

Abstract
Pulsed-field gradient (PFG) diffusion NMR spectroscopy studies were conducted with several helix-loop-helix regulatory Ca2+-binding proteins to characterize the conformational changes associated with Ca2+-saturation and/or binding targets. The calmodulin (CaM) system was used as a basis for evaluation, with similar hydrodynamic radii (Rh) obtained for apo- and Ca2+-CaM, consistent with previously reported Rh data. In addition, conformational changes associated with CaM binding to target peptides from myosin light chain kinase (MLCK), phosphodiesterase (PDE), and simian immunodeficiency virus (SIV) were accurately determined compared with small-angle X-ray scattering results. Both sets of data demonstrate the well-established collapse of the extended Ca2+-CaM molecule into a globular complex upon peptide binding. The Rh of CaM complexes with target peptides from CaM-dependent protein kinase I (CaMKI) and an N-terminal portion of the SIV peptide (SIV-N), as well as the anticancer drug cisplatin were also determined. The CaMKI complex demonstrates a collapse analogous to that observed for MLCK, PDE, and SIV, while the SIV-N shows only a partial collapse. Interestingly, the covalent CaM–cisplatin complex shows a near complete collapse, not expected from previous studies. The method was extended to related calcium binding proteins to show that the Rh of calcium and integrin binding protein (CIB), calbrain, and the calcium-binding region from soybean calcium-dependent protein kinase (CDPK) decrease on Ca2+-binding to various extents. Heteronuclear NMR spectroscopy suggests that for CIB and calbrain this is likely because of shifting the equilibrium from unfolded to folded conformations, with calbrain forming a dimer structure. These results demonstrate the utility of PFG-diffusion NMR to rapidly and accurately screen for molecular size changes on protein–ligand and protein–protein interactions for this class of proteins.
Keywords: Calmodulin, calcium and integrin binding protein (CIB), calcium-dependent protein kinase (CDPK), PFG diffusion, protein–ligand interactions, protein–protein interactions, protein conformational change
The helix-loop-helix class of regulatory Ca2+-binding proteins are a varied and pervasive group involved in numerous cellular functions such as signalling and ion buffering (Kawasaki et al. 1998; Lewit-Bentley and Rety 2000). Although numerous subfamilies exist, one of the best structurally characterized is the CaM subfamily, which is often used as a model system. Three-dimensional studies of the apo- (Kuboniwa et al. 1995; Zhang et al. 1995) and holo-CaM proteins (Babu et al. 1988) demonstrate that the binding of Ca2+ causes almost no change in the amount of secondary structure, but leads to a significant rearrangement of the helices surrounding the calcium-binding sites. The Ca2+ form takes on a dumbbell shape, although the linker between the two domains has been shown to be flexible by NMR relaxation studies (Barbato et al. 1992) and molecular dynamics calculations (van der Spoel et al. 1996). Canonically, CaM has been shown to collapse around target binding regions such as those from myosin light chain kinase (MLCK) (Ikura et al. 1992; Meador et al. 1992), CaM-dependent kinase II-α (Meador et al. 1992), and CaM-dependent kinase-kinase (Osawa et al. 1999) to form a globular, compact complex. The binding mode involving the collapse of CaM around targets has recently been shown to be a broad but specialized situation (Hoeflich and Ikura 2002) with reports of CaM complex structures in an extended conformation, as seen for complexes with anthrax adenylyl cyclase (Drum et al. 2002) and a Ca2+-activated K+ channel (Schumacher et al. 2001).
The variability in the molecular size characteristics of the CaM system provides an interesting case study in the use of molecular size to characterize signalling and conformational change. Several techniques have been used to quantify global resizing based on target or ligand binding independent of complete structural determination. These include small-angle X-ray scattering (SAXS), which provides radius of gyration (Rg) information (Seaton et al. 1985; Heidorn and Trewhella 1988; Heidorn et al. 1989; Trewhella 1992; Yoshino et al. 1996; Izumi et al. 2001), while other studies have been used to obtain information on the solution-state radius of hydration (Rh) such as gel permeation chromatography (GPC) (Crouch and Klee 1980; Sorensen and Shea 1996), and dynamic light scattering (DLS) (Papish et al. 2002). In all cases the molecular size has been used to monitor the signalling capabilities of CaM with respect to either Ca2+-binding or target binding.
In general, numerous methods are available to elucidate particular interactions between specific proteins for signalling networks (e.g., see Zhu and Snyder 2002). The next level of understanding entails structural characterization of these interactions, for which a myriad of biophysical techniques have evolved to measure various spatial parameters as described above for CaM. Among these, NMR spectroscopy is typically thought to have a place in the characterization of the contact interface on an atomic level of structural determination (Zuiderweg 2002).
In addition, measurements based on molecular diffusion coefficients have been extensively reported to demonstrate binding of relatively small molecules to proteins, primarily for pharmaceutical type screening but also for studies of protein ligand interactions (Liu et al. 1997; Gonnella et al. 1998; Gounarides et al. 1999; Otto and Larive 2001). Measurements of protein diffusion and the hydrodynamic radius (Rh) by 1H homonuclear NMR have been gaining in interest to monitor protein folding (Jones et al. 1997; Pan et al. 1997; Wilkins et al. 1999; Morgan et al. 2000; Bhattacharjya et al. 2001; Morar et al. 2001; Choy et al. 2002). Interestingly, this method has also been applied to demonstrate the heme-dependent folding of cytochrome c (Wain et al. 2001), as well as the dimerization and multimerization of proteins and peptides (Altieri et al. 1995; Ekiel and Abrahamson 1996; Mansfield et al. 1998; Schibli et al. 2002). These studies demonstrate that pulse-field gradient diffusion NMR can be used in a manner similar to other techniques that provide solution-state Rh information such as GPC and DLS.
In this study, we demonstrate that the methodology developed to monitor protein folding by PFG-diffusion NMR can equally well be applied to monitor global size changes for CaM–ligand (Ca2+) and CaM–protein interactions for CaM (for binding targets, Table 1). The apo-CaM and fully Ca2+-saturated CaM species, as well as CaM complexed binding targets from skeletal myosin light chain kinase (skMLCK) (Blumenthal et al. 1985; Ikura et al. 1992; Zhang et al. 1993), cyclic nucleotide phosophodiesterase (PDE) (Charbonneau et al. 1991; Yuan et al. 1999), and simian immunodeficiency virus (SIV) (Yuan et al. 1995, 2001), for which structural information is available from SAXS, DLS, and/or GPC are examined. Based upon successful reproduction of these confirmed molecular sizes, we extend this analysis to three CaM–target systems for which the molecular size is not well characterized at this time, namely CaM complexed with CaM-dependent kinase I (CaMKI) (Gomes et al. 2000), an N-terminal portion of the SIV peptide (SIV-N) (Yuan et al. 2001), and the anticancer drug cisplatin (H. Ouyang and H.J. Vogel, in prep.). In addition, we characterize the Ca2+-binding changes of three related helix-loop-helix Ca2+-binding proteins, calcium, and integrin-binding protein (CIB) (Naik et al. 1997; Hwang and Vogel 2000), calbrain (CaB) (Yamaguchi et al. 1999), and CLD from calmodulin-like domain protein kinase (Yoo and Harmon 1996). Moreover, collapse of the CLD when bound to its junction domain (JD), and the possible dimerization of CaB were also studied.
Table 1.
Sequences of CaM-binding peptides and junction domain peptide used in this study
| Peptide/Ligand | Sequence | Ref |
| MLCK | KRRWKKNFIAVSAANRFKKISS | (Blumenthal et al. 1985) |
| CaMKI-α | AKSKWKQAFNATAVVRHMRKLQ | (Gomes et al. 2000) |
| PDE | Ac-QTEKMWQRLKGILRCLVKQL-NH2 | (Charbonneau et al 1991) |
| SIV | DLWETLRRGGRWILAIPRRIRQGLELTL-NH2 | (Yuan et al 1995, 2001) |
| SIV-N | DLWETLRRGGRWI-NH2 | (Yuan et al 2001) |
| JD | Ac-CAV LSR LKQ FSA XNK LKK MAL RVIA-NH2 | (Yoo and Harmon 1996) |
Results and Discussion
Diffusion of apo- and Ca2+-CaM
The measurement of Rh for the apo- and Ca2+-CaM samples was relatively straightforward in that the 1H spectra consisted of only protein resonances that decayed essentially simultaneously with increasing PFG strength. The hydrodynamic radii determined for apo- and Ca2+-CaM are shown in Table 2, with lysozyme used to calibrate the diffusion of the internal dioxane standard as described in Material and Methods. The value of 24.8 ± 0.9 Å for apo-CaM is in excellent agreement with previous determinations by DLS (25 ± 1 Å; Papish et al. 2002) and GPC (24.91 ± 0.12 Å; Sorensen and Shea 1996) under similar conditions of pH and ionic strength.
Table 2.
Hydrodynamic radii and compactions factors of various EF-hand proteins
| Rh (Å) | Cf | #AA | Rg (Å) | |
| Lysozyme | 19.8 ± 0.3 | 1.02 ± 0.02 | 144 | |
| apo-CaM | 24.8 ± 0.9 | 0.74 ± 0.05 | 148 | 21.2 ± 0.3a |
| Ca2+-CaM | 24.5 ± 0.4 | 0.76 ± 0.02 | 148 | 21.5 ± 0.3b |
| CaM-MLCK | 21.8 ± 0.3 | 0.97 ± 0.01 | 170 | 17.9c |
| CaM-PDE | 22.3 ± 0.3 | 0.94 ± 0.02 | 168 | 18.8 |
| apo-CaM-PDE | N.D. | N.D. | 168 | 23.5 |
| CaM-SIV | 22.5 ± 0.9 | 0.94 ± 0.04 | 176 | 18.9 |
| CaM-CaMKI | 21.2 ± 1.2 | 1.00 ± 0.06 | 159 | |
| CaM-SIV-N | 23.1 ± 0.7 | 0.88 ± 0.03 | 161 | |
| CaM-CisPlat | 21.4 ± 0.2 | 0.94 ± 0.01 | 148 | |
| apo-CIB | 32.1 ± 1.7 | 0.57 ± 0.07 | 201 | |
| Ca-CIB | 24.5 ± 1.5 | 0.90 ± 0.06 | 201 | |
| apo-CLD | 25.1 ± 0.4 | 0.83 ± 0.02 | 181 | |
| Ca-CLD | 21.3 ± 1.0 | 1.01 ± 0.05 | 181 | |
| JD-CLD | 20.2 ± 0.7 | 1.08 ± 0.03 | 204 | |
| apo-CaB | 25.0 ± 0.5 | 0.37 ± 0.05 | 93 | |
| Ca2+-CaB | 21.0 ± 0.3 | 0.71 ± 0.02 | 93 | |
| 2x Ca2+-CaB | 1.03 ± 0.01 | 186 |
The Ca2+-CaM result of 24.5 ± 0.4 Å also agrees reasonably well with the value reported by GPC (23.95 ± 0.09; Sorensen and Shea 1996), but is smaller than that determined by DLS (30 ± 1; Papish et al. 2002). The decrease of ∼0.3 Å in the hydrodynamic radius on calcium binding in this study is also reflected in an early gel filtration study in which the Rh of Ca2+-CaM was ∼0.5 Å less than the apo form (Crouch and Klee 1980). SAXS data has also been reported for the Rg of apo- and Ca2+-CaM (Seaton et al. 1985; Heidorn and Trewhella 1988; Matsushima et al. 1989; Trewhella 1992; Izumi et al. 2001). In these studies, Ca2+-CaM is consistently larger than the apo-protein, although the range of the enhancement varies from +0.3 Å (Matsushima et al. 1989; Izumi et al. 2001) to +1.1 Å (Heidorn and Trewhella 1988) when the data is analyzed using the Guinier method. Detailed analysis of the implications of these results has been reported elsewhere (Mehler et al. 1991; Sorensen and Shea 1996); however, it is clear that our data is consistent with previously reported molecular size changes on CaM calcium binding in solution.
CaM–target interactions—skMLCK peptide
The hydrodynamic radii of CaM complexed with skMLCK was determined to be 21.8 ± 0.3 Å (Table 1), in close agreement with the DLS determination of 22 ± 1 Å (Papish et al. 2002). This indicates that the hydrodynamic radius of calcium-saturated CaM decreases by 2.7 Å in the presence of the skMLCK peptide. The NMR structure of the CaM–skMLCK complex (Ikura et al. 1992) demonstrates a similar collapse in which the two lobes of CaM surround the target peptide to form a compact globular complex. Likewise, previous SAXS studies have determined a collapse, with the Rg decrease measured at 4.9 Å (Heidorn et al. 1989) and 3.6 Å. (Yoshino et al. 1996) (Table 2). It should be noted that the interpretation of the SAXS values relative to the current PFG-diffusion measurements is dependent upon the Ca2+-CaM Rh and Rg, which remains a point for further investigation (Sorensen and Shea 1996).
Taken together, the diffusion of the apo-, Ca2+-CaM, CaM–MLCK proteins in the current study suggest that the PFG-diffusion technique is quantitatively sensitive to changes in the conformation of CaM, and can be extended to other systems.
CaM–target interactions—PDE and SIV
The determined hydrodynamic radii of the CaM–PDE and CaM–SIV complexes were 22.3 ± 0.3 Å and 22.5 ± 0.9 Å, respectively (Table 2). These results suggest that similar to the MLCK peptide, there is a significant collapse of 2.2 Å (PDE) and 2.0 Å (SIV). The result obtained for PDE was similar to that reported by DLS of 23 ± 1 Å (Papish et al. 2002). The CaM–PDE value of this study is also within the error of the measurement of the Rh by DLS of the CaM–SIV complex (24 ± 2 Å). We have previously shown that the interactions of both peptides (PDE; Yuan et al. 1999; SIV; Yuan et al. 2001) with Ca2+-CaM occurs in a manner generally analogous to that of MLCK. The spectroscopic evidence of these studies suggest that the peptides bind with α-helical secondary structure and the peptide tryptophan residue is sequestered into the C-terminal lobe of the protein.
The binding of these two peptides is energetically unique from other CaM–target complexes, however, and falls into a separate binding category (Brokx et al. 2001). Thus, to confirm that the collapse observed with the PDE and SIV peptides was indeed akin to that observed with MLCK, SAXS analysis was conducted on the CaM complexes as shown in Figure 1 ▶. Subsequent Guinier analysis indicates that the Rg values of the Ca2+-saturated complexes are 18.8 Å (PDE) and 18.9 Å (SIV) (Table 2). Comparison with the Ca2+-CaM (21.5 Å) and CaM–MLCK (17.9 Å) molecular sizes obtained under similar conditions (Yoshino et al. 1996), demonstrates that the collapses observed in all three peptide bound forms with Ca2+-CaM are of a similar magnitude as indicated by the PFG-diffusion NMR measurements.
Figure 1.

SAXS P(r) functions for the titration of CaM and (A) SIV peptide or (B) PDE peptide with Ca2+. Note that in (B), as the calcium concentration increases above a 2:1 Ca2+:CaM ratio, there is a substantial decrease in the number of longer vector lengths with a final molecular size of 18.8 Å. This behavior has been reported before for the CaM-MLCK complex (Heidorn et al. 1989). A similar but less dramatic effect is noticed for (LA), with a measure Rg of 18.9 Å.
We note with interest that in the case of PDE, the Rg for the apo complex of PDE and CaM has increased over apo-CaM (see Table 2). This effect is not seen for MLCK (data not shown). This observation is consistent with earlier reports by Yuan et al. (1999), showing that the PDE peptide specifically interacts with the C-terminal lobe of CaM in the absence of calcium. A similar increase in the radius of gyration has recently been reported for other CaM-binding peptides binding to apo-calmodulin from SAXS measurements (Izumi et al. 2001). Thus, in agreement with other studies, the SAXS data shows that PDE, but not SIV is capable of forming a complex with apo-CaM.
CaM–target interactions—CaMKI- α, SIV-N, CisPlat
The hydrodynamic radii of several other CaM-complexed species are given in Table 2. The collapse seen in the measurement of CaMKI-α (21.2 ± 1.1 Å) is also reflected in DLS data, for which an Rh value of 22 ± 1 Å has been reported. Similar to the PDE and SIV peptides, previous fluorescence and CD data are in support of a binding mode for this peptide analogous to MLCK, which is again consistent with the observed data (Gomes et al. 2000).
For the CaM–SIV-N complex (23.1 ± 0.6 Å), there has been no previous reported molecular size characterization of the CaM–SIV-N complex. Previous biochemical evidence is somewhat conflicting. NMR data suggests that there is relatively strong binding to the C-terminal lobe in a 1:1 ratio (Yuan et al. 2001), and further weak binding to the N-terminal lobe in a 2:1 ratio. On the other hand, the CD spectra demonstrate little change when the SIV-N peptide is titrated into CaM, in contrast to the canonical CaM–target result of increased helical content. Our results suggest that there may be a partial collapse of the CaM–SIV-N complex of ∼1.4 Å, about half that seen with the CaM–MLCK complex (2.7 Å). The interaction of this peptide with a fragment encompassing the C-terminal lobe of CaM would be an interesting target for further structural characterization, as the apparent lack of secondary structure indicated by the CD data, combined with the clear evidence for binding, suggest a noncanonical CaM-binding motif, perhaps similar to that seen for the N-terminal portion of the CaM-binding domain of the plasma membrane calcium pump (Elshorst et al. 1999).
The value of the Rh measured of the CaM–CisPlat complex, 21.4 ± 0.2 Å is quite intriguing, as it suggests a near-complete collapse of the CaM–CisPlat complex. Work in our laboratory (H. Ouyang and H.J. Vogel, in prep.) suggests that the binding of the drug to CaM occurs through the S atoms of the Met side chains in a 9:1 ratio, and inhibits the ability of CaM to bind the MLCK CaM-binding peptide. The unexpected complete collapse of the complex raises the possibility that there may be some tertiary interaction between the two lobes of CaM, which is not obvious from previous data.
Apo- and Ca2+-CIB
The measured hydrodynamic radius of CIB in the apo- (32.1 ± 1.6 Å) and Ca2+-form (24.5 ± 1.5 Å) reflects a collapse of approximately 7.6 Å. The relative error in the measurements for these samples is higher than in the CaM determinations most likely because of the significantly lower protein concentration used (∼0.15 mM for CIB versus ∼1.0 mM for CaM). Despite the error, it is readily apparent that there is a significant increase in the diffusion rate of Ca2+-CIB compared to the apo-form. This contrasts starkly with the Rh measurements for CaM in which there was little change in both these PFG-diffusion measurements and previous GPC studies (Sorensen and Shea 1996).
Possible explanations for a slower diffusion rate (and, hence, larger apparent Rh) include intermolecular aggregation, and/or the presence of unfolded protein regions. In the case of intermolecular aggregation, one would expect the slower diffusion to be caused by a large complex, which in turn, would also have slower molecular tumbling. In a typical NMR spectrum, a significant increase in the molecular tumbling rate causes an increase in the linewidth of protein resonances. In the case of CIB, the aromatic region of the 1H spectrum exhibited the opposite effect, with the Ca2+-CIB lines significantly broader than in the apo-CIB spectra (not shown). Such a result is consistent with the notion that the apo form is significantly disordered and extended, while the Ca2+ form is more compact. To test this hypothesis, 2D 1H -15N HSQC spectra were acquired of 15N-labeled CIB (Fig. 2 ▶). The spectra of Figure 2a ▶ is indicative of a highly unfolded protein, because of the lack of dispersion and close clustering of peaks around random coil values. In contrast, Figure 2b ▶ shows dispersion symptomatic of a folded protein with the amide resonance appreciably scattered. Hence, we conclude that it is likely that the increase in diffusion, and decrease in Rh, is primarily because of an intramolecular collapse upon binding Ca2+, although we cannot completely rule out the possibility that there may be some intermolecular aggregation.
Figure 2.
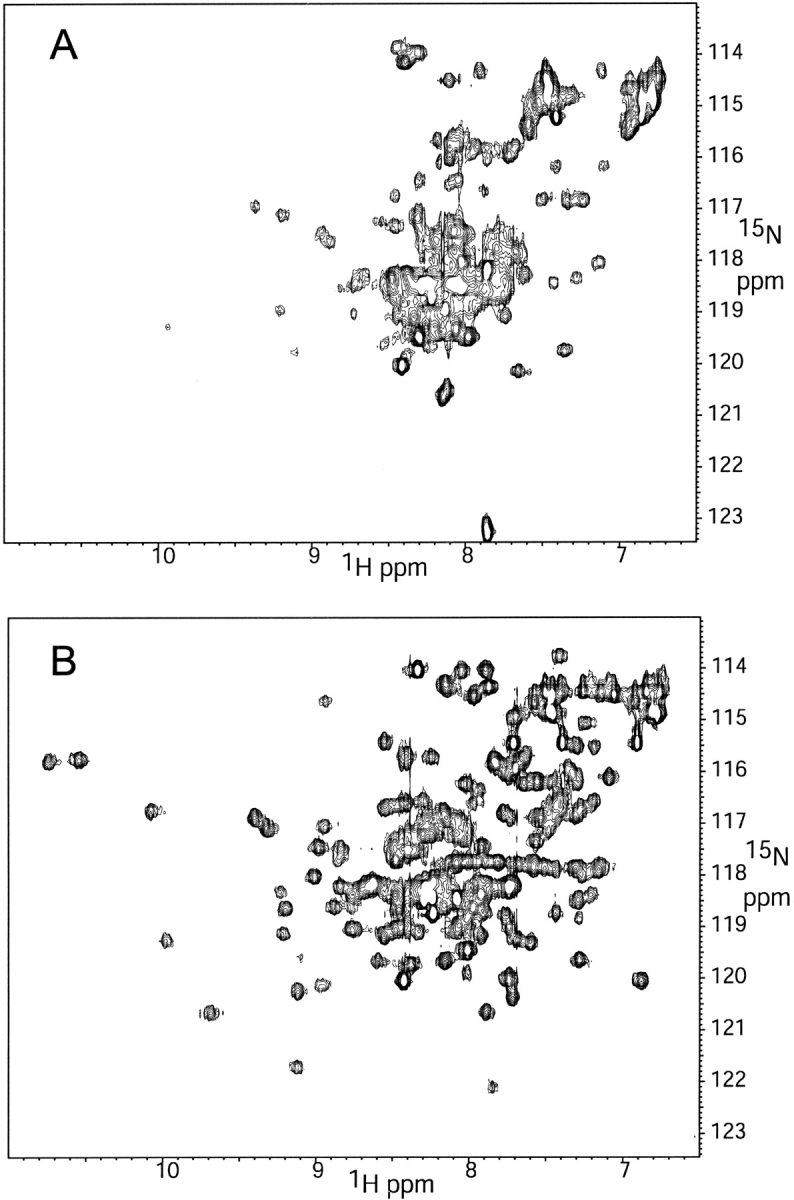
1H-15N-HSQC NMR spectra of (A) apo- and (B) Ca2+-saturated CIB. The dramatic increase in the number of visible peaks and dispersion is characteristic of transition from a chemically degenerate state in (A) to a more compact, folded form in (B).
Apo-, Ca2+-, and JD–CLD
The results for apo-CLD (25.1 ± 0.4) and Ca2+-CLD (21.3 ± 1.0) suggest that this protein domain acts in a manner analogous to CIB on binding calcium. There is a collapse of 3.8 Å, not quite as dramatic as CIB, but significantly on the order of 15%. Previous biochemical work characterizing the properties of the intact CDPK protein (Putnam-Evans et al. 1990; Harper et al. 1991; Yoo and Harmon 1996) has shown that the protein maintains monomeric characteristics in solution. As a result, we expect the change in the protein PFG-diffusion to be related to the structured (or unstructured) state, and not oligomerization. 1H 1D NMR and 1H-15N-HSQC spectra of the CLD (not shown) support this conclusion, as discussed in the above CIB section.
The CLD of soybean CDPKα is also thought to bind another region of the CDPK known as the JD (Table 1) (Yoo and Harmon 1996). The hydrodynamic radius of a complex of Ca2+-CLD–JD was measured to be 20.2 ± 0.7 Å. Although this might be indicative of a slight collapse on the order of 1 Å compared with the Ca2+ state, it is not possible to assert this with confidence because of the error in the two results. Titration of peptide encompassing the JD into Ca2+-CLD produces noticeable change in the 1H 1D spectrum of the aromatic region of the protein (not shown) highly indicative of binding. Evidence of direct binding between the CLD and JD in a manner analogous to CaM–target peptides, has also been reported previously (Yoo and Harmon 1996; Weljie et al. 2000). Interestingly, the results reported here suggest that in CLD the greatest conformational change occurs in the transition from the apo- to Ca2+- bound form, and not on target binding, again contrasting the CaM result of negligible size change upon calcium binding.
Apo- and Ca2+-CaB
The hydrodynamic radius for apo-CaB (25.0 ± 0.3 Å) was slightly larger than CaM, despite the fact that it has half the residues. On binding Ca2+, the Rh decreased by about 16% to 21.0 ± 0.6 Å (Table 2). This trend follows that observed for both CIB and CLD, and the 1H-15N HSQC data indicates that the protein exists primarily in an unfolded conformation in the apo-form (not shown). The hydrodynamic radius of the Ca2+ form was on the order seen for the CaM complex samples (Table 2), which would be unlikely if Ca2+-CaB existed as a fully folded monomeric protein, and hence, further analysis was conducted to assess this result as described below.
Analysis of the degree of compaction
The hydrodynamic radius alone can be a very useful value, especially when comparing relative conformational states such as the changes measured in this study on binding of Ca2+ ions. However, in cases where the effective radius of protein–protein interactions is being measured, it is useful to have a basis for comparison between a fully “compact” or globular state, and extended or “unfolded” states for various systems. We have found it useful to extend the formalism introduced by Wilkins et al. (1999) to examine protein folding. In their work, an empirical relationship was established using the PFG-diffusion technique for proteins in a globular, native state (RhN = (4.75 ± 1.11)N0.29 ± 0.02, where N is the number of residues), and in an unfolded, denatured state (RhD = (2.21 ± 1.07)N0.57 ± 0.02). Using these “extreme” values for a protein (or protein complex) consisting of N resides as well as the experimental Rh value, one can establish a dimensionless “Compaction factor” Cf:
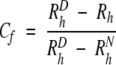 |
1 |
Accordingly, a fully denatured monomeric protein would have a Cf value of 0, and a fully globular protein a value of 1. For the purposes of the current study, a fully globular complex such as CaM–MLCK should have a Cf value near 1.00, and an extended molecule such as Ca2+-CaM should have a value between 0.00 and 1.00. Table 2 gives the Cf values for all the systems examined in this study. The advantages of this analysis is apparent by examining the CaM–skMLCK and CaM–CisPlat complexes. The hydrodynamic radii, Cf are 21.8 Å, 0.97 (skMLCK) and 21.4 Å, 0.94 (CisPlat), so while the overall CaM–CisPlat complex is smaller than that of CaM–skMLCK, when the contribution from the peptide (22 residues) is taken into account, the skMLCK complex is found to be more compact (Table 2). Not surprisingly, the apo- and Ca2+ forms show similar Cf values near 0.75, and the CaM complexes are beween 0.94 and 0.99, with the exception of CaM–SIV-N which has a Cf value near 0.87, highlighting the partial degree of collapse on target binding in this case.
The compaction factors for the other Ca2+-binding proteins examined in this study highlight the varying degrees to which Ca2+-binding causes conformational change. In the case of CIB, the change is quite dramatic (from 0.56 to 0.90), reflecting the degree to which folding of the protein is dependent upon Ca2+-binding. Interestingly, the Ca2+-form is much more compact relative to Ca2+-CaM, which may reflect the fact that the calcium-binding region is one domain of the protein, and/or that both lobes of CIB interact with each other (Hwang and Vogel 2000).
The difference in Ca2+-binding for the CLD protein is not nearly as dramatic, changing from 0.83 to 1.00 on Ca2+-binding. The fact that the Ca2+-form has a value indicative of a fully folded protein is intriguing, especially considering that the JD–CLD complex has an even larger value of 1.08. However, it should be noted that the Cf is dependent on the empirical equations for the extended and globular states, and the globular state equation [(RhN = (4.75 ± 1.11)N0.29 ± 0.02] is sensitive to anisotropic structures (Wilkins et al. 1999). It is likely that the CLD does collapse slightly on binding JD, but that the Ca2+ and complex forms are not completely globular, which indeed corresponds to 3D structural evidence from our laboratory (A.M. Weljie and H.J. Vogel, in prep.).
The Cf values for apo- (0.19) and Ca2+-CaB (0.59) are also quite intriguing as they suggest that the apo form of the protein is almost entirely unfolded, but that upon binding Ca2+ there is only a partial refolding of the protein. However, NMR evidence indicates that the protein is fully folded (Dr. R.T. McKay, pers. comm.). As a result, when another compaction factor was calculated assuming dimerization of the Ca2+-CaB, a new value of 0.93 is obtained that is much closer to what would be expected for a globular folded protein. This is not completely conclusive evidence of dimerization as the monomer Cf value of 0.59 may be because of nonspecific aggregation as opposed to “pure” dimerazation. Further studies are required for verification, for example, determining the concentration dependence of the apparent Rh, or through the use of another technique such as sedimentation equilibrium analysis (Hicks et al. 2002). Combined with such information, we and others have previously successfully used the PFG-diffusion technique to characterize the multimerization of various proteins and peptides (Altieri et al. 1995; Ekiel and Abrahamson 1996; Mansfield et al. 1998; Hunter et al. 2002; Schibli et al. 2002).
Materials and methods
Protein and peptide samples for NMR spectroscopy
Wild-type CaM was biosynthetically expressed and subsequently purified using double hydrophobic phenyl Sepharose chromatography as described previously (Waltersson et al. 1993; Zhang and Vogel 1993), as was CLD (Weljie et al. 2000). CaB was expressed from a pET19b vector (Novagen) in Escherichia coli and purified by Nickel chelate affinity chromatography by Dr. R.T.McKay (University of Alberta). CIB was expressed with a 10 Histidine N-terminal tag from an optimized synthetic gene construct in E. coli, purified by Nickel chelate affinity chromatograpy, and then dialyzed extensively against 8 mM ammonium bicarbonate pH 7.5 before lyophilization (A.P. Yamniuk, T. Huang, and H.J. Vogel, unpubl.). Peptides used in this study (Table 1) were synthesized by the Queen’s University Core Facility for Protein/DNA Chemistry, and were >95% pure as determined by HPLC and mass spectrometry.
Typical sample conditions for CaM samples were 1 mM CaM, 100 mM KCl, unbuffered at pH 7.3 in 99.96% D2O (no correction for pD was made), with either 3 mM EDTA for apo samples or 10 mM CaCl2 for Ca2+- and target complex samples. CaM–target peptides were quantitated based on a single Trp ɛ280 of 5500 M−1 (or 11,000 M−1 in the case of the SIV peptide), and added to a ratio of 1.2:1 (peptide:protein); 0.50 μL of neat dioxane was added to the samples, with 1D 1H spectra acquired before and after to ensure that the dioxane did not change the protein resonances.
Samples for the other protein were similar, with the following exceptions. The concentration of CaB and CLD samples was 0.20 mM, and CIB, 0.15 mM. KCl (200 mM) was added to the CaB and CLD samples, and the pH of the CaB samples was 7.6. For these three proteins apo-spectra were first acquired with 1.0 mM EDTA, and subsequently CaCl2 added to 3.5 mM for the Ca2+ samples. The JD peptide was initially quantitated by the method of Lowry, and subsequently taken to be 60% of the dry weight. Peptide was titrated into the protein until no change was observed in the aromatic region of the 1D 1H spectrum. The lysozyme sample was 1 mM in protein, 100 mM KCl, pH 6.0.
NMR spectroscopy
All spectra were acquired on a Bruker shielded magnet having a 1H frequency of 700 MHz equipped with a 1H, X, 13C probe with triple axis gradients. The variable gradient diffusion data was acquired based on the PG-SLED pulse sequence of Wilkins et al. (1999) modified to acquire as a 2D series file. Sixty-four gradient experiments were acquired for each dataset, with the gradient strengths augmented linearly through the acquisition from 0 to 40 G/cm, and all other delays and pulses held constant. Gradient pulses were applied for 6.3 msec with a recovery time of 0.7 msec, and diffusion delay of 100 msec. This was found to be adequate for all proteins to give a total decay of ∼90%. Either 32 transients (CaM samples) or 256 transients (CIB, CLD, and CaB) were acquired per gradient experiment. Each experiment was acquired with a spectral width of 10,000 Hz, and 8-k complex points. The data were zero filled to 16-k complex points, and apodized with a sine-squared function. The baseline was corrected with two separate polynomial functions, 0.15 ppm upfield and 0.15 ppm downfield of the water peak.
Data analysis
Data were analyzed using the variable gradient fitting routines in XWINNMR 3.0 from Bruker, and in all cases protein resonances were fit with a single exponential decay function using peak intensities (Wilkins et al. 1999). The dioxane peak intensity decay was fitted to either a mono- or bi-exponential decay depending on overlap with protein resonances. The signal from the dioxane peak was in all cases much greater than any protein resonances, and thus both the mono- and bi-exponential decay curves showed excellent fits with the experimental data. The use of highly purified D2O also allowed us to use the HOD peak as another internal standard to verify the observed trends in the data, although it was more sensitive to salt and pH effects than dioxane and the results are not reported here. The protein decay values are an average value obtained based on intensities measured from a minimum of five separate peaks in both the aromatic and aliphatic regions. The reported error is the range of values obtained by fitting these individual resonances within samples, as this resulted in significantly greater error than sample to sample differences of the average values. The hydrodynamic radius was determined by the ratio of (ddioxane/dprotein) with increasing gradient strength, which eliminated the need to determine absolute diffusion coefficients (Jones et al. 1997; Wilkins et al. 1999), and thus the high degree of reproducibility between samples. The hydrodynamic radius of internal dioxane standard was calibrated based upon the known structure and size of lysozyme (Wilkins et al. 1999) to provide a value of 1.87 Å. The Rh values of the proteins and complexes were then calculated based on the relationship Rhprot = (ddioxane/dprotein)Rhref, where Rhref is the calibrated dioxane radius.
Small-angle X-ray scattering
Data collection and analysis of the small-angle X-ray scattering data was performed exactly as described before (Yoshino et al. 1996; Izumi et al. 2001).
Acknowledgments
This work was supported by an operating grant from the Canadian Institute of Health Research. H.J.V. holds a senior scientist award from the Alberta Heritage Foundation for Medical Research (AHFMR), while A.M.W. is the recipient of an AHFMR Studentship award. We are indebted to Dr. Ryan McKay for providing the sample of calbrain, to Dr. Hui Ouyang for providing the CaM cisplatin sample, and to Dr. V. Leontiev and Dr. T. Hoang for establishing the expression vectors for calbrain and CIB, respectively. We greatly acknowledge helpful comments made by Dr. H.N. Hunter. The 700-MHz NMR spectrometer in the Bio-NMR center was purchased through grants provided by the Canada Foundation for Innovation, with matching support from the Alberta Science and Research Council (ASRA), the Alberta Intellectual Infrastructure Parnership program (IIPP), and the Alberta Heritage Foundation for Medical Research (AHFMR). Maintenance of the Bio-NMR Center is supported by the Canadian Institutes for Health Research and the University of Calgary.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CaM, calmodulin
Rh, hydrodynamic radius
Rg, radius of gyration
PFG, pulse field gradient
NMR, nuclear magnetic resonance
GPC, gel permeation chromatography
DLS, dynamic light scattering
skMLCK, skeletal muscle myosin light-chain kinase
PDE, cyclic nucleotide phosphodiesterase
SIV, simian immunodeficiency virus
CaMKI, calmodulin-dependent protein kinase I
CIB, calcium and integrin binding protein
CaB, calbrain
CLD, calmodulin-like domain
CDPK, calmodulin-like domain protein kinase
JD, junction domain
SAXS, small-angle X-ray scattering
HSQC, heteronuclear single quantum coherence
Cf, compaction factor
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0226203.
References
- Altieri, A.S., Hinton, D.P., and Byrd, R.A. 1995. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 117 7566–7567. [Google Scholar]
- Babu, Y.S., Bugg, C.E., and Cook, W.J. 1988. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 204 191–204. [DOI] [PubMed] [Google Scholar]
- Barbato, G., Ikura, M., Kay, L.E., Pastor, R.W., and Bax, A. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: The central helix is flexible. Biochemistry 31 5269–5278. [DOI] [PubMed] [Google Scholar]
- Bhattacharjya, S., Xu, P., Xiang, H., Chretien, M., Seidah, N.G., and Ni, F. 2001. pH-induced conformational transitions of a molten-globule-like state of the inhibitory prodomain of furin: Implications for zymogen activation. Protein Sci. 10 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal, D.K., Takio, K., Edelman, A.M., Charbonneau, H., Titani, K., Walsh, K.A., and Krebs, E.G. 1985. Identification of the calmodulin-binding domain of skeletal muscle myosin light chain kinase. Proc. Natl. Acad. Sci. 82 3187–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokx, R.D., Lopez, M.M., Vogel, H.J., and Makhatadze, G.I. 2001. Energetics of target peptide binding by calmodulin reveals different modes of binding. J. Biol. Chem. 276 14083–14091. [DOI] [PubMed] [Google Scholar]
- Charbonneau, H., Kumar, S., Novack, J.P., Blumenthal, D.K., Griffin, P.R., Shabanowitz, J., Hunt, D.F., Beavo, J.A., and Walsh, K.A. 1991. Evidence for domain organization within the 61-kDa calmodulin-dependent cyclic nucleotide phosphodiesterase from bovine brain. Biochemistry 30 7931–7940. [DOI] [PubMed] [Google Scholar]
- Choy, W.Y., Mulder, F.A., Crowhurst, K.A., Muhandiram, D.R., Millett, I.S., Doniach, S., Forman-Kay, J.D., and Kay, L.E. 2002. Distribution of molecular size within an unfolded state ensemble using small-angle x-ray scattering and pulse field gradient NMR techniques. J. Mol. Biol. 316 101–112. [DOI] [PubMed] [Google Scholar]
- Crouch, T.H. and Klee, C.B. 1980. Positive cooperative binding of calcium to bovine brain calmodulin. Biochemistry 19 3692–3698. [DOI] [PubMed] [Google Scholar]
- Drum, C.L., Yan, S.Z., Bard, J., Shen, Y.Q., Lu, D., Soelaiman, S., Grabarek, Z., Bohm, A., and Tang, W.J. 2002. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415 396–402. [DOI] [PubMed] [Google Scholar]
- Ekiel, I. and Abrahamson, M. 1996. Folding-related dimerization of human cystatin C. J. Biol. Chem. 271:1314–1321. [DOI] [PubMed] [Google Scholar]
- Elshorst, B., Hennig, M., Forsterling, H., Diener, A., Maurer, M., Schulte, P., Schwalbe, H., Griesinger, C., Krebs, J., Schmid, H., et al. 1999. NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+ pump. Biochemistry 38 12320–12332. [DOI] [PubMed] [Google Scholar]
- Gomes, A.V., Barnes, J.A., and Vogel, H.J. 2000. Spectroscopic characterization of the interaction between calmodulin-dependent protein kinase I and calmodulin. Arch. Biochem. Biophys. 379 28–36. [DOI] [PubMed] [Google Scholar]
- Gonnella, N., Lin, M., Shapiro, M.J., Wareing, J.R., and Zhang, X. 1998. Isotope-filtered affinity NMR. J. Magn. Reson. 131 336–338. [DOI] [PubMed] [Google Scholar]
- Gounarides, J.S., Chen, A., and Shapiro, M.J. 1999. Nuclear magnetic resonance chromatography: Applications of pulse field gradient diffusion NMR to mixture analysis and ligand-receptor interactions. J. Chromatogr. B Biomed. Sci. Appl. 725 79–90. [DOI] [PubMed] [Google Scholar]
- Harper, J.F., Sussman, M.R., Schaller, G.E., Putnam-Evans, C., Charbonneau, H., and Harmon, A.C. 1991. A calcium-dependent protein kinase with a regulatory domain similar to calmodulin. Science 252 951–954. [DOI] [PubMed] [Google Scholar]
- Heidorn, D.B. and Trewhella, J. 1988. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry 27 909–915. [DOI] [PubMed] [Google Scholar]
- Heidorn, D.B., Seeger, P.A., Rokop, S.E., Blumenthal, D.K., Means, A.R., Crespi, H., and Trewhella, J. 1989. Changes in the structure of calmodulin induced by a peptide based on the calmodulin-binding domain of myosin light chain kinase. Biochemistry 28 6757–6764. [DOI] [PubMed] [Google Scholar]
- Hicks, L.D., Alattia, J.R., Ikura, M., and Kay, C.M. 2002. Multiangle laser light scattering and sedimentation equilibrium. Methods Mol. Biol. 173 127–136. [DOI] [PubMed] [Google Scholar]
- Hoeflich, K.P. and Ikura, M. 2002. Calmodulin in action. Diversity in target recognition and activation mechanisms. Cell 108 739–742. [DOI] [PubMed] [Google Scholar]
- Hunter, H.N., Fulton, D.B., Ganz, T., and Vogel, H.J. 2002. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J. Biol. Chem. 277 37597–37603. [DOI] [PubMed] [Google Scholar]
- Hwang, P.M. and Vogel, H.J. 2000. Structures of the platelet calcium- and integrin-binding protein and the αIIb-integrin cytoplasmic domain suggest a mechanism for calcium-regulated recognition; homology modelling and NMR studies. J. Mol. Recognit. 13 83–92. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Clore, G.M., Gronenborn, A.M., Zhu, G., Klee, C.B., and Bax, A. 1992. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256 632–638. [DOI] [PubMed] [Google Scholar]
- Izumi, Y., Kuwamoto, S., Jinbo, Y., and Yoshino, H. 2001. Increase in the molecular weight and radius of gyration of apocalmodulin induced by binding of target peptide: Evidence for complex formation. FEBS Lett. 495 126–130. [DOI] [PubMed] [Google Scholar]
- Jones, J.A., Wilkins, D.K., Smith, L.J., and Dobson, C.M. 1997. Characterization of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 10 199–203. [Google Scholar]
- Kawasaki, H., Nakayama, S., and Kretsinger, R.H. 1998. Classification and evolution of EF-hand proteins. Biometals 11 277–295. [DOI] [PubMed] [Google Scholar]
- Kuboniwa, H., Tjandra, N., Grzesiek, S., Ren, H., Klee, C.B., and Bax, A. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2 768–776. [DOI] [PubMed] [Google Scholar]
- Lewit-Bentley, A. and Rety, S. 2000. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 10 637–643. [DOI] [PubMed] [Google Scholar]
- Liu, M., Nicholson, J.K., and Lindon, J.C. 1997. Analysis of drug-protein binding using nuclear magnetic resonance based molecular diffusion measurements. Anal. Commun. 34 225–228. [Google Scholar]
- Mansfield, S.L., Jayawickrama, D.A., Timmons, J.S., and Larive, C.K. 1998. Measurement of peptide aggregation with pulsed-field gradient nuclear magnetic resonance spectroscopy. Biochim. Biophys. Acta 1382 257–265. [DOI] [PubMed] [Google Scholar]
- Matsushima, N., Izumi, Y., Matsuo, T., Yoshino, H., Ueki, T., and Miyake, Y. 1989. Binding of both Ca2+ and mastoparan to calmodulin induces a large change in the tertiary structure. J. Biochem. (Tokyo) 105 883–887. [DOI] [PubMed] [Google Scholar]
- Meador, W.E., Means, A.R., and Quiocho, F.A. 1992. Target enzyme recognition by calmodulin: 2.4 Å structure of a calmodulin-peptide complex. Science 257 1251–1255. [DOI] [PubMed] [Google Scholar]
- ———. 1993. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 262 1718–1721. [DOI] [PubMed] [Google Scholar]
- Mehler, E.L., Pascual-Ahuir, J.L., and Weinstein, H. 1991. Structural dynamics of calmodulin and troponin C. Protein Eng. 4 625–637. [DOI] [PubMed] [Google Scholar]
- Morar, A.S., Olteanu, A., Young, G.B., and Pielak, G.J. 2001. Solvent-induced collapse of α-synuclein and acid-denatured cytochrome c. Protein Sci. 10 2195–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, C.J., Wilkins, D.K., Smith, L.J., Kawata, Y., and Dobson, C.M. 2000. A compact monomeric intermediate identified by NMR in the denaturation of dimeric triose phosphate isomerase. J. Mol. Biol. 300 11–16. [DOI] [PubMed] [Google Scholar]
- Naik, U.P., Patel, P.M., and Parise, L.V. 1997. Identification of a novel calcium-binding protein that interacts with the integrin αIIb cytoplasmic domain. J. Biol. Chem. 272 4651–4654. [DOI] [PubMed] [Google Scholar]
- Osawa, M., Tokumitsu, H., Swindells, M.B., Kurihara, H., Orita, M., Shibanuma, T., Furuya, T., and Ikura, M. 1999. A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat. Struct. Biol. 6 819–824. [DOI] [PubMed] [Google Scholar]
- Otto, W.H. and Larive, C.K. 2001. Improved spin-echo-edited NMR diffusion measurements. J. Magn. Reson. 153 273–276. [DOI] [PubMed] [Google Scholar]
- Pan, H., Barany, G., and Woodward, C. 1997. Reduced BPTI is collapsed. A pulsed field gradient NMR study of unfolded and partially folded bovine pancreatic trypsin inhibitor. Protein Sci. 6 1985–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papish, A.L., Tari, L.W., and Vogel, H.J. 2002. Dynamic light scattering study of calmodulin-target peptide complexes. Biophys. J. 83 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam-Evans, C.L., Harmon, A.C., and Cormier, M.J. 1990. Purification and characterization of a novel calcium-dependent protein kinase from soybean. Biochemistry 29 2488–2495. [DOI] [PubMed] [Google Scholar]
- Schibli, D.J., Hunter, H.N., Aseyev, V., Starner, T.D., Wiencek, J.M., McCray, Jr., P.B., Tack, B.F., and Vogel, H.J. 2002. The solution structures of the human β-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 277 8279–8289. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Rivard, A.F., Bachinger, H.P., and Adelman, J.P. 2001. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410 1120–1124. [DOI] [PubMed] [Google Scholar]
- Seaton, B.A., Head, J.F., Engelman, D.M., and Richards, F.M. 1985. Calcium-induced increase in the radius of gyration and maximum dimension of calmodulin measured by small-angle X-ray scattering. Biochemistry 24 6740–6743. [DOI] [PubMed] [Google Scholar]
- Sorensen, B.R. and Shea, M.A. 1996. Calcium binding decreases the Stokes radius of calmodulin and mutants R74A, R90A, and R90G. Biophys. J. 71 3407–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewhella, J. 1992. The solution structures of calmodulin and its complexes with synthetic peptides based on target enzyme binding domains. Cell Calcium 13 377–390. [DOI] [PubMed] [Google Scholar]
- van der Spoel, D., de Groot, B.L., Hayward, S., Berendsen, H.J., and Vogel, H.J. 1996. Bending of the calmodulin central helix: A theoretical study. Protein Sci. 5 2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain, R., Pertinhez, T.A., Tomlinson, E.J., Hong, L., Dobson, C.M., Ferguson, S.J., and Smith, L.J. 2001. The cytochrome c fold can be attained from a compact apo state by occupancy of a nascent heme binding site. J. Biol. Chem. 276 45813–45817. [DOI] [PubMed] [Google Scholar]
- Waltersson, Y., Linse, S., Brodin, P., and Grundstrom, T. 1993. Mutational effects on the cooperativity of Ca2+ binding in calmodulin. Biochemistry 32 7866–7871. [DOI] [PubMed] [Google Scholar]
- Weljie, A.M., Clarke, T.E., Juffer, A.H., Harmon, A.C., and Vogel, H.J. 2000. Comparative modeling studies of the calmodulin-like domain of calcium-dependent protein kinase from soybean. Proteins 39 343–357. [DOI] [PubMed] [Google Scholar]
- Wilkins, D.K., Grimshaw, S.B., Receveur, V., Dobson, C.M., Jones, J.A., and Smith, L.J. 1999. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38 16424–16431. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, K., Yamaguchi, F., Miyamoto, O., Sugimoto, K., Konishi, R., Hatase, O., and Tokuda, M. 1999. Calbrain, a novel two EF-hand calcium-binding protein that suppresses Ca2+/calmodulin-dependent protein kinase II activity in the brain. J. Biol. Chem. 274 3610–3616. [DOI] [PubMed] [Google Scholar]
- Yoo, B.C. and Harmon, A.C. 1996. Intramolecular binding contributes to the activation of CDPK, a protein kinase with a calmodulin-like domain. Biochemistry 35 12029–12037. [DOI] [PubMed] [Google Scholar]
- Yoshino, H., Izumi, Y., Sakai, K., Takezawa, H., Matsuura, I., Maekawa, H., and Yazawa, M. 1996. Solution X-ray scattering data show structural differences between yeast and vertebrate calmodulin: Implications for structure/function. Biochemistry 35 2388–2393. [DOI] [PubMed] [Google Scholar]
- Yuan, T., Mietzner, T.A., Montelaro, R.C., and Vogel, H.J. 1995. Characterization of the calmodulin binding domain of SIV transmembrane glycoprotein by NMR and CD spectroscopy. Biochemistry 34 10690–10696. [DOI] [PubMed] [Google Scholar]
- Yuan, T., Walsh, M.P., Sutherland, C., Fabian, H., and Vogel, H.J. 1999. Calcium-dependent and -independent interactions of the calmodulin-binding domain of cyclic nucleotide phosphodiesterase with calmodulin. Biochemistry 38 1446–1455. [DOI] [PubMed] [Google Scholar]
- Yuan, T., Tencza, S., Mietzner, T.A., Montelaro, R.C., and Vogel, H.J. 2001. Calmodulin binding properties of peptide analogues and fragments of the calmodulin-binding domain of simian immunodeficiency virus transmembrane glycoprotein 41. Biopolymers 58 50–62. [DOI] [PubMed] [Google Scholar]
- Zhang, M. and Vogel, H.J. 1993. Determination of the side chain pKa values of the lysine residues in calmodulin. J. Biol. Chem. 268 22420–22428. [PubMed] [Google Scholar]
- Zhang, M., Yuan, T., and Vogel, H.J. 1993. A peptide analog of the calmodulin-binding domain of myosin light chain kinase adopts an α-helical structure in aqueous trifluoroethanol. Protein Sci. 2 1931–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M., Tanaka, T., and Ikura, M. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2 758–767. [DOI] [PubMed] [Google Scholar]
- Zhu, H. and Snyder, M. 2002. “Omic” approaches for unraveling signaling networks. Curr. Opin. Cell Biol. 14 173–179. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R. 2002. Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry 41 1–7. [DOI] [PubMed] [Google Scholar]