

Abstract
Unlike the heme cd1-based nitrite reductase enzymes, the molecular mechanism of copper-containing nitrite reductases remains controversial. A key source of controversy is the productive binding mode of nitrite in the active site. To identify and characterize the molecular determinants associated with nitrite binding, we applied a combinatorial mutagenesis approach to generate a small library of six variants at position 257 in nitrite reductase from Alcaligenes faecalis S-6. The activities of these six variants span nearly two orders of magnitude with one variant, I257V, the only observed natural substitution for Ile257, showing greater activity than the native enzyme. High-resolution (> 1.8 Å) nitrite-soaked crystal structures of these variants display different modes of nitrite binding that correlate well with the altered activities. These studies identify for the first time that the highly conserved Ile257 in the native enzyme is a key molecular determinant in directing a catalytically competent mode of nitrite binding in the active site. The O-coordinate bidentate binding mode of nitrite observed in native and mutant forms with high activity supports a catalytic model distinct from the heme cd1 NiRs.
(The atomic coordinates for I257V[NO2−], I257L[NO2−], I257A[NO2−], I257T[NO2−], I257M[NO2−] and I257G[NO2−] AfNiR have been deposited in the Protein Data Bank [PDB identification codes are listed in Table 2].)
Keywords: Nitrite reductase, copper, denitrification, X-ray crystallography
In some facultative anaerobic microorganisms, bioenergetic respiration is coupled to the reduction of nitrate and nitrite to gaseous nitrogen oxide compounds (NO, N2O) and dinitrogen (N2) during dissimilatory dentrification (Averill 1996; Cutruzzola 1999). The first committed step in the dissimilatory pathway is catalyzed by NiR, which converts a mineral form of nitrogen (nitrite–NO2−) to a gaseous form (nitric oxide–NO). NiRs are common to many facultative anaerobic bacteria and occur in two genetically distinct forms that contain either heme c and heme d1 prosthetic groups or multiple copper centers (CuNiRs) (for review, see Cutruzzola 1999). Many of these NiRs are not yet purified but were identified using DNA hybridization techniques (Smith and Tiedje 1992; Ye et al. 1993) or PCR-based methods (Braker et al. 1998). Interestingly, the nitrite reductase activity of a heme cd1 NiR deficient strain of Pseudomonas stutzeri can be complemented with a CuNiR demonstrating that both classes of enzymes perform the same function in vivo (Glockner et al. 1993).
Cytochrome cd1 denitrifying nitrite reductases are soluble homodimers of 120 kD. Each monomer binds covalently a heme c prosthetic center that accepts an electron from soluble cytochrome c551 or azurin (Averill 1996), and is noncovalently associated with a heme d1 group where nitrite binding and catalysis occurs (Hochstein and Tomlinson 1989; Weeg-Aerssens et al. 1991). The mechanism of heme cd1 nitrite reductases is well characterized from spectroscopic measurements, crystallographic and isotopic exchange studies (Averill 1996) and proceeds with nitrite binding in an N-coordinate fashion to a reduced heme d1 group followed by formation of an electrophilic nitrosyl intermediate (Fulop et al. 1995).
CuNiRs are soluble periplasmic homotrimers of ∼110 kD with each monomer comprising two Greek key β-barrel domains (Godden et al. 1991; Kukimoto et al. 1994) and coordinating two spectroscopically different copper atoms. Crystallographic, spectroscopic, and functional studies of CuNiRs from several species have revealed that the type I copper center is the site of electron transfer from a proteaceous electron donor, and the type II copper is the site of nitrite reduction (Libby and Averill 1992; Abraham et al. 1993; Kukimoto et al. 1994). The two copper sites are ∼12.5 Å apart and are intimately linked through a Cys-His bridge incorporating the type I ligand Cys1364 and the type II ligand His135.
In the native nitrite-soaked structure of AfNiR, nitrite binds in a bidentate fashion and forms a hydrogen bond with Asp98, which in turn connects to His255 through a solvent-bridged hydrogen bond (Murphy et al. 1997). Since this original observation, several kinetic, spectroscopic, and structural studies have elucidated further the role of Asp98 and His255 in CuNiRs (Prudencio et al. 1999; Boulanger et al. 2000; Kataoka et al. 2000; Zhang et al. 2000; Boulanger and Murphy 2001). The hydrogen bond between Asp98 and the bound nitrite is key for a fully functional enzyme, likely acting as a direct conduit for the transfer of protons during catalysis (Boulanger et al. 2000; Kataoka et al. 2000; Boulanger and Murphy 2001). Although no direct interaction is observed in the native enzyme between His255 and the bound nitrite, structural evidence suggests that His255 plays a role in directing the mode of nitrite binding through an active site hydrogen bond network (Boulanger and Murphy 2001). Despite the extensive characterization of these catalytically essential residues, the role of the blanket of hydrophobic residues that form the nitrite-binding pocket is largely unexplored. Little is known about the molecular determinants of nitrite binding, contributing to the controversy associated with the proposed mechanisms for CuNiRs.
The first catalytic model for CuNiRs proposed by Hulse and Averill (1989) and revised by Dodd and coworkers (Dodd et al. 1997) is similar to that of the heme cd1 NiRs where nitrite binds N-coordinate to a reduced metal center and the reaction proceeds through the formation of an electrophilic nitrosyl intermediate. The chemical intermediate proposed in this mechanism requires the substrate to be N-coordinate to the type II copper. The chemical relevance of this binding mode for nitrite is supported by studies where nitrite bound in an N-coordinate fashion to copper in biomimetic complexes results in stoichiometric production of NO (Halfen and Tolman 1994).
A distinct second model describing the catalytic process for CuNiRs has been proposed where nitrite binds in an O-coordinate fashion to an oxidized type II copper atom prior to electron transfer from the type I copper (Murphy et al. 1997; Boulanger et al. 2000; Boulanger and Murphy 2001). The chemically unusual O-coordinate binding mode for nitrite originally inferred by ENDOR spectroscopy (Howes et al. 1994) was later confirmed with EXAFS spectroscopy (Strange et al. 1995, 1999) and crystallography (Murphy et al. 1997). The O-dependent coordination of nitrite limits the formation of a nitrosyl species suggested by the first mechanism. Further support for this model is provided by cyclic voltametry and EPR spectroscopic (Olesen et al. 1998) studies on NiR from Rhodobacter sphaeroides, which shows that intramolecular electron transfer is favored following nitrite binding to an oxidized type II copper center.
To understand the basis of substrate recognition by CuNiRs, we probed the role of a highly conserved active site Ile257 as a key molecular determinant in directing the mode of nitrite binding in CuNiRs. Functional and structural characterization of six variants of Ile257 reveals an intimate role for Ile257 in directing a catalytically productive bidentate mode of nitrite binding in the active site. These observations provide a unique insight into the molecular mechanism of CuNiRs and are discussed with respect to the current mechanistic models.
Results
Characterization of AfNiR variants
A small combinatorial library of six mutations was generated at position 257 in the active site of AfNiR (Table 1). All mutations were confirmed by DNA sequence analysis, which showed that no one mutation was dominant. Mass spectrometry showed each purified variant protein to be within 4 Da of the expected molecular mass following removal of the N-terminal methionine (Table 2). Absorbance maxima and ratios from UV-Vis spectroscopy are at the same three characteristic wavelengths of 458, 595, and 680 nm as seen in previous preparations of native AfNiR (Kakutani et al. 1981; Kukimoto et al. 1994).
Table 1.
Degenerate primers used in the combinatorial mutagenesis of Ile257
| % X1 | % X2 | % X3 | |||||||||||
| Forward primer |
5′ GAG ACC ACA TCG 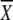 1 1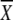 2 2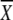 3 GG 3 GG |
A | G | T | C | A | G | T | C | A | G | T | C |
| GGG GCA TGG GGA TTA TG 3′ | a21 | 46 | 0 | 33 | 0 | 17 | 41 | 42 | 33 | 33 | 33 | 0 | |
| Reverse primer |
5′ CAT AAT CCC GCC C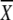 1 1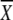 2 2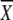 3 3
|
A | G | T | C | A | G | T | C | A | G | T | C |
| CA GAT GTG GTC TC 3′ | 33 | 33 | 0 | 33 | 41 | 42 | 0 | 17 | 0 | 33 | 21 | 46 | |
a These numbers represent the % by weight of each nucleotide at the X1,2 and 3 positions within the mutated codon.
Table 2.
Characterization of native and Ile257 variant AfNiRs
| AfNiR | A458/595 | A277/458 | Mass (Da)a | Specific activity (U/mg)b |
| Native | 1.30 | 16.6 | 36847 (36843) | 417 (44)c |
| I257V | 1.33 | 18.5 | 15 (14) | 521 (50) |
| I257L | 1.26 | 19.1 | 1 (1) | 108 (14) |
| I257M | 1.35 | 17.0 | 22 (18) | 17.9 (3.4) |
| I257A | 1.32 | 17.9 | 43 (42) | 15.6 (0.8) |
| I257G | 1.36 | 17.0 | 57 (56) | 10.4 (0.9) |
| I257T | 1.29 | 17.1 | 12 (12) | 5.90 (0.7) |
a The absolute mass of the native is given along with the theoretical mass in parentheses. The values for the mutants are the absolute observed and theoretical mass (in parentheses) differences relative to the native.
b Specific activity values are reported as an average of at least three trials.
c Values in parentheses represent the standard error.
Using dithionite-reduced methyl viologen as the artificial electron donor, the specific activity measured for native AfNiR is within experimental error (10%) of that measured previously (Boulanger et al. 2000). The activities of the variant AfNiR species span nearly two orders of magnitude (Table 2). The replacement of Ile257 with a valine results in an elevated specific activity relative to the native enzyme, while the conservative mutation, I257L, shows a three to fourfold decrease in activity. A reduction of 25- to 40-fold in specific activity is observed for the I257A, I257G, and I257M variants. The replacement of Ile257 with a threonine results in the most significant loss of activity of > 98% relative to the native enzyme.
Overall structures
Each of the nitrite-soaked variant AfNiR crystal structures is refined to a resolution of at least 1.8 Å facilitating a detailed comparison between the observed binding modes of nitrite. Each structure shows excellent stereochemistry as indicated by over 90% of the residues adopting the most favorable conformations as determined by a Ramachandran plot (Laskowski et al. 1993) with the remaining residues in the allowed regions. Data collection and refinement statistics are presented in Table 3.
Table 3.
Data collection and refinement statistics
| Crystal | I257L [NO2−] | I257V [NO2−] | I257A [NO2−] | I257G [NO2−] | I257M [NO2−] | I257T [NO2−] |
| Cell | a = 61.95 | a = 61.73 | a = 62.00 | a = 62.05 | a = 61.77 | a = 61.48 |
| Dimensions (Å) | b = 102.5 | b = 102.3 | b = 102.4 | b = 102.4 | b = 102.4 | b = 102.0 |
| c = 146.2 | c = 145.9 | c = 146.2 | c = 146.2 | c = 146.0 | c = 146.1 | |
| Resolution (Å) | 1.70 | 1.l75 | 1.70 | 1.75 | 1.78 | 1.78 |
| R-merge | 4.7 (21.6)a | 6.2 (26.2) | 6.8 (21.6) | 5.7 (32.4) | 8.2 (30.6) | 6.6 (27.4) |
| {I}/{σ(I)}b | 23.6 (4.4) | 18.3 (3.6) | 16.8 (4.1) | 19.6 (2.8) | 16.0 (3.2) | 15.1 (4.1) |
| Completeness (%) | 91 (64.2) | 95 (83.5) | 93 (74.5) | 98 (90.7) | 95 (77.6) | 87 (71.9) |
| Unique reflections | 93,657 | 89,868 | 95,952 | 92,678 | 84,883 | 77,077 |
| Redundancy | 7.9 | 11.7 | 7.9 | 7.6 | 9.6 | 9.8 |
| Rwork | 0.156 | 0.166 | 0.158 | 0.159 | 0.154 | 0.157 |
| Rfree | 0.188 | 0.208 | 0.192 | 0.194 | 0.197 | 0.197 |
| No. solvent atoms | 1345 | 1324 | 1352 | 1340 | 1465 | 1428 |
| R.m.s.d. bonds (Å) | 0.011 | 0.010 | 0.011 | 0.010 | 0.010 | 0.009 |
| B-factor (Å2) | 21.3 | 26.6 | 21.1 | 23.0 | 26.1 | 23.8 |
| PDB entry code | IL9Q | IL9T | IL9O | IL9P | IL9R | IL9S |
a Values in parentheses are for the highest resolution shell.
b {I}/{σ(I)} is the average intensity divided by the average estimated error in intensity.
Each refined structure contains the assembled physiological homotrimer of a total of 1005 residues and six copper atoms in the asymmetric unit of the orthorhombic cell. The variant structures begin at residue Ala4 and end at Gly339. At the C terminus, five residues (TIEGR) that include part of the factor Xa cleavage site remain unmodeled as a result of disorder. Generally, the surface loops of the variants are well ordered with the exception of two loops extending from Gly165 to Ala169 and Asp188 to Gly191 that show average B-factors of 30–40 Å2. The overall r.m.s.d. of the α-carbon atoms between the variant and the native enzyme structures does not exceed 0.21 Å, indicating that no global structural rearrangement has resulted from the mutations. The dark green color of the crystals indicates that copper is the primary occupant of the type I site and a comparison of crystallographic B-factors between the copper atoms and their ligands are consistent with both the type I and type II copper sites being nearly fully occupied.
A least-squares superposition of the α-carbon atoms of the three histidine ligands and the catalytically essential residues Asp98 and His255 to the native structure show an r.m.s.d. of < 0.14 Å for each variant. Also conserved in the six nitrite-soaked variant structures is a hydrogen bond bridge through Wat1098 that connects Asp98 with His255, likely required for activity in the native enzyme (Boulanger et al. 2000; Boulanger and Murphy 2001). The positions of the side-chains of the active site residues that lie within 8 Å of the copper (Leu308, Phe312, Val142, Ala137, and Val304), with the exception of 257, are equally well conserved conformationally.
Bidentate mode of nitrite binding
In the nitrite-soaked I257V[NO2−] and I257L[NO2−] crystal structures well-defined omit electron density maps (Fig. 1 ▶) indicate that nitrite is coordinated to the type II copper in an asymmetric bidentate fashion via the oxygen atoms. Ligand bond distances are presented in Table 4. Thermal motion parameters ranging from 30–35 Å2 further indicate that the nitrite is ordered. In both structures, nitrite is oriented in a bent conformation with the nitrite nitrogen being displaced ∼20° out of the plane defined by the type II copper atom and the oxygen atoms of nitrite. By comparison, the nitrite nitrogen in the native nitrite-soaked structure shows a displacement of ∼45° out of the analogous plane.
Figure 1.
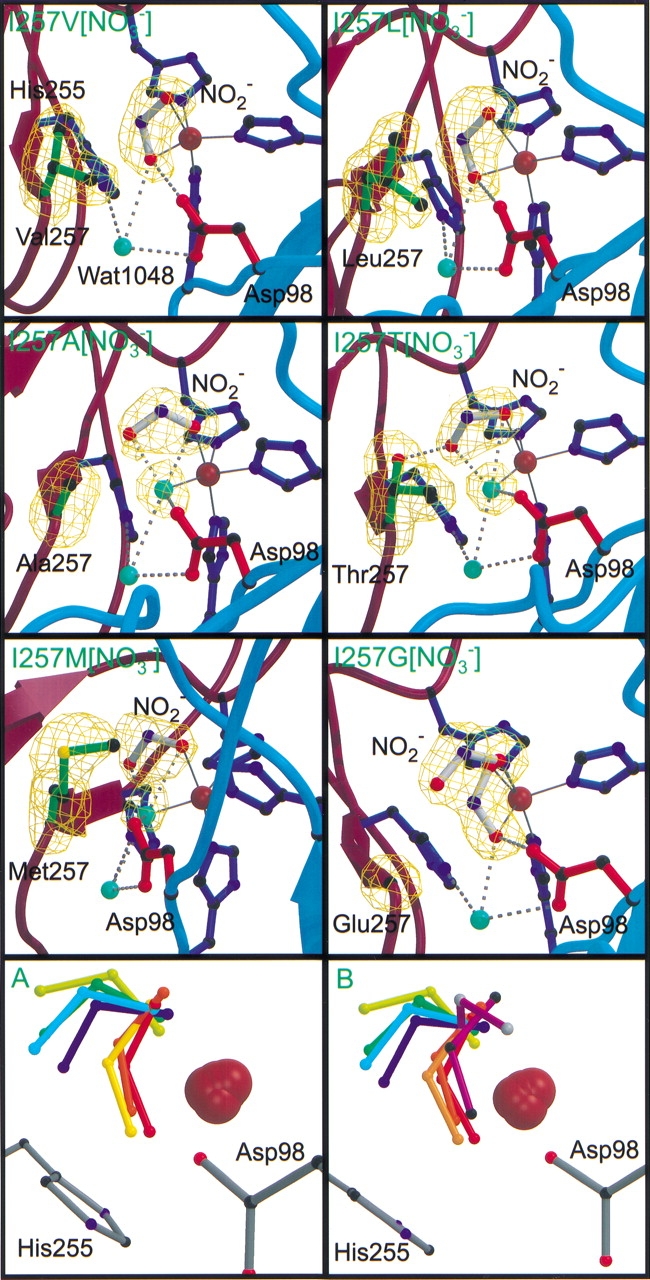
Crystal structures of the six nitrite-soaked I257 variants in the upper six panels. Hydrogen bonds are shown as dashed gray lines with ligand bonds drawn in solid, dark gray lines. Water molecules are drawn as aquamarine spheres. Copper atoms are gray; nitrogen atoms are dark blue, oxygen atoms are red, and sulfur atoms are yellow. The backbone of monomers B and C are shown in burgundy and in teal, respectively. Bonds of the nitrite molecule bound in the active site are white. Omit Fo–Fc electron density maps are contoured at 4σ and drawn as a green wire mesh. Panels A and B depict the multiple conformations of the nitrite bound in the active site of the Ile257 variants and including nitrite bound to the oxidized D98N (black atoms) and H255N (gray atoms) crystal structures (Boulanger and Murphy 2001). With the exception of the purple nitrite molecules in panel B, nitrite molecules are blue to red with increasing specific activity: Red, I257V[NO2−]; orange, native NiR from Alcaligenes faecalis S-6; light orange, I257L[NO2−]; yellow, I257M[NO2−]; green, I257A[NO2−]; cyan, I257G[NO2−]; blue, I257T[NO2−].
Table 4.
Type II copper—ligand bond lengths
| Parameter | I257L [NO2−] | I257V [NO2−] | I257A [NO2−] | I257G [NO2−] | I257M [NO2−] | I257T [NO2−] |
| I. Atomic distances (Å) | ||||||
| a504O1–Cu | 2.3 (0.1)b | 2.4 (0.1) | 2.0 (0) | 2.0 (0) | 2.1 (0.3) | 2.2 (0.2) |
| 2.2 (0.1)c | ||||||
| 504O2–Cu | 2.1 (0.1) | 2.0 (0.1) | 3.4 (0.2) | 3.8 (0.1) | 4.0 (0.4) | 3.1 (0.1) |
| 2.0 (0) | ||||||
| 504O2–98Oδ2 | 2.6 (0) | 2.5 (0.1) | N/A | 2.5 (0.2) | N/A | N/A |
| 503O–98Oδ2 | N/A | N/A | 2.5 (0) | 2.6 (0.1) | 2.5 (0.0) | 2.4 (0.1) |
| 503O–Cu | N/A | N/A | 2.1 (0.2) | 2.3 (0.2) | 2.2 (0.1) | 2.1 (0.1) |
a Residues 503 and 504 represent the solvent ligand and bound nitrite, respectively.
b Values in parentheses represent the range of measurements of the three monomers.
c Values in italics represent alternate conformation of nitrite in I257G [NO2−] structure.
In the I257V[NO2−] structure (Fig. 1 ▶), the nitrite is rotated ∼15° such that the O2 atom is shifted toward the side-chain of Asp98 and the nitrite nitrogen is displaced 0.5 Å from the analogous position in the native AfNiR structure (Murphy et al. 1997). Despite the positional shift in nitrite, no significant displacement of the side-chain of Asp98 is observed. The O2 atom of the bound nitrite forms a hydrogen bond (2.65 Å) with the Oδ2 atom of Asp98. The Cγ2 atom of Val257 is closest to the nitrite nitrogen (3.27 Å).
The χ1 angle of Leu257 adopts an alternate rotameric conformation (Δ60°) from Val257 (I257V[NO2−]) and Ile257 in native AfNiR. Attempts to model Leu257 such that the Cδ1 atom is positioned analogously to the Cδ1 atom of Ile257 in the native structure were unsuccessful because of steric clashes with the side-chain of His306 (2.7 Å) and the bound nitrite (1.6 Å). In the appropriately modeled conformation, the Cγ atom of Leu257 is positioned ∼0.5 Å from the analogous Cγ1 atom of Val257 (I257V[NO2−]). Overall, the Cδ1 atom of Leu257 approaches to within 3.1 Å from the nitrite nitrogen atom and 3.4 Å of the bridging water (Wat1098). The side-chain of Asp98 and the bound nitrite in the I257L[NO2−] structure are well defined with average B-factors of 22 Å2 and 29 Å2, respectively.
Monodentate mode of nitrite binding
Replacement of Ile257 with an alanine, threonine, methionine, or glycine results in an unusual reorientation of the bound nitrite (Fig. 1 ▶). In these variants, nitrite adopts a monodentate coordination through the O1 atom to the type II copper with the O2 atom directed approximately toward residue 257. This reorientation of nitrite allows a solvent molecule (Wat503) to ligate to the copper and occupy the approximate position of the O2 atom of nitrite observed in the bidentate coordination. In the I257G[NO2−] structure, Wat503 was not included in the refinement because of positional disorder in the binding of nitrite. The positions of the catalytically important active site residues, Asp98 and His255, are not shifted significantly in the monodentate nitrite bound structures, and the active site hydrogen bond network remains largely unperturbed.
In the I257A[NO2−] crystal structure the nonligand O2 atom of nitrite (avg. B-factor 29.1 Å2) is positioned 3.0 Å from the β-carbon atom of Ala257. The solvent ligand (Wat503) also displays low average B-factor of 23.5 Å2 and sits 2.12 Å from the type II copper (Table 3). Wat503 may form four potential hydrogen bonds, one with the Oδ2 atom of Asp98, two with the oxygen atoms of nitrite, and one with Wat1098 that bridges Asp98 with His255 (Fig. 1 ▶). The β-strand incorporating residues Tyr301, Ala302, and Tyr303 that packs against residue 257 is shifted toward the active site resulting in the displacement of the α-carbon of Ala257 nearly 0.5 Å toward the bound nitrite.
The repositioned α-carbon atom of Ala257 is within 3.7 Å of the side-chain of His255, which shows a 30° rotation about the χ2 angle and a shift of the Nɛ2 atom of 0.35 Å. The side-chain of Asp98 also shows a rotation about the χ2 angle of ∼15° such that the Oδ atom is also displaced 0.5 Å from the analogous position in the native structure. Despite the reorientation of the side-chains of His255 and Asp98, the active site hydrogen bond network including the copper ligand (Wat503) and bridging (Wat1098) waters is maintained.
The monodentate binding mode of nitrite in the I257T[NO2−] structure is surprisingly similar to that of the I257A[NO2−] rather than the bidentate mode of binding observed in the I257V variant (Fig. 1 ▶). The O1 atom of nitrite is directed toward Thr257 and is oriented to form a hydrogen bond (2.45 Å) with the Oγ1 atom of the threonine side-chain. The conformation of the threonine side-chain in the I257T[NO2−] crystal structure was assigned based on omit electron density maps. The bound nitrite is well-defined with clear omit electron density maps and an average B-factor of 28 Å2. The solvent ligand (Wat503, B-factor 17.2 Å2) maintains the hydrogen bond network with the oxygen atoms of nitrite, the Asp98 Oδ2 atom and Wat1098 observed in the I257A[NO2−] structure. The side-chains of Asp98 and His255 display no significant reorientation.
Despite the introduction of the extended side-chain methionine or the truncated side-chain of glycine at position 257, omit electron density maps indicate clearly that nitrite is capable of binding to the type II copper (Fig. 1 ▶). In the I257M variant, high B-factors averaging > 40 Å2, suggests that the monodentate nitrite is disordered and likely binds at a reduced occupancy. The side-chain of Met257 also shows disorder with B-factors averaging nearly 50 Å2 between the three noncrystallographically related active sites in the asymmetric unit. In the I257G[NO2−] structure, nitrite is modeled in both a monodentate and bidentate coordination (Fig. 1 ▶). Refinement of the structure indicates that the occupancy of the two conformations varies between the three noncrystallographically related active sites; although the monodentate form always predominates. A solvent molecule was refined as the fifth ligand to the type II copper analogous to that of the I257A[NO2−] structure when nitrite adopts the monodentate coordination. In this conformer, the O2 atom of nitrite, is positioned 3.8 Å from the α-carbon atom of Gly257. In the bidentate binding mode, nitrite forms a 2.60 Å hydrogen bond with the Oδ2 atom of Asp98. Steric clashes arising from inappropriate ligand-metal interactions between the carbon atoms and the type II copper eliminated acetate as a possible ligand.
Discussion
Despite numerous experiments characterizing CuNiRs, no clearly defined molecular mechanism exists. One of the major sources of controversy is evidence for the formation of an electrophilic nitrosyl intermediate, which requires the nitrogen oxide species to be bound N-coordinate to the type II copper (Hulse and Averill 1989). The high-resolution crystal structures of the six Ile257 variants presented here provide significant insight into the molecular determinants associated with substrate recognition and taken together identify the catalytically productive mode of nitrite binding.
The active site pocket in native AfNiR lies at the bottom of a 16.5 Å cavity formed at the interface between two adjacent monomers. Several bulky residues form a hydrophobic blanket that surrounds the type II copper and restrict access to the active site. Of these residues, Ile257 is positioned within 5 Å of the type II copper and directly occludes the nitrite-binding site. The isoleucine at position 257 is highly conserved amongst almost all CuNiRs suggesting an important role in catalysis (Fig. 2 ▶). The only two known exceptions, Bacillus stearothermophilus and Corynebacterium diphtheriae, are from the recently defined class II family of CuNiRs (Boulanger and Murphy 2002), which both encode a valine at this position.
Figure 2.

Partial amino-acid sequence alignment of copper-containing nitrite reductases generated with CLUSTALW (Higgins et al. 1996). The outline box highlights analogous residues to Ile257 in NiR from Alcaligenes faecalis S-6. (See Boulanger and Murphy 2002 for the complete alignment and source of each sequence.)
Role of Ile257: Bidentate vs. monodentate
The altered activities of the I257 AfNiR variants correlate strongly with the distinct binding modes of nitrite observed and collectively indicate an intimate catalytic role for Ile257. The bidentate mode of nitrite binding observed in the I257V and I257L mutants is similar to that of the native enzyme and accordingly, these mutants display the smallest variations in specific activity. Interestingly, the substitution of Ile257 with valine, which is the only identified natural substitution (Fig. 2 ▶), produces a more catalytically active enzyme than native AfNiR. From the I257V[NO2−] and native structures, the mode of nitrite binding differs primarily in the degree of bend (Fig. 1A ▶), which may be responsible for the increased activity. The slight reorientation of nitrite in the mutant results in a hydrogen bond with the side-chain of Asp98 that is ∼0.5 Å shorter than in the native enzyme, which may facilitate proton transfer during catalysis. Alternatively, the reduction in steric hindrance resulting from the substitution of an isoleucine with the smaller valine may facilitate access to the copper site while maintaining a sufficient presence to direct the catalytically productive bidentate binding mode for nitrite.
Although substitution of Ile257 with a leucine is conservative like the mutation to valine, the change in the rotameric conformation of Leu257 directed by the side-chain of His306 likely disrupts nitrite binding as shown by the somewhat diffuse electron density (Fig. 1 ▶). Additionally, this new conformation results in a close contact with the side-chain of the catalytically essential Asp98, which may also contribute to the reduced activity.
The unusual monodentate mode of nitrite binding observed in the nitrite-soaked crystal structures of the alanine, glycine, threonine, and methionine variants provide a unique perspective from which to characterize the role of Ile257 in directing the catalytically competent binding mode of nitrite. In this binding mode, the nitrite oxygen atom that ligates to the copper and participates in a hydrogen bond with the side-chain of Asp98 in native AfNiR is repositioned such that it is directed toward residue 257 (Fig. 1 ▶). This unusual nitrite binding mode is likely catalytically less productive consistent with a reduced specific activity of >25 fold for these variant AfNiRs.
The stability of the monocoordinate nitrite-binding mode and the associated reduction in catalytic activity is clear in the I257T variant. Despite the structural similarity between the side-chains of threonine and valine, which differ only in the nature of the γ1 atom, the specific activity for I257T is the lowest of all the six variants, whereas I257V variant is more active than the native enzyme (Table 2). Analysis of the nitrite-soaked crystal structure revealed the presence of a hydrogen bond between the Oγ1 atom of Thr257 and the O1 atom of the bound nitrite. This interaction appears to stabilize the monodentate coordination of nitrite that impairs catalytic activity.
The structural data suggest that in the absence of an appropriate residue at position 257, nitrite prefers to bind in a monodentate fashion via a single oxygen atom. Importantly, the hydrogen bond between nitrite and the side-chain of Asp98 known to be essential for catalytic activity (Zhang et al. 2000; Boulanger and Murphy 2001) is not conserved in this unusual binding mode. Instead, a solvent molecule ligand (Wat503) completes the hydrogen bond with Asp98. Overall, the role of Ile257 is to direct the catalytically productive bidentate mode of nitrite binding to the type II copper maintaining the essential hydrogen bond with Asp98.
Mechanistic implications
Two distinct catalytic mechanisms have been reported for copper-containing nitrite reductases where catalysis proceeds via either an N-coordinate nitrite, followed by formation of a nitrosyl intermediate (Hulse and Averill 1989) similar to the heme cd1 NiR mechanism, or an O-coordinate nitrite with poorly defined intermediates (Adman et al. 1995; Murphy et al. 1997; Boulanger et al. 2000; Boulanger and Murphy 2001). Functional studies with biomimetic synthetic copper complexes support the N-coordinate mechanism (Halfen and Tolman 1994; Halfen et al. 1996; Monzani et al. 2000), whereas studies with the enzyme are consistent with an O-coordinate binding mode for nitrite (Howes et al. 1994; Strange et al. 1995, 1999; Murphy et al. 1997). Additionally, the formation of a brown color in the native AfNiR nitrite-soaked crystals suggests that the O-coordination of nitrite is catalytically competent (Murphy et al. 1997).
Modeling studies with the native nitrite-soaked AfNiR structure suggests that the topology of the active site, in particular the presence of the side-chain of Ile257, precludes nitrite binding in an N-coordinate fashion (Murphy et al. 1997). Despite enlargement of the active site with mutations at position 257, no N-coordinate binding of nitrite is observed in the high-resolution crystal structures presented here. At most, a monodentate O-coordination is observed that correlates with a reduced specific activity. The O-coordination of nitrite is also observed in the nitrite-soaked D98N and H255N AfNiR (Fig. 1B ▶) variant structures published previously (Boulanger and Murphy 2001). The specific activities of D98N and H255N mutants are 1% and 0.06% of the wild-type protein, respectively. Interestingly, nitrite in the H255N variant is bound in a monodentate fashion; however, the free oxygen of nitrite is directed nearly 120° away from the analogous oxygen in the Ile257 variant structures. Collectively, these structural and functional studies are inconsistent with the proposed requirement for an N-coordinate nitrite as suggested by the biomimetic copper complexes.
Conclusion
Asp98 and His255 alone are insufficient to direct a catalytically productive mode of nitrite binding. Instead, the residue at position 257 plays a significant role in orienting the bound nitrite in such a manner as to maintain the catalytically essential hydrogen bond with the side-chain of Asp98. The observed contribution of Ile257 in the native enzyme is consistent with a mechanism that proceeds with nitrite binding to an oxidized type II copper in the active site of CuNiRs in an O-coordinate fashion. Further studies are required to determine if during catalysis, NO bound to the type II copper is N-coordinate.
Materials and methods
Combinatorial mutagenesis and protein expression
A combinatorial mutagenesis procedure based on the “Quick Change” system (Stratagene) was developed to generate a small library of six mutations at position 257 in the A. faecalis nirK gene cloned in pET28a (Boulanger et al. 2000). The degenerate primers (Table 1) were designed using the Combinatorial Codons program (Wolf and Kim 1999) to maximize the likelihood of replacing Ile257 with aliphatic amino acids. The PCR mixture contained 5 ng of double-stranded template DNA, 125 ng of each mutagenic primers, 1 μL of a 10-mM dNTPs, 5 μL reaction buffer supplied by Stratagene, DMSO to a final concentration of 0.5 %, 1 μL of PFU Turbo DNA polymerase (Stratagene) and distilled water to a final volume of 50 μL. Mutations of Ile257 to Leu, Val, Ala, Gly, Met, and Thr were confirmed by DNA sequence analysis. Protein was expressed and purified as described previously (Boulanger et al. 2000), which results in a highly purified protein with full copper occupation.
Spectral analysis and activity assays
Samples of each of the variant AfNiR proteins (I257L, V, A, G, T, M) were prepared to a concentration of 10 mg/mL in 10 mM Tris pH 7.0, prior to injection onto a reverse-phase column interfaced to an electrospray mass spectrometer (Boulanger et al. 2000). Optical scanning spectra for each AfNiR variant were recorded on a Varian Cary 50 Bio UV-visible spectrophotometer. Protein samples were analyzed in 10 mM Tris buffer pH 7.0.
Nitrite reductase activity was measured at 30°C in 5 mL test tubes with a final reaction volume of 1 mL as described by Kakutani et al. (1981). Methylviologen reduced with an excess of dithionite buffered in 100 mM sodium bicarbonate was used as the artificial electron donor to AfNiR. The starting solution (900 μL) contains 2 mM sodium nitrite, 0.1 mM methylviologen, and 20 mM potassium phosphate (KPO4) buffer pH 7.0. AfNiR (100 μL) was added such that the final nitrite concentration was between 0.4 mM (20%) and 1.6 mM (80%) after 5 min. The reaction was started with the addition of sodium dithionite to a final concentration of 5 mM. Residual nitrite was detected in a 15-μL sample from the first step using the Griess reagents [500 μL each of 0.02% N-(1-naphthyl) ethylenediamine and 1% sulfanilic acid in 25% HCl]. For the control, 1.95 mM (98%) nitrite remained. Units are defined as the amount of AfNiR required to reduce 1 μmole of NO2− per minute. All buffers were degassed prior to use.
Crystallization, soaking and data collection
Variant AfNiR crystals were grown at 19°C by the hanging drop vapor diffusion method using a reservoir of 0.1 M sodium acetate pH 4.7, 6%–10% polyethylene glycol (PEG) 4000 supplemented with 1–5 mM cupric chloride. Each drop was made from an equal volume of reservoir and a 15 mg mL−1 protein stock solution buffered in 10 mM Tris pH 7.0. All variant AfNiR protein samples produced crystals that grew in an orthorhombic lattice with space group P212121 that are isomorphous with previous AfNiR crystals (Kukimoto et al. 1994)
Nitrite-soaked oxidized crystals were obtained by placing crystals in reservoir solution supplemented with 5 mM sodium nitrite over 45 min. at room temperature. The crystals were then transferred to fresh soaking solution supplemented with 5 mM nitrite and glycerol as the cryo-protectant. A step gradient of glycerol from 10% to 30% was required to avoid cracking of the crystals. Crystals were looped directly into a cryo stream at 100 K generated by a cryostat (Oxford Cryo Systems). X-ray data was collected on a Rigaku R-AXIS IIc image plate system with CuKα radiation generated by a Rigaku RU 300 rotating anode operating at 100 mA and 50 kV and focused with Osmic confocal optical mirrors and processed with DENZO (Otwinowski and Minor 1997). Data collection statistics are summarized in Table 3.
Structure solution and refinement
The variant AfNiR crystals used for the soaking experiments grew in a primitive orthorhombic lattice and contain the assembled trimer in the asymmetric unit. The nitrite-soaked native AfNiR structure (Murphy et al. 1997) was used as the starting model for the nitrite-soaked AfNiR variant structures following removal of the mutated residues, active site solvent atoms, and the nitrite. Each nitrite-soaked variant crystal structure was refined to a resolution of at least 1.78 Å by standard CNS (Brunger et al. 1998) maximum likelihood positional and B-refinement. Solvent was added with the WATERPICK procedure resulting in an Rwork <18% and an Rfree of <21% in each of the six structures. During refinement, the interaction between the copper and nitrite and their interactions with the polypeptide were not restrained. Manual intervention was accomplished using the visualization program O (Jones et al. 1991). Final refinement statistics are presented in Table 3.
Acknowledgments
We thank Dr. Shouming He and Dr. Stephen Withers for mass spectroscopy analysis. This work is supported by a research grant from the National Science and Engineering Research Council of Canada (to M.E.P.M). M.J.B. was supported by a university graduate fellowship, and M.E.P.M. is a Canadian Institute of Health research scholar.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
NiR, nitrite reductase
CuNiR, copper-containing nitrite reductase
AfNiR, NiR from Alcaligenes faecalis S-6
AcNiR, NiR from Achromobacter cycloclastes
[NO2−], nitrite-soaked crystal structure
FT-IR, Fourier transform infrared spectroscopy
EPR, electron paramagnetic resonance
EXAFS, extended X-ray fine absorption structure spectroscopy
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0224503.
The amino acid numbering is that of AcNiR based on the sequence alignment by Nishiyama et al. (Nishiyama et al. 1993).
References
- Abraham, Z.H., Lowe, D.J., and Smith, B.E. 1993. Purification and characterization of the dissimilatory nitrite reductase from Alcaligenes xylosoxidans subsp. xylosoxidans (N.C.I.M.B. 11015): Evidence for the presence of both type 1 and type 2 copper centres. Biochem. J. 295 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adman, E.T., Godden, J.W., and Turley, S. 1995. The structure of copper-nitrite reductase from Achromobacter cycloclastes at five pH values, with NO2− bound and with type II copper depleted. J. Biol. Chem. 270 27458–27474. [DOI] [PubMed] [Google Scholar]
- Averill, B.A. 1996. Dissimilatory nitrite and nitric oxide reductases. Chem. Rev. 96 2951–2964. [DOI] [PubMed] [Google Scholar]
- Boulanger, M.J. and Murphy, M.E.P. 2001. Alternate substrate binding modes to two mutant (D98N and H255N) forms of nitrite reductase from Alcaligenes faecalis S-6: Structural model of a transient catalytic intermediate. Biochemistry 40 9132–9141. [DOI] [PubMed] [Google Scholar]
- ——2002. Crystal structure of the soluble domain of the major anaerobically induced outer membrane protein (AniA) from pathogenic Neisseria: A new class of copper-containing nitrite reductases. J. Mol. Biol. 315 1111–1127. [DOI] [PubMed] [Google Scholar]
- Boulanger, M.J., Kukimoto, M., Nishiyama, M., Horinouchi, S., and Murphy, M.E.P. 2000. Catalytic roles for two water bridged residues (Asp98 and His255) in the active site of copper-containing nitrite reductase. J. Biol. Chem. 275 23957–23964. [DOI] [PubMed] [Google Scholar]
- Braker, G., Fesefeldt, A., and Witzel, K.P. 1998. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64 3769–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, N., Pannu, N.S., et al. 1998. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta. Cryst. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Cutruzzola, F. 1999. Bacterial nitric oxide synthesis. Biochim. Biophys. Acta. 1411 231–249. [DOI] [PubMed] [Google Scholar]
- Dodd, F.E., Hasnain, S.S., Abraham, Z.H., Eady, R.R., and Smith, B.E. 1997. Structures of a blue-copper nitrite reductase and its substrate-bound complex. Acta. Cryst. D53 406–418. [DOI] [PubMed] [Google Scholar]
- Fulop, V., Moir, J.W., Ferguson, S.J., and Hajdu, J. 1995. The anatomy of a bifunctional enzyme: Structural basis for reduction of oxygen to water and synthesis of nitric oxide by cytochrome cd1. Cell 81 369–377. [DOI] [PubMed] [Google Scholar]
- Glockner, A.B., Jungst, A., and Zumft, W.G. 1993. Copper-containing nitrite reductase from Pseudomonas aureofaciens is functional in a mutationally cytochrome cd1 free background (NirS−) of Pseudomonas stutzeri. Arch. Microbiol. 160 18–26. [DOI] [PubMed] [Google Scholar]
- Godden, J.W., Turley, S., Teller, D.C., Adman, E.T., Liu, M.Y., Payne, W.J., and LeGall, J. 1991. The 2.3 Å X-ray structure of nitrite reductase from Achromobacter cycloclastes. Science 253 438–442. [DOI] [PubMed] [Google Scholar]
- Halfen, J.A. and Tolman, W.B. 1994. Synthetic model of the substrate adduct to the reduced active site of copper nitrite reductase. J. Amer. Chem. Soc. 116 5475–5476. [Google Scholar]
- Halfen, J.A., Mahapatra, S., Wilkinson, E.C., Gengenbach, A.J., Young, V.G., Que, L., and Tolman, W.B. 1996. Synthetic modeling of nitrite binding and activation by reduced copper proteins. Characterization of copper(I)-nitrite complexes that evolve nitric oxide. J. Amer. Chem. Soc. 118 763–776. [Google Scholar]
- Higgins, D.G., Thompson, J.D., and Gibson, T.J. 1996. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 266 383–409. [DOI] [PubMed] [Google Scholar]
- Hochstein, L.I. and Tomlinson, G.A. 1989. The enzymes associated with denitrification. Annu. Rev. Microbiol. 42 231–261. [DOI] [PubMed] [Google Scholar]
- Howes, B.D., Abraham, Z.H.L., Lowe, D.J., Brüser, T., Eady, R.R., and Smith, B.E. 1994. EPR and electron nuclear double resonance (ENDOR) studies show nitrite binding to the type 2 copper centers of the dissimilatory nitrite reductase of Alcaligenes xylosoxidans (NCIMB 11015). Biochemistry 33 3171–3177. [DOI] [PubMed] [Google Scholar]
- Hulse, C.L. and Averill, B.A. 1989. Evidence for a copper-nitrosyl intermediate in denitrification by the copper-containing nitrite reductase of Achromobacter cycloclastes. J. Amer. Chem. Soc. 111 2322–2323. [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Cryst. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kakutani, T., Watanabe, H., Arima, K., and Beppu, T. 1981. Purification and properties of copper-containing nitrite reductase from a denitrifying bacterium Alcaligenes faecalis strain S-6. J. Biochem. 89 453–461. [DOI] [PubMed] [Google Scholar]
- Kataoka, K., Furusawa, H., Takagi, K., Yamaguchi, K., and Suzuki, S. 2000. Functional analysis of conserved aspartate and histidine residues located around the type 2 copper site of copper-containing nitrite reductase. J Biochem 127 345–350. [DOI] [PubMed] [Google Scholar]
- Kukimoto, M., Nishiyama, M., Murphy, M.E.P., Turley, S., Adman, E.T., Horinouchi, S., and Beppu, T. 1994. X-ray structure and site-directed mutagenesis of a nitrite reductase from Alcaligenes faecalis S-6: Roles of two copper atoms in nitrite reduction. Biochemistry 33 5246–5252. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Libby, E. and Averill, B.A. 1992. Evidence that the type 2 copper centers are the site of nitrite reduction by Achromobacter cycloclastes nitrite reductase. Biochem. Biophys. Res. Comm. 187 1529–1535. [DOI] [PubMed] [Google Scholar]
- Monzani, E., Anthony, G., Koolhaas, A., Spandre, A., Leggieri, E., Casella, L., Gullotti, M., Nardin, G., Randaccio, L., Fontani, M., et al. 2000. Binding of nitrite and its reductive activation to nitric oxide at biomimetic copper centers. J. Biol. Inorg. Chem. 5 251–261. [DOI] [PubMed] [Google Scholar]
- Murphy, M.E., Turley, S., and Adman, E.T. 1997. Structure of nitrite bound to copper-containing nitrite reductase from Alcaligenes faecalis. Mechanistic implications. J. Biol. Chem. 272 28455–28460. [DOI] [PubMed] [Google Scholar]
- Nishiyama, M., Suzuki, J., Kukimoto, M., Ohnuki, T., Horinouchi, S., and Beppu, T. 1993. Cloning and characterization of a nitrite reductase gene from Alcaligenes faecalis and its expression in Escherichia coli. J. Gen. Micro. 139 725–733. [DOI] [PubMed] [Google Scholar]
- Olesen, K., Veselov, A., Zhao, Y., Wang, Y., Danner, B., Scholes, C.P., and Shapleigh, J.P. 1998. Spectroscopic, kinetic, and electrochemical characterization of heterologously expressed wild-type and mutant forms of copper-containing nitrite reductase from Rhodobacter sphaeroides 2.4.3. Biochemistry 37 6086–6094. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Prudencio, M., Eady, R.R., and Sawers, G. 1999. The blue copper-containing nitrite reductase from Alcaligenes xylosoxidans: Cloning of the nirA gene and characterization of the recombinant enzyme. J. Bacteriol. 181 2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, G. and Tiedje, J. 1992. Isolation and characterization of a nitrite reductase gene and its use as a probe for denitrifying bacteria. Appl. Environ. Microbiol. 58 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange, R.W., Dodd, F.E., Abraham, Z.H.L., Grossman, J.G., Brüser, T., Eady, R.R., Smith, B.E., and Hasnain, S.S. 1995. The substrate-binding site in Cu nitrite reductase and its similarity to Zn carbonic anhydrase. Nat. Struct. Biol. 2 287–292. [DOI] [PubMed] [Google Scholar]
- Strange, R.W., Murphy, L.M., Dodd, F.E., Abraham, Z.H., Eady, R.R., Smith, B.E., and Hasnain, S.S. 1999. Structural and kinetic evidence for an ordered mechanism of copper nitrite reductase. J. Mol. Biol. 287 1001–1009. [DOI] [PubMed] [Google Scholar]
- Weeg-Aerssens, E., Wu, W., Ye, R.W., Tiedje, J.M., and Chang, C.K. 1991. Purification of cytochrome cd1 nitrite reductase from Pseudomonas stutzeri JM 300 and reconstitution with native heme d1. J. Biol. Chem. 266 7496–7502. [PubMed] [Google Scholar]
- Wolf, E. and Kim, P.S. 1999. Combinatorial codons: A computer program to approximate amino acid probabilities with biased nucleotide usage. Protein Sci. 8 680–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, R., Fries, M., Bezborodnikov, S., Averill, B., and Tiedje, J. 1993. Characterization of the structural gene encoding a copper-containing nitrite reductase and homology of this gene to DNA of other denitrifiers. Appl. Environ. Microbiol. 59 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Boulanger, M.J., Mauk, A.G., and Murphy, M.E.P. 2000. Carbon monoxide binding to copper-containing nitrite reductase from Alcaligenes faecalis. J. Phys. Chem. B. 104 10738–10742. [Google Scholar]