exclusion chromatography was used as an initial screen of oligomeric state and homogeneity of both peptides. At two loading concentrations (460 and 45 μM) WSPLB eluted as a single symmetrical peak from a G75 Superose column previously calibrated with globular molecular weight standards (data not shown). The slope of the peak suggested that it formed a single molecular species. The observed mass for WSPLB was 23,500 ± 4000 versus a calculated monomer mass of 6294. Deviations of WSPLB molecular weights from those expected are, for the pentameric helical bundle, assumed to be due to the nonglobular structure of helical bundles. The truncated WSPLB (21–52) peptide was run at two concentrations with and without boiling. In this case, a concentration dependent mixture of two species eluting at 22 and 31 mL (MWapp 16,000 and 11,500, respectively) was observed, versus a calculated monomer mass of 3956. The mass difference between these two species observed is 4520, roughly one monomer. Thus, two associating species need to be considered.
Sedimentation equilibrium was used to rigorously determine the association state and thermodynamics of the observed oligomers. Because WSPLB eluted as a single homogenous species from size exclusion chromatography, and its CD spectra were concentration independent above 100 μM, we initially determined its association state at 113 μM centrifuged at 35,000 rpm. In these conditions, WSPLB sedimented as a single species with a molecular weight of approximately 31,600 ± 200, or a pentamer (n = 5.04). Because CD spectra showed a concentration dependence below 50 μM, peptide samples of 113, 39, and 15 μM were then centrifuged at 30,000, 35,000, and 40,000 rpm, and the nine data sets were used to globally determine the monomer–pentamer dissociation constant (Fig. 3A ▶). The monomer–pentamer dissociation constant was determined to be 6.34 × 10−21 M4 with a P(1/2) = 6 μM, where P(1/2) is the midpoint of the monomer–nmer isotherm in total peptide concentration.
Figure 3.

Sedimentation equilibrium of WSPLB in 100 mM NaCl, 25 mM MOPS pH 7.5, 1 mM EDTA, 1 mM TCEP-HCl at 35,000 rpm. Equilibrium A280-radius profiles for three cell compartments containing peptide concentrations of 15, 39, and 113 μM are shown. (A) Shows the data fit to a monomer–pentamer equilibrium, while (B) shows a less satisfactory data fit to a monomer–tetramer equilibrium.
Sedimentation equilibrium was also used to determine the molecular weights of species present in the truncated WSPLB (21–52). WSPLB (21–52) (97 μM initial loading concentration) was centrifuged at 43,000 rpm, and a single species fit yielded a molecular weight of ∼17,300 (n = 4.4), a molecular weight intermediate between tetramers and pentamers. Subsequently, two lower concentrations (14 and 46 μM) of this peptide were also centrifuged, at 43,000 rpm, and all three data sets were used to determine the monomer-nmer dissociation constant. Various equilibrium models were used to fit the parameters including monomer–tetramer (Fig. 4A ▶), monomer–pentamer (Fig. 4B ▶), and monomer–tetramer–pentamer equilibrium (Fig. 4C ▶). The best fit was to a monomer–tetramer–pentamer. The monomer–tetramer–pentamer fit yielded a monomer–tetramer dissociation constant of 1 × 10−21 M3 and an overall monomer–pentamer dissociation constant of 1 × 10−28 M4.
Figure 4.



Sedimentation equilibrium of WSPLB (21–52) in 15 mM MOPS pH 7, 50 mM NaCl, 1 mM EDTA, 1 mM TCEP-HCl at 48,000 rpm. Equilibrium A280-radius profile for three cells containing 14, 46, and 97 μM. (A) Shows the data fit to a monomer–tetramer, (B) a monomer–pentamer, and (C) a monomer–tetramer–pentamer equilibrium. Although all three schemes provide a good fit to the data, the presence of multiple peaks on size-exclusion chromatography requires the use of a monomer–tetramer–pentamer scheme.
Stability of WSPLB constructs
All of the peptides were thermally unfolded, monitoring the loss of signal at 222 nm by CD spectroscopy. The unfolding curve of WSPLB shows a single transition whose midpoint depends on concentration, as expected for a monomer– oligomer equilibrium (Fig. 5 ▶). Previously it has been shown that global fitting of such curves obtained at multiple protein concentrations can be used to confirm the aggregation state and obtain highly accurate measures of the free energy of association. Application of these procedures to WSPLB allows an excellent fit for a monomer–pentamer equilibrium, providing the thermodynamic parameters described in Table 1. The predicted midpoint concentration for the monomer–pentamer isotherm was 4.9 μM (30 kcal/mole) at 25°C, in good agreement with the value of 6 μM (28 kcal/mole) obtained from sedimentation equilibrium. Thermal denaturation of phosphorylated WSPLB (pWSPLB) showed that phosphorylation of Ser16 significantly increased the stability of the pentamer (Fig. 6 ▶, Table 1). At these concentrations the ΔTm was approximately 8°C, and the ΔΔGunf was 4 kcal/mole.
Figure 5.

Thermal denaturation of WSPLB, measuring the loss of signal at 222 nm with increasing temperature by circular dichroism. Scans taken with 60-s signal averaging time and equilibration of 4 min at each temperature. Concentrations of peptide are 50 and 125 μM. Data were fit to a two-state model (theoretical curves are shown) with a monomer–pentamer equilibrium.
Table 1.
Thermodynamic parameters derived from global fitting of thermal denaturation curves as measured by cirular dichroism (CD)
| Construct | Tm at 50 μM (°C) | ΔGunf (kcal/mole) | ΔHunf (kcal/mole) | ΔCp (kcal/mole/°C) |
| WSPLB | 55 | 19.2 | 229 | 1.9 |
| pWSPLB | 63 | 23.1 | 231 | 1.9 |
Parameters are shown for full-length WSPLB and phosphorylated pWSPLB, including ΔGunf, ΔHunf, and ΔCp, all at 1 M standard state, and a reference temperature of 350 K. Tm at 50 μM is also shown. ΔCp was a global fitting parameter for WSPLB, but held fixed for pWSPLB at the same value as WSPLB.
Figure 6.

Thermal denaturation of WSPLB and pWSPLB from 2 to 94 °C, normalized to % unfolded peptide. Conditions are identical to those in Figure 2 ▶. Scans taken with 60-s signal averaging time and equilibration of 4 min at each temperature. Concentrations of both peptides are 50 μM.
Thermal unfolding curves of WSPLB (21–52) are illustrated in Figure 7 ▶. Unlike WSPLB, this peptide showed a pretransition at low temperature that was independent of peptide concentration. At higher temperatures a main transition is observed, which depends on the concentration of the peptide. Because the first transition has a small amplitude and does not appreciably depend on concentration, it may corespond to a switch between tetramer and pentamer aggregation states that should show a very weak concentration dependence. In contrast, the main transition corresponds to dissociation of the oligomer to unfolded monomer. The increase in stability of WSPLB (21–52) to WSPLB, also seen in sedimentation equilibrium, is apparent in the increase in Tm, which is 76°C for WSPLB (21–52) versus 55°C for WSPLB at 50 μM.
Figure 7.

Thermal denaturation of WSPLB (21–52) from 2 to 94°C. Scans taken with 60-s signal averaging time and equilibration of 4 min at each temperature. Conditions and peptide concentrations are identical to those in Figure 2 ▶.
Discussion
To produce a water-soluble variant of PLB, we have computationally mutated the transmembrane lipid-exposed residues from native, hydrophobic, amino acids to polar amino acids. In the absence of a three-dimensional structure of the PLB pentamer, indirect methods were used to infer which of the transmembrane residues are lipid exposed. In the case of PLB, a large amount of mutagenesis data has been amassed by other groups (Arkin et al. 1994; Simmerman et al. 1996; Kimura et al. 1997), which was used to guide the building of a structural model of the pentamer, allowing us to choose those amino acids that might be mutated to introduce water solubility.
Although a great deal of information is known about the membrane protein PLB, there remain several key pieces of information that have not been obtained until now. The effect of phosphorylation on WSPLB oligomerization has been studied by SDS-PAGE (Wegener and Jones 1984; Wegener et al. 1989; Arkin et al. 1994; Reddy et al. 1995), electron paramagnetic resonance (Cornea et al. 1997), and on the catalytic activity of SERCA2a (Sham et al. 1991; Cantilina et al. 1993), but its energetic contribution to PLB pentamer stabilization has heretofore not been measured. Phosphorylation at Ser16 and Thr17 has been suggested to have several effects on the system. It may decrease the electrostatic repulsion between positively charged monomers (Jones et al. 1985), or destabilize the interaction of PLB with SERCA2a as well as with the negatively charged membrane (Plank et al. 1983; Tada and Inui 1983; Inui et al. 1986; Kirchberger et al. 1986; Suzuki and Wang 1986), shifting the monomer–pentamer equilibrium toward the pentamer. We have shown that phosphorylation increases the stability of WSPLB by ∼0.9 kcal/mole/monomer, demonstrating that some part of the role of phosphorylation is exclusively an effect on PLB itself. The fact that phosphorylation has a similar effect on both PLB and WSPLB suggests also that the core and interhelical packing interactions are maintained in our design.
Both size-exclusion chromatography in SDS followed by laser light scattering (Watanabe et al. 1991) and sucrose density centrifugation with octyl glucoside (Harrer and Kranias 1994) show that phospholamban is a mix of mainly monomers and pentamers in the membrane. Our studies show that WSPLB exists in a monomer–pentamer equilibrium when the protein is full length. However, when the regulatory region of phosphorylation is removed (1–20), the oligomeric state is actually stabilized, with the production of a significant population of tetramers. Although the full-length WSPLB peptide is more specifically a pentamer, it is less stable than WSPLB (21–52). Thus, the region (1–20) of WSPLB acts as a negative design element, destabilizing any oligomer formed, but specifying pentamer over tetramer in the full-length peptide. Trading stability for specificity by burying polar sidechains has been seen in other model peptides (Hill et al. 1998, 1999; Lumb and Kim 1998; Hill and DeGrado 2000), and suggests that the presence of the PLB cytoplasmic domain allows intermoleculcar interactions within the oligomer to favor pentamers.
The propensity for tetramers in WSPLB, and possibly PLB, is consistent with previous results on the model system GCN4-pLI (Harbury et al. 1993). This tetrameric peptide contains the same hydrophobic core repeat as PLB, with salt bridges between residues at the “e” and “g” positions. It is possible that the nature of the residues at these “e” and “g” positions influence the agregation state. For example, in PLB and WSPLB, Q29 (a g position) and N34 (an e position) might form an interhelical interaction analagous to a hydrogen bond between N41 and E36′ in the five-helix bundle COMP (Malashkevich et al. 1996). Also, if the helical bundle in PLB extends beyond the hydrophobic transmembrane region, Q26 and N30 would occupy buried d and a positions. In the crystal structure of COMP (Malashkevich et al. 1996) Gln54 is buried in the hydrophobic core at a d position.
Our studies present the first experimentally verified, fully automated design procedure for introducing water solubility to membrane-spanning α-helical proteins. Using the model system PLB, we have studied the determinants of WSPLB oligomerization and quantified the effect of phosphorylation on its stability. By making the PLB transmembrane helix water soluble, we designed a system that has a similar structure and behavior as membrane-soluble PLB, and may contain all of its essential features. Although PLB and WSPLB have different solubilities, and therefore different forces stabilizing their folded states, both proteins showed the same oligomerization and response to phosphorylation. With a thermodynamic characterization of PLB, it may be found that the absolute values of stabilities measured of WSPLB may be different from PLB because of the introduction of the hydrophobic effect upon water solubilization. However, we have shown that the process of water solubilization of certain membrane-spanning helices will not alter their global properties and structure.
Materials and methods
Protein design
The energy function used in this work can be written as the sum of the energy due to intrinsic helical propensities of the amino acids, the intrahelical pairwise residue interaction energies, the interaction energy between the residues and helix macrodipole, the interhelical electrostatic interaction energy, a solubility term to enforce a low overall hydrophobicity, and a sequence entropy term as follows:
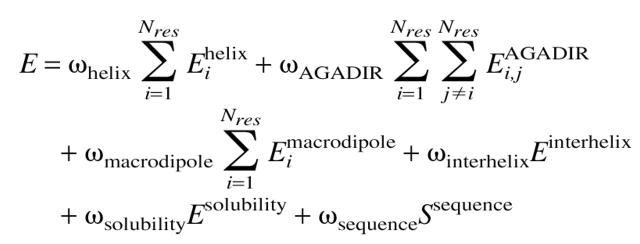 |
2 |
Each term has weight (ω) that can be used to tune the relative strengths of the energy terms. In this study, each weighting term has been set to 1.0 unless otherwise noted. The first term in equation 2 is the α-helix partition energy, taken from the analysis of helical propensities of O’Neil and DeGrado (1990). Because one of our goals was to maintain the helical nature of the transmembrane helices of phospholamban, this term was applied such that amino acids with higher α-helical propensity should contribute favorably to the energy term.
The second term in equation 2 represents intrahelical i to i + 3 and i to i + 4 interaction energies. The values used to represent these energies were taken from the program AGADIR (Muñoz and Serrano 1994). This set of intrahelical interaction energies was originally derived to predict the percent helicity of a peptide of a given sequence, but here we are using the interaction energies as a function that can be searched in order to find a sequence with an optimal energy (Villegas et al. 1996). An update to AGADIR (Muñoz and Serrano 1995) contains a term that accounts for interaction of charged residues with the helix macrodipole, represented by the third term in equation 2.
To measure interhelical electrostatic interactions, amino acids were assigned a designation of either +, −, or 0 according to their charge at neutral pH, and interacting pairs were scored as previously reported (Summa et al. 2002). These scores are a simplified representation of experimentally measured interhelical electrostatic interaction energies (Krylov et al. 1994). Because the energies were intended only as an approximation to find an optimal pattern of charge, and not to calculate binding energies, the authors felt that this simplification was justified. All e–g′ (an interaction between the e residue of a given helix, and the nearest g residues of a neighboring helix), b–g′, e–c′, and b–c′ pairs were assumed to be interacting. These interactions were summed over all contacts i giving the fourth term in equation 2 as follows:
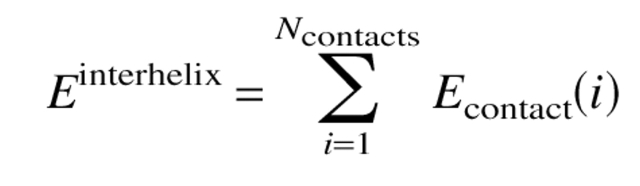 |
3 |
We enforced an upper limit on the hydrophobicity of the coiled coil region by adding a term that penalized sequences that have a hydrophobicity that is greater than that of the water-soluble pentameric peptide, COMP. The average per-residue hydrophobicity of the COMP structure for the sequence analogous to the transmembrane domain was calculated to be 0.372 kcal/mole (using the octanol–water transfer free energies of the amino acids) (Fauchere and Pliska 1983). If the per-residue hydrophobicity of any sequence exceeded this value, then the solubility energy term was defined as:
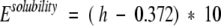 |
4 |
Otherwise, this parameter was set to a value of 0.0. This has the effect of preventing all sequences with a higher hydrophobicity score than the COMP sequence from appearing in the optimal sequence set.
To force sequence diversity, we added a term that we called the sequence entropy, which we have defined as follows:
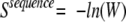 |
5 |
where
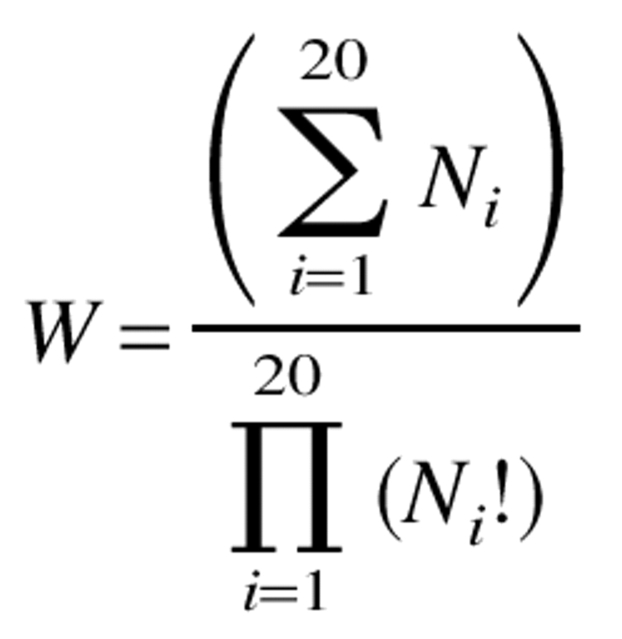 |
6 |
Ni represents the number of residues in the full sequence with an amino acid identity of type i. In the calculations below, this term was given a scaling factor of 0.1 so that its absolute value was roughly equal to those of the other terms in equation 2.
The sequence was optimized using a Monte Carlo/simulation annealing algorithm run from 700 to 10 K, with linear decrements over 700,000 steps. This process was repeated 500 times, and the sequences were then ranked and analyzed. For each energy calculation the entire sequence (the variable residues as well as the nonvariable residues) of the transmembrane domain was considered in the calculation. The top scoring sequence was built onto the backbone structure and analyzed. Amino acid side chains were modeled on an SGI Indigo2 workstation running the program InsightII (Molecular Simulations, Inc.). The sequence that was eventually produced differs slightly from the automatically designed sequence because of steric clashes that could not have been predicted with a residue-based energy function. This highlights the need for consideration of atomic level detail in protein design algorithms, but does not diminish the usefulness of residue-based functions for initial screening of possible sequences.
Modeling of perturbation index data
The parameters were fit to the data on an Apple Powerbook G3 using the program Kaliedagraph (Synergy software). The data points for residue numbers greater than 49 were not evaluated. Data for these residues seemed to contradict pentamer sensitivity data from another study (Simmerman et al. 1996), and were therefore excluded.
Expression and purification of WSPLB
The gene for WSPLB (MW = 6293.4) was synthesized by PCR overlap extension using four primers. This gene product was cloned at BamHI and EcoRI sites into an HT-UK vector (Gregory VanDuyne Lab), a variant of pET21a (Novagen), which contains a tobacco etch virus (TEV) protease cleavage site N-terminal to a six-histidine tag. The protein was expressed in BL21 (DE3) cells (Novagen) in terrific broth for 4 h after induction at OD 0.6 with 0.5 mM isopropyl-β-D-thiogalactoside. After expression, cells were harvested at 4°C by centrifugation at 5000 rpm. Cells pellets were lysed in denaturing lysis buffer by resuspension in 6 M Gdn/GdnHCl, 0.1 M sodium phosphate, 0.01 M Tris-HCl pH 8.0, and stirring for 3 h. This mixture was then sonicated and centrifuged at 13,000 rpm in preparation for purification.
For purification, the lysed cells were loaded onto a 25 mL Ni2+-Superose (Quiagen) column in the lysis buffer. The column was washed with lysis buffer, previously adjusted to pH 6.3. WSPLB-6His was eluted isochratically with the same buffer adjusted to pH 4.5, plus 1 mM imidazole. Fractions of the eluate were collected, and the presence of WSPLB-6His was verified by gel electrophoresis on a 12% Bis-Tris SDS-PAGE reducing gel (Invitrogen). Fractions containing WSPLB-6His were then pooled, concentrated, and diluted with TEV protease cleavage buffer to a final component mixture of 200 mM NaCl, 1 M Gdn/GdnHCl, 0.1 M sodium phosphate buffer, 0.01 M Tris-HCl pH 8.0, 1 mM ethylenediaminetetracaetic acid (EDTA), and 1 mM dithiothreitol (DTT). Cleavage with 2000 U TEV protease (Life Technologies) proceeded in this buffer for 3 days at 30°C, until ∼80% of the peptide was cleaved. Finally, WSPLB was purified by reverse-phase HPLC on a Vydac C4 preparative column using a linear gradient of water and acetonitrile containing 0.1% trifluoroacetic acid (TFA). Purity was assessed by analytical reverse-phase HPLC, MALDI-TOF mass spectrometry, and 12% Bis-Tris reducing SDS-PAGE gels.
Peptide synthesis of WSPLB (21–52)
WSPLB (21–52, MW. 3956) was chemically synthesized as a C-terminal carboxyamide on a 0.25-mmole scale using an Applied Biosystems model 433A solid phase peptide synthesizer (Perkin-Elmer) with standard FMOC amino acid chemistry. Peptides were washed on the resin with DMF and ether, cleaved for 1.5 h with TFA:water:thioanisole:ethanedithiol:phenol (40/2/2/1/3 v/v/v/v/w), and subsequently precipitated with cold ether. Purification proceeded by reverse-phase HPLC using a preparative C4 column (Vydac) and a linear gradient of the appropriate buffers. The purity of samples was then verified by MALDI-TOF mass spectrometry, analytical reverse-phase HPLC, and 12 % Bis-Tris reducing SDS-PAGE gels.
Phosphorylation of WSPLB
Expressed and purified WSPLB was phosphorylated enzymatically using cAMP-dependent protein kinase (PKA) catalytic subunit (New England Biologicals). To determine the efficiency of phosphorylation of WSPLB, initial screens using ATP γ32 P were conducted containing 10 μM WSPLB, 1 mM ATP, 16.6 nM ATP γ32 P, 5 U PKA, 150 mM NaCl, 50 mM Tris-HCl pH 7.5, and 10 mM MgCl2. The reaction was allowed to proceed for 1.5 h at 30°C, and was separated from reaction contents by gel electrophoresis using 12% Bis-Tris reducing SDS-PAGE. After drying the gel, the extent of phosphorylation was visualized by exposure on a Molecular Dynamics Storm 280, and analyzed using Image Quant v1.11 software. The extent phosphorylation was determined to be greater than 90% (data not shown). The reaction was repeated on a preparative scale with 334 μM WSPLB, 3 mM ATP, and 15 U PKA at 30°C. Phosphorylated protein was separated from the reaction contents by gel filtration chromatography using G-25 resin, and monitored at 280 nm.
Circular dichroism spectroscopy
All CD spectra were collected on an AVIV 62DS spectropolarimeter, using a 1-mm pathlength quartz cuvette. CD spectra of WSPLB and pWSPLB were collected in 10 mM sodium phosphate pH 7.5, 50 mM NaCl, 1 mM Tris (2-carboxyethyl)-phosphine hydrochloride (TCEP-HCl), and 1 mM EDTA, while those for WSPLB (21–52) in the same buffer substituted with 50 mM MOPS pH 7. Each wavelength scan from 200–260 nm is an average of four scans with 4-sec averaging time per data point at 25°C. All thermal melting curves were collected in the same buffers as their corresponding wavelength scans, with 60-sec averaging time and 4-min equilibration time. The elipticity was measured at 222 nm as a function of increasing temperature (2–94°C). WSPLB and pWSPLB melting curves were analyzed with Igor Pro® 3.16, assuming a two-state unfolding pathway. These data were treated as a monomer-nmer two-state system, using the functional form of the Gibbs-Helmholtz formula described previously (Boice et al. 1996):
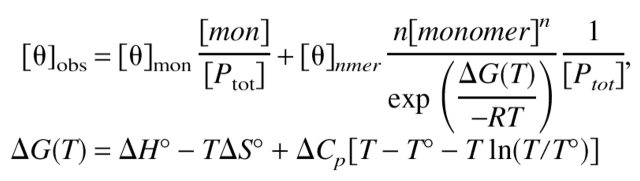 |
7 |
where [θ] is mean residue elipticity (deg cm2 dmole−1), T° is the midpoint of the transition, ΔH° is the van’t Hoff enthalpy at the midpoint (kcal mole−1), ΔS° is the standard state entropy (kcal mole−1 K−1), and ΔCP is the change in heat capacity over the temperature range of the experiment (kcal mole−1 K−1). In fitting, the floating parameters are the initial and final [θ] values, the slopes of the folded and unfolded baselines, ΔH°, ΔCp, and Tm. The parameters were globally fit to three equilibria schemes, from monomer–tetramer to hexamer, using data collected at 49 and 126 μM. Only the monomer–pentamer scheme provided an adequate fit to the data. ΔCp was held constant for pWSPLB at the value obtained for WSPLB.
Gel filtration chromatography
Analytical gel filtration chromatography was used to assess the distribution of oligomeric states in solution at various concentrations using a Superose G75 column (Amersham Biosciences). The dilution of the peak over the column was calculated to be 10-fold. All runs were performed in running buffer 25 mM sodium phosphate pH 7.0, 100 mM NaCl, 1 mM TCEP-HCl, and 1 mM EDTA using an FPLC (Amersham Biosciences). The column was calibrated using a 10 mg/mL solution of ovalbumin (43 kD), chymotrypsin (25 kD), cytochrome C (12.5 kD), and aprotinin (6.5 kD). WSPLB was loaded both at 458 (3.8 mg/mL) and 50 μM (0.31 mg/mL). WSPLB (21–52) was run at 2.5 mM (10 mg/mL) and 250 μM (1 mg/mL) both boiled and unboiled. Elution from the column was monitored at 280 nm wavelength with a UVM-II monitor (Amersham Biosciences).
Analytical ultracentrifugation
Measurements were made at 25°C using a Beckman XL-I analytical ultracentrifuge. Samples of WSPLB (15, 39, and 113 μM in 100 mM NaCl, 25 mM MOPS pH 7.5, 1 mM EDTA, and 1mM TCEP-HCl) were centrifuged to equilibrium at 30, 35, and 40,000 rpm in six-channel, carbon-epoxy composite centerpieces supplied by Beckman. WSPLB (21–52) samples (14, 46, and 97.5 μM in 15 mM MOPS pH 7.0, 50 mM NaCl, 1 mM TCEP-HCl, and 1 mM EDTA) were centrifuged at 48,000 rpm. Concentrations were monitored using absorption optics at a wavelength of 275 nm, and equilibrium was assessed by the absence of significant change in radial concentration gradients in scans separated by a few hours. Peptide partial specific volumes, solvent densities, monomer molecular masses, and molar extinction coefficients were calculated using the program SEDINTERP (Laue et al. 1992) modified to use the amino acid partial specific volumes and molecular weights reported by Kharakoz (1997). We estimate an uncertainty of about ±10% in the calculated molecular weight of the protein, arising largely from uncertainty in the partial specific volume, which is calculated from a weight average of individual amino acids. Calculated values were held constant, and data at both initial concentrations and the different speeds were analyzed by global curve-fitting of error-weighted optical absorption data to the sedimentation equilibrium equation for monomer–nmer equilibrium. To obtain the oligomer size present, the molecular weight was fit to data from the most concentrated samples with a single molecular weight species fit using Igor-Pro® (Wavemetrics) programs developed from a previous version (Brooks et al. 1993). In these fits, baselines, signal values, and the molecular weight were allowed to vary. Association constants were determined similarly, keeping the monomer molecular weight and oligomer number constant, and allowing the equilibrium constant to float. After fitting, a species plot was calculated representing the contribution of each species to total signal as a function of concentration.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0226603.
References
- Arkin, I.T., Adams, P.D., MacKenzie, K.R., Lemmon, M.A., Brünger, A.T., and Engelman, D.M. 1994. Structural organization of the pentameric transmembrane α-helices of phospholamban, a cardiac ion channel. EMBO J. 13 4757–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin, I.T., Rothman, M., Ludlam, C.F., Aimoto, S., Engelman, D.M., Rothschild, K.J., and Smith, S.O. 1995. Structural model of the phospholamban ion channel complex in phospholipid membranes. J. Mol. Biol. 248 824–834. [DOI] [PubMed] [Google Scholar]
- Boice, J.A., Dieckmann, G.R., DeGrado, W.F., and Fairman, R. 1996. Thermodynamic analysis of a designed three-stranded coiled coil. Biochemistry 35 14480–14485. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272 780–789. [DOI] [PubMed] [Google Scholar]
- Brittsan, A.G., Carr, A.N., Schmidt, A.G., and Kranias, E.G. 2000. Maximal inhibition of SERCA2 Ca2+ affinity by phospholamban in transgenic hearts overexpressing a non-phosphorylatable form of phospholamban. J. Biol. Chem. 275 12129–12135. [DOI] [PubMed] [Google Scholar]
- Brooks, I.S., Soneson, K.K., and Hensley, P. 1993. Development and use of a mac based data-analysis package for equilibrium sedimentation data from the analytical ultracentrifuge. Biophys. J. 64 a244. [Google Scholar]
- Cantilina, T., Sagara, Y., Inesi, G., and Jones, L.R. 1993. Comparative studies of cardiac and skeletal sarcoplasmic reticulum ATPases: Effect of a phospholambn antibody on enzyme activation. J. Biol. Chem. 268 17018–17025. [PubMed] [Google Scholar]
- Chakrabartty, A., Schellman, J.A., and Baldwin, R.L. 1991. Large differences in the helix propensities of alanine and glycine. Nature 351 586–588. [DOI] [PubMed] [Google Scholar]
- Chu, G.X., Lester, J.W., Young, K.B., Luo, W.S., Zhai, J., and Kranias, E.G. 2000. A single site (Ser(16)) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J. Biol. Chem. 275 38938–38943. [DOI] [PubMed] [Google Scholar]
- Cornea, R.L., Jones, L.R., Autry, J.M., and Thomas, D.D. 1997. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry 36 2960–2967. [DOI] [PubMed] [Google Scholar]
- Cornea, R.L., Autry, J.M., Chen, Z.H., and Jones, L.R. 2000. Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the Ca2+ pump of cardiac sarcoplasmic reticulum. J. Biol. Chem. 275 41487–41494. [DOI] [PubMed] [Google Scholar]
- Crick, F. 1953. The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 6 689–697. [Google Scholar]
- Dieckmann, G.R. and DeGrado, W.F. 1997. Modeling transmembrane helical oligomers. Curr. Opin. Struct. Biol. 7 486–494. [DOI] [PubMed] [Google Scholar]
- Eilers, M., Patel, A.B., Liu, W., and Smith, S.O. 2002. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys. J. 82 2702–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauchere, J.-L. and Pliska, V. 1983. Hydrophobic parameters p of amino acid side chains from the partitioning of N-aceyl-amino acid amides. Eur. J. Med. Chem. 18 369–375. [Google Scholar]
- Frank, S., Kammerer, R.A., Hellstern, S., Pegoraro, S., Stetefeld, J., Lustig, A., Moroder, L., and Engel, J. 2000. Toward a high-resolution structure of phospholamban: Design of soluble transmembrane domain mutants. Biochemistry 39 6825–6831. [DOI] [PubMed] [Google Scholar]
- Fujii, J., Kadoma, M., Tada, M., Toda, H., and Sakiyama, F. 1986. Characterization of structural unit of phospholamban by amino acid sequencing and electrophoretic analysis. Biochem. Biophys. Res. Commun. 138 1044–1050. [DOI] [PubMed] [Google Scholar]
- Fujii, J., Ueno, A., Kitano, K., Tanaka, S., Kadoma, M., and Tada, M. 1987. Complete complementary DNA-derived amino acid sequence of canine cardiac phospholamban. J. Clin. Invest. 79 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury, P.B., Zheng, T., Kim, P.S., and Alber, T. 1993. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262 1401–1407. [DOI] [PubMed] [Google Scholar]
- Harrer, J.M. and Kranias, E.G. 1994. Characterization of the molecular form of cardiac phospholamban. Mol. Cell. Biochem. 140 185–193. [DOI] [PubMed] [Google Scholar]
- Hill, R.B. and DeGrado, W.F. 1998. Solution structure of α2D, a nativelike de novo designed protein. J. Am. Chem. Soc. 120 1138–1145. [Google Scholar]
- Hill, R.B. and DeGrado, W.F. 2000. A polar, solvent-exposed residue can be essential for native protein structure. Struct. Fold. Design 8 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R.B., Hong, J-.K., and DeGrado, W.F. 1999. Hydrogen bonding cluster can specify the unique conformation of a protein. J. Am. Chem. Soc. 122 746–747. [Google Scholar]
- Hubbard, J.A., Maclachlan, L.K., Meenan, E., Salter, C.J., Reid, D.G., Lahouratate, P., Humphries, J., Stevens, N., Bell, D., Neville, W.A., et al. 1994. Conformation of the cytoplasmic domain of phospholamban by NMR and CD. J. Mol. Membr. Biol. 11 263–269. [DOI] [PubMed] [Google Scholar]
- Inui, M., Chamberlain, B.H., Saito, A., and Fleischer, S. 1986. The nature of the modulation of Ca2+ transport as studied by reconstitution of cardiac sarcoplasmic reticulum. J. Biol. Chem. 261 1794–1800. [PubMed] [Google Scholar]
- Jones, L.R. and Field, L.J. 1993. Residues 2–25 of phospholamban are insufficient to inhibit Ca2+ transport ATPase of cardiac sarcoplasmic-reticulum. J. Biol. Chem. 268 11486–11488. [PubMed] [Google Scholar]
- Jones, L.R., Simmerman, H.K.B., Wilson, W.W., Gurd, F.R.N., and Wegener, A.D. 1985. Purification and characterization of phospholamban from canine cardiac sarcoplasmic reticulum. J. Biol. Chem. 260 7721–7730. [PubMed] [Google Scholar]
- Kharakoz, D.P. 1997. Partial volumes and compressibilities of extended polypeptide chains in aqueous solution: Additivity scheme and implication of protein unfolding at normal and high pressure. Biochemistry 36 10276–10285. [DOI] [PubMed] [Google Scholar]
- Kimura, Y., Kurzydlowski, K., Tada, M., and MacLennan, D.H. 1997. Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 272 15061–15064. [DOI] [PubMed] [Google Scholar]
- Kirchberger, M.A., Borchman, D., and Ckasinathan, C. 1986. Proteolytic activation of the canine cardiac sarcoplasmic reticulum calcium pump. Biochemistry 25 5484–5492. [DOI] [PubMed] [Google Scholar]
- Kovacs, R.J., Nelson, M.T., Simmerman, H.K.B., and Jones, L.R. 1988. Phospholamban forms Ca2+-selective channels in lipid bilayers. J. Biol. Chem. 263 18364–18368. [PubMed] [Google Scholar]
- Krylov, D., Mikhailenko, I., and Vinson, C. 1994. A thermodynamic scale for leucine zipper stability and dimerization specificity: e and g interhelical interactions. EMBO J. 13 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue, T., Shaw, B.D., Ridgeway, T.M., and Pelletier, S.L. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. The Royal Society of Chemistry, Cambridge, UK.
- Lee, C. and Subbiah, S. 1991. Prediction of protein side-chain conformation by packing optimization. J. Mol. Biol. 217 373–388. [DOI] [PubMed] [Google Scholar]
- Li, H., Cocco, M.J., Steitz, T.A., and Engelman, D.E. 2001. Conversion of phospholamban into a soluble pentameric helical bundle. Biochemistry 40 6636–6645. [DOI] [PubMed] [Google Scholar]
- Li, M., Cornea, R.L., Autry, J.M., Jones, L.R., and Thomas, D.D. 1998. Phosphorylation-induced structural change in phospholamban and its mutants, detected by intrinsic fluorescence. Biochemistry 37 7869–7877. [DOI] [PubMed] [Google Scholar]
- Louis, C.F., Maffitt, M., and Jarvis, B. 1982. Factors that modify the molecular-size of phospholamban, the 23,000-Dalton cardiac sarcoplasmic-reticulum phosphoprotein. J. Biol. Chem. 257 5182–5186. [PubMed] [Google Scholar]
- Lumb, K.J. and Kim, P.S. 1998. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled coil. Biochemistry 37 10342. [DOI] [PubMed] [Google Scholar]
- Malashkevich, V.N., Kammerer, R.A., Efimov, V.P., Schulthess, T., and Engel, J. 1996. The crystal structure of a five-stranded coiled coil in COMP: A prototype ion channel? Science 274 761–765. [DOI] [PubMed] [Google Scholar]
- Mortishiresmith, R.J., Pitzenberger, S.M., Burke, C.J., Middaugh, C.R., Garsky, V.M., and Johnson, R.G. 1995. Solution structure of the cytoplasmic domain of phospholamban-phosphorylation leads to a local perturbation in secondary structure. Biochemistry 34 7603–7613. [DOI] [PubMed] [Google Scholar]
- Muñoz, V. and Serrano, L. 1994. Elucidating the folding problem of helical peptides using empirical parameters. Nature 1 399–409. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 245 275– 296. [DOI] [PubMed] [Google Scholar]
- North, B., Summa, C.M., Ghirlanda, G., and DeGrado, W.F. 2001. D(n)-symmetrical tertiary templates for the design of tubular proteins. J. Mol. Biol. 311 1081–1090. [DOI] [PubMed] [Google Scholar]
- O’Neil, K.T. and DeGrado, W.F. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250 646–651. [DOI] [PubMed] [Google Scholar]
- Plank, B., Pifl, C., Hellmann, G., Wyskovsky, W., Hoffmann, R., and Suko, J. 1983. Correlation between calmodulin-dependent increase in the rate of calcium transport and calmodulin-dependent phosphorylation of cardiac sarcoplasmic reticulum. Eur. J. Biochem. 136 215–221. [DOI] [PubMed] [Google Scholar]
- Quirk, P.G., Patchell, V.B., Colyer, J., Drago, G.A., and Gao, Y. 1996. Conformational effects of serine phosphorylation in phospholamban peptides. Eur. J. Biochem. 263 85–91. [DOI] [PubMed] [Google Scholar]
- Reddy, L.G., Jones, L.R., Cala, S.E., O’Brian, J.J., Tatulian, S.A., and Stokes, D.L. 1995. Functional reconstitution of recombinant phospholamban with rabbit skeletal Ca2+-ATPase. J. Biol. Chem. 270 9390–9397. [DOI] [PubMed] [Google Scholar]
- Rees, D.C., DeAntonio, L., and Eisenberg, D. 1989. Hydrophobic organization of membrane proteins. Science 245 510–513. [DOI] [PubMed] [Google Scholar]
- Sabine, F., Kammerer, R.A., Hellstern, S., Pegoraro, S., Stetefeld, J., Lustig, A., Moroder, L., and Engel, J. 2000. Toward a high-resolution structure of phospholamban: Design of soluble transmembrane domain mutants. Biochemistry 39 6825–6831. [DOI] [PubMed] [Google Scholar]
- Sham, J.S.K., Jones, L.R., and Morad, M. 1991. Phospholamban mediates the β-adrenergic-enhanced Ca2+ uptake in mammalian ventricular myocytes. Am. J. Physiol. 261 H1344–H1349. [DOI] [PubMed] [Google Scholar]
- Simmerman, H.K., Lovelace, D.E., and Jones, L.R. 1989. Secondary structure of detergent-solubilized phospholamban, a phosphorylatable, oligomeric protein of cardiac sarcoplasmic reticulum. Biochim. Biophys. Acta 997 322–329. [DOI] [PubMed] [Google Scholar]
- Simmerman, H.K.B., Collins, J.H., Theibert, J.L., Wegener, A.D., and Jones, L.R. 1986. Sequence-analysis of phospholamban —Identification of phosphorylation sites and 2 major structural domains. J. Biol. Chem. 261 3333–3341. [PubMed] [Google Scholar]
- Simmerman, H.K.B., Kobayashi, Y.M., Autry, J.M., and Jones, L.R. 1996. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271 5941–5946. [DOI] [PubMed] [Google Scholar]
- Stevens, T.J. and Arkin, I.T. 2000. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins 39 417–420. [DOI] [PubMed] [Google Scholar]
- Summa, C.M., Rosenblatt, M.M., Hong, J.-K., Lear, J.D., and DeGrado, W.F. 2002. Computational de novo design, and characterization of an A2B2 diiron protein. J. Mol. Biol. (in press). [DOI] [PubMed]
- Suzuki, T. and Wang, J.H. 1986. Stimulation of bovine cardiac sarcoplasmic reticulum Ca2+ pump and blocking of phospholamban phosphorylation and dephosphorylation by a phospholamban monoclonal antibody. J. Biol. Chem. 261 7018–7023. [PubMed] [Google Scholar]
- Tada, M. and Inui, M. 1983. Regulation of calcium transport by the ATPase-phospholamban system. J. Mol. Cell. Cardiol. 15 565–575. [DOI] [PubMed] [Google Scholar]
- Terzi, E., Poteur, L., and Trifilieff, E. 1992. Evidence for a phosphorylation-indiced conformational change in phospholamban cytoplasmic domain by CD analysis. FEBS Lett. 309 413–416. [DOI] [PubMed] [Google Scholar]
- Torres, J., Adams, P.D., and Arkin, I.T. 2000. Use of a new label, 13C-18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 300 677–685. [DOI] [PubMed] [Google Scholar]
- Toyofuku, T., Kurzydlowski, K., Tada, M., and Maclennan, D.H. 1994. Amino-acids Lys-Asp-Asp-Lys-Pro-Val(402) in the Ca2+−ATPase of cardiac sarcoplasmic-reticulum are critical for functional association with phospholamban. J. Biol. Chem. 269 22929–22932. [PubMed] [Google Scholar]
- Villegas, V., Viguera, A.R., Aviles, F.X., and Serrano, L. 1996. Stabilization of proteins by rational design of α-helix stability using helix-coil transition theory. Fold. Design 1 29–34. [PubMed] [Google Scholar]
- Vorherr, T., Chiesi, M., Schwaller, R., and Carafoli, E. 1992. Regulation of the calcium ion pump of sarcoplasmic reticulum: Reversible inhibition by phospholamban and by the calmodulin binding domain of the plasma membrane calcium ion pump. Biochemistry 31 371–376. [DOI] [PubMed] [Google Scholar]
- Watanabe, Y., Kijima, Y., Kadoma, M., Tada, M., and Tagaki, T. 1991. Molecular weight determination of phospholamban oligomer in the presence of sodium dodecyl sulfate: Application of low-angle laserlight scattering photometry. J. Biochem. 110 40–45. [DOI] [PubMed] [Google Scholar]
- Wegener, A.D. and Jones, L.R. 1984. Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels—Evidence for a protein-structure consisting of multiple identical phosphorylatable subunits. J. Biol. Chem. 259 1834–1841. [PubMed] [Google Scholar]
- Wegener, A.D., Simmerman, H.K., Liepnieks, J., and Jones, L.R. 1986. Proteolytic cleavage of phospholamban purified from canine cardiac sarcoplasmic reticulum vesicles. Generation of a low resolution model of phospholamban structure. J. Biol. Chem. 261 5154–5159. [PubMed] [Google Scholar]
- Wegener, A.D., Simmerman, H.B.K., Liepnieks, J., and Jones, L.R. 1989. Phospholamban phosphorylation in intact ventricles: Phosphorylation of serine 16 and threonine 17 in response to b-adrenergic stimulation. J. Biol. Chem. 264 11468–11474. [PubMed] [Google Scholar]
- Zhao, S., Goodsell, D.S., and Olson, A.J. 2001. Analysis of a data set of paired uncomplexed protein structures: New metrics for side-chain flexibility and model evaluation. Proteins 43 271–279. [DOI] [PubMed] [Google Scholar]