

Abstract
Coenzyme A (CoA) is an essential cofactor used in a wide variety of biochemical pathways. The final step in the biosynthesis of CoA is catalyzed by dephosphocoenzyme A kinase (DPCK, E.C. 2.7.1.24). Here we report the crystal structure of DPCK from Escherichia coli at 1.8 Å resolution. This enzyme forms a tightly packed trimer in its crystal state, in contrast to its observed monomeric structure in solution and to the monomeric, homologous DPCK structure from Haemophilus influenzae. We have confirmed the existence of the trimeric form of the enzyme in solution using gel filtration chromatography measurements. Dephospho-CoA kinase is structurally similar to many nucleoside kinases and other P-loop-containing nucleotide triphosphate hydrolases, despite having negligible sequence similarity to these enzymes. Each monomer consists of five parallel β-strands flanked by α-helices, with an ATP-binding site formed by a P-loop motif. Orthologs of the E. coli DPCK sequence exist in a wide range of organisms, including humans. Multiple alignment of orthologous DPCK sequences reveals a set of highly conserved residues in the vicinity of the nucleotide/CoA binding site.
Keywords: Crystal structure, dephosphocoenzyme A kinase, trimer, nucleotide triphosphate hydrolase
Coenzyme A (CoA) is an essential cofactor used in numerous biochemical pathways. It is synthesized from pantothenate (vitamin B5), cysteine, and ATP in five enzymatic steps (Abiko 1975). The final two steps in the biosynthetic pathway are the coupling of phosphopantetheine with ATP to form dephosphocoenzyme A (dephospho-CoA), followed by phosphorylation of the 3′-hydroxyl group to form CoA. Genes encoding the enzymes for these steps, phosphopantetheine adenylyltransferase (PPAT, E.C. 2.7.7.3) and dephospho-coenzyme A kinase (DPCK, E.C. 2.7.1.24), have been identified in Escherichia coli by Geerlof et al. (1999) and Mishra et al. (2001), respectively. The sequences of both enzymes are highly conserved among various bacterial species. In mammalian cells the activity of PPAT and DPCK is carried by a single 62-kD enzyme (Worral and Tubbs 1983). Recently, the human gene that encodes this bifunctional enzyme (CoA synthase) was identified by Aghajanian and Worral (2002) and Daugherty et al. (2002). The sequence of human CoA synthase contains a 208-amino-acid region homologous to E. coli DPCK (24% sequence identity) and a region of 157 residues attributed to PPAT activity, based on weak sequence identity to the corresponding bacterial enzyme. The crystal structure of PPAT from E. coli has been determined by Izard and Geerlof (1999), and residues identified as involved in dephospho-CoA binding are conserved in the PPAT portion of human CoA synthase. The E. coli gene identified by Mishra and co-workers (2001) as responsible for DPCK activity, coaE, encodes a 206-amino-acid protein. Sequence analysis reveals several proteins homologous to E. coli DPCK, including mammalian orthologs (Mishra et al. 2001). The enzyme and its homologs contain the highly conserved P-loop or Walker A sequence motif (GXXXXGKT/S, where X is any residue) responsible for nucleotide binding (Walker et al. 1982). Recently the crystal structure of a dephospho-CoA kinase from Haemophilus influenzae has been reported (Oblomova et al. 2001) showing that this enzyme has a structure similar to many P-loop-containing nucleotide triphosphate (NTP) hydrolases, such as the nucleotide and nucleoside kinases and shikimate kinase. Here we report our independent determination of the crystal structure of DPCK from E. coli by multiple anomalous dispersion (MAD) phasing at 1.8 Å resolution. Unlike the monomeric H. influenzae enzyme and other members of its structural family, E. coli DPCK forms trimers in the crystal, stabilized by sulfate ions. The presence of sulfate ions also induces the formation of trimers in solution, resulting in a monomer–-trimer equilibrium.
Results and discussion
Quality of the model
The structure of E. coli DPCK has been refined to a final R-factor of 0.217 (Rfree = 0.246) at 1.8 Å resolution. There are three molecules of DPCK in the asymmetric unit. Molecules B and C contain all 206 amino acid residues; however, the segment Ile-50–Asn-77 in molecule A is disordered and was not included in the final model due to poor electron density. Residues in chain A adjacent to this region have temperature factors considerably higher than the average for this monomer. The temperature factors of the corresponding regions Arg-67–Asn-82 (helix α4 + loop + beginning of helix α5) in monomers B and C also show an accentuated increase that correlates with the disorder seen in monomer A. Another region of higher temperature factors common to all monomers is Thr-141–Ile-155 (end of helix α7 +loop + beginning of helix α8). As discussed later, the observed mobility of these segments is related to the enzyme’s function as these segments are involved in catalysis. The final model also contains 459 water molecules and 10 sulfate ions. Validation with the program PROCHECK (Laskowski et al. 1993) indicates good stereochemistry for the model. The Ramachandran plot shows 93.5% of residues in the most favored regions, whereas three residues are in disallowed regions. Each of the residues in disallowed regions (Glu-79 in chain A, Ala-76 in chain B, and His-151 in chain C) is in a poorly ordered region of the structure and exhibit poor electron density. Refinement statistics are shown in Table 1.
Table 1.
Statistics for data collection, processing, and refinement
| Data seta | Inflection | Peak |
| Wavelength (Å) | 0.97957 | 0.97941 |
| Resolution limits (last shell, Å) | 40.0–1.80 (1.87–1.80) | 40.0–1.80 (1.87–1.80) |
| I/σ(I) after merging | 17.7 | 18.9 |
| Completeness (%) | 88.7 (71.1) | 89.2 (74.3) |
| Rsymb | 0.063 (0.546) | 0.064 (0.566) |
| No. of reflections | 380533 | 377803 |
| No. of unique reflections | 56459 | 56361 |
| Rwork (# reflections)c | 0.217 (1024 05) | |
| Rfree (# reflections)d | 0.246 (5518) | |
| r.m.s.d. bonds (Å) | 0.008 | |
| r.m.s.d. bond angles (°) | 1.26 | |
| Number of atoms (B-factor [Å2]) | ||
| Overall protein atoms | 4785 (28.5) | |
| Main chain | 2472 (26.7) | |
| Side chains | 2313 (30.3) | |
| Waters | 454 (34.9) | |
| Sulfate groups | 10 (61.1) |
a Diffraction data were collected with 1° oscillations at a crystal to detector distance of 170 mm, with a total of 360 frames for the inflection point and 354 frames for the peak datasets, respectively.
bRsym = ∑h∑i|(I(h,i) − 〈I(h)〉|/∑h∑iI(h,i), where I(h,i) is the intensity of the ith measurement of h, and 〈I(h)〉 is the corresponding mean value of h over all i measurements of h, with the summation being over all measurements.
cRwork = ∑|Fobs − Fcalc|/∑Fobs.
dRfree = Rwork, but for a subset of 5518 reflections not included in refinement.
Monomer structure
Each monomer of DPCK (Fig. 1 ▶) consists of five parallel β-strands flanked by α-helices in a similar arrangement to that observed for other P-loop-containing nucleotide kinases. A DALI (Holm and Sander 1995) search of the Protein Data Bank shows that the most similar (Z scores in excess of 7) structures are members of the structural superfamily of P-loop-containing NTP hydrolases, according to the SCOP hierarchy (Murzin et al. 1995). This superfamily includes many nucleotide and nucleoside kinases, as well as shikimate kinase and chloramphenicol phosphotransferase. With the exception of the short sequence corresponding to the P-loop motif and a small number of key residues involved in nucleotide binding, none of these proteins exhibit significant sequence homology to DPCK. On the other hand, the sequence comparison with the DPCK from H. influenzae shows 48% of sequence identity between the two enzymes. Therefore, as expected, the two structures show very high similarity (r.m.s. deviation of 1.5 Å for 205 Cα atom pairs; DALI Z score = 25.4).
Figure 1.

Ribbon diagram depicting the E. coli DPCK monomer structure. The LID domain is red and the CoA-binding domain is blue, whereas the rest of the molecule is green. The nucleotide-binding P-loop motif is marked in pink. Secondary structural elements are marked. Figures 1 ▶, 2 ▶, and 4 ▶ were produced with Molscript (Kraulis 1991) and Raster3D (Merrit and Bacon 1997).
Oligomeric state
The trimeric organization observed for E. coli DPCK in the crystal structure (Fig. 2 ▶) is unusual among structurally related proteins, most of which are monomers in their solution and crystal forms. The DPCK from H. influenza conforms to the general trend and is also a monomer. A notable exception to the monomeric organization is a trimeric adenylate kinase from Sulfolobus acidocaldarius (Vonrhein et al. 1998, PDB code 1NKS), which exhibits high structural similarity to the present enzyme (DALI score Z = 10.1, corresponding to an r.m.s. deviation of 3.5 Å for 149 Cα atom pairs). The subunits of this adenylate kinase bury 15% of their solvent-accessible surface area upon trimerization. The structural results prompted these investigators to question previous gel filtration chromatographic studies, which had indicated a dimeric state in solution for this enzyme. Subsequent ultracentrifugation and matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectroscopy measurements confirmed a trimer as the dominant oligomeric form in solution for this protein (Vonhrein et al. 1998).
Figure 2.

Stereo ribbon diagram of the E. coli DPCK trimer structure, looking down the pseudo-threefold axis. The molecule is colored according to the temperature factors of the Cα atoms in each residue, from blue (B = 14 Å2) to orange (B = 60 Å2). Sulfate ions in the structure are displayed, as well as the side chains of residues Ser-119, Tyr-121, and Lys-122 from each monomer. Sulfates bound to the P-loop motif are shown as space-filling models in green.
In the structure of E. coli DPCK, chains A, B, and C bury 1544, 1501, and 1542 Å2, respectively, in their trimeric association with the other two subunits. This corresponds to a burial through intramolecular contacts of ∼12.5% of the full surface area of each monomer. The trimer as a whole makes 10 intermolecular crystal packing contacts with an average surface contact area of only 533 Å2, corresponding to a 17% burial of the total trimer surface in crystal packing, which is a typical value for proteins at this packing density (Vm = 2.55 Å3/Da). These parameters are characteristic for proteins designated as “functional” trimers based on studies of the buried surface areas resulting from oligomerization versus crystal contacts (e.g., Henrick and Thornton 1998). However, both gel filtration analysis by Mishra et al. (2001) and in our laboratory dynamic light scattering measurements on the purified protein before crystallization indicated a monomeric state in solution.
A careful inspection of the trimer-forming interactions in the crystal showed the presence of a sulfate ion in the center of the trimer interface surrounded by the residues Ser-119, Tyr-121, and Lys-122 from each monomer. Only Tyr-121 from each monomer participates in hydrogen-bonding interactions with the sulfate, forming a strong hydrogen bond (∼2.6 Å) between Tyr-121OH and an oxygen atom of the sulfate. In addition, three other sulfates were identified at equivalent positions in the interfaces between chains A/C, B/C, and C/A, each possessing somewhat higher temperature factors (average ∼80 Å2). The presence of sulfate ions at subunit interfaces suggests a role for sulfate in trimer stabilization. To confirm this hypothesis we carried out gel filtration chromatography experiments with a Superdex 75 column using the protein in its purification buffer with and without 0.2 M ammonium sulfate. In the absence of sulfate, the protein eluted in a single peak with a volume equivalent to a monomeric molecular mass of ∼22 kD, whereas in the presence of ammonium sulfate, an additional peak was observed corresponding to a molecular mass of ∼66 kD (Fig. 3 ▶), indicating the formation of trimers in the presence of sulfate. The trimeric form of the enzyme was thus shown to exist in solution in equilibrium with the monomeric form when sulfate ions are present. A similar sulfate-dependent oligomerization was observed for the structurally similar enzyme chloramphenicol phosphotransferase (DALI score Z = 10.2 corresponding to an r.m.s. deviation of 3.4 Å for 139 Cα atom pairs), which is dimeric or tetrameric in solution in the absence or presence of sulfate, respectively (Izard and Ellis 2000).
Figure 3.

Gel filtration chromatogram of purified DPCK. The dashed line depicts a gel filtration chromatogram from a Superdex 75 column with the DPCK protein in its purification buffer (see Materials and Methods). The solid line depicts a run using the buffer containing 0.2 M ammonium sulfate. The peak marked A is due to the trimeric form of the enzyme. The peaks B correspond to the monomer. The arrow in the figure marks the center of the observed elution peak for the molecular mass standard bovine serum albumin (molecular mass, 67 kD).
Comparison of E. coli and H. influenzae DPCK proteins
Although there is an overall close correspondence between the structures of DPCK from E. coli and the recently published ortholog from H. influenzae (Obmolova et al. 2001, PDB code 1JJV), some regions of divergence are noteworthy. Figure 4 ▶ shows the superposition of the Cα backbones of the two structures, as well as a structure-based sequence alignment of the corresponding sequences. DPCK from H. influenzae has a four-residue insertion at position 176. This insertion forms an extended loop between strand β5 and helix α10 absent in the E. coli enzyme. On the other hand, the DPCK from E. coli has a five residue extension at the carboxyl terminus extending beyond helix α10. These residues contribute to the trimerization by wrapping around the neighboring molecule in the trimer. Another cluster of residues at which the sequences diverge lies within the second half of the loop between strands β3 and β4. These residues are also involved in the trimer interface and contain the three residues (Ser-119, Tyr-121, Lys-122) in the vicinity of the central sulfate ion in the E. coli structure. These areas of sequence divergence between the two proteins appear to account for their differing states of oligomerization, particularly when one bears in mind that the DPCK enzyme from H. influenzae was crystallized in the presence of 2 M ammonium sulfate.
Figure 4.


(A) Stereo view of the superposition of Cα backbone structures of DPCK from E. coli (blue) and H. influenzae (red). Residues at the trimer interface for the E. coli structure are marked with black dots. Every 25th residue of the E. coli structure is labeled. (B) Structure-based alignment of E. coli (EC) and H. influenzae (HI) DPCK sequences. Identical residues are shown in white lettering on a black background and conserved residues are shown in black lettering on a gray background. Secondary structure elements of the E. coli enzyme are marked above the alignment. Residues at the trimer interface for the E. coli structure are marked with asterisks below the alignment.
Active site
The mechanism of phosphoryl transfer in the structural group of proteins to which DPCK belongs has been studied extensively (Cox et al. 1994; Matte et al. 1998). General features of substrate binding and the domain structure can be inferred by qualitative comparison with structurally related enzymes (e.g., Gerstein et al. 1993; Vonrhein et al. 1998). The β-phosphate of ATP can be assumed to bind at the P-loop motif, with the rest of the molecule bound by residues in the central five-stranded β-sheet that forms a nucleotide-binding domain. The homologous structure of Oblomova et al. (2001), which is in complex with ATP, confirms this prediction. In the present structure, without bound ATP, sulfate ions reside at the approximate positions of the β-phosphate of ATP in all three chains of the trimer. Such mimicking of the ATP phosphate ions by sulfate ions is frequently observed in related crystal structures, for example, thymidine monophosphate kinase (Li de la Sierra et al. 2001) and chloramphenicol phosphotransferase (Izard and Ellis 2000).
In comparison with related structures and the H. influenzae DPCK structure, the insertion after strand β2 containing the four helices α2, α3, α4, and α5 can be designated a CoA-binding domain. The alpha helices α7 and α8 comprise a LID domain (Fig. 1 ▶). Coenzyme A is expected to bind within the deep cleft between the LID domain and the CoA-binding domain. In related structures the LID and acceptor substrate-binding domains close over the active site during catalysis to prevent phosphate transfer to water. The sizes of the LID and acceptor-binding domains vary between proteins in this structural family of kinases, as do their degrees of conformational flexibility. As noted earlier, residues in the LID and CoA-binding domains in the E. coli structure have higher temperature factors than other parts of the structure due to their flexibility (Fig. 2 ▶). In the absence of either donor or acceptor substrates it is reasonable to assume that the present structure adopts an “open” conformation. The superposition between the H. influenzae DPCK structure, which contains a bound ATP molecule, and the present structure (Fig. 3 ▶) shows that the LID domain is a little closer to the active site in H. influenzae than in E. coli DPCK. A portion of the LID domain in H. influenzae DPCK was not modeled due to the lack of well-defined electron density, but, for example, the Cα position for the H. influenzae Asn-148 is 5.7 Å closer to the proposed active site than the corresponding Cα coordinates of the E. coli Thr-148. Obmolova et al. (2001) present a model for the binding of dephospho-CoA to their structure. Because those parts of the E. coli and H. influenzae structures involving catalysis, particularly the nucleotide and CoA-binding domains, do not differ greatly between the two structures the reader is referred to the analysis of the Obmolova et al. model for more details of the proposed catalytic mechanism for this enzyme.
Possible biological role of the trimer
The question of the biological relevance of the trimeric arrangement remains open. The central sulfate ion in the trimer, which is assumed to play an essential role in its formation, could perhaps be replaced by a phosphate ion in vivo. The three subunits are arranged in such a way as to leave the proposed active site cleft oriented to the exterior of the trimer, whereas the mobile LID and CoA-binding domains are free to close over the catalytic domain (Fig. 2 ▶). A qualitatively similar organization was found for the trimeric adenylate kinase from Sulfolobus acidocaldarius (Vonrhein et al. 1998). Another feature of the trimerization that is of potential functional importance is the stabilization it affords the loop between strands β3 and β4. Many residues in this region participate in the trimer interface (Fig. 4B ▶), and correspond to the portion of the E. coli DPCK sequence containing the residues Ser-119, Tyr-121, and Lys-122, which coordinate with the central sulfate ion in the trimer. These residues differ from the H. influenzae sequence, in which they are Lys-119, Thr-121, and Ala-122, and are not conserved among DPCK sequences from other sources. Obmolova et al. (2001) highlighted the conformation of this loop as important for the proposed model of CoA binding to DPCK, as it forms a bridge separating two pockets in which parts of coenzyme A were predicted to bind.
Alignment of DPCK sequences
More than 100 homologs of the E. coli coaE gene product can be found in the Swiss-Prot and TrEMBL (Bairoch and Apweiler 2000) protein sequence databases from a wide variety of organisms. Most of the orthologs from bacterial genomes have been assigned the dephospho-CoA kinase function by homology. Using the program Clustal W (Thompson et al. 1994), we aligned the 25 most homologous DPCK sequences, all from bacterial genomes (alignment not shown). Surface residues of the E. coli DPCK structure were colored according to the level of conservation using the program GRASP (Nicholls et al. 1991), as illustrated in Figure 5 ▶. Clearly, highly conserved residues are clustered in the cleft expected to form the active site, based on comparisons with structurally related enzymes and the observed ATP and modeled CoA-binding positions for the H. influenzae DPCK structure. There are 19 strictly conserved residues among these 25 bacterial orthologous sequences. Seven (Gly-9, Gly-10, Ile-11, Gly-14, Lys-15, Arg-140, Asn-175) are expected to be involved in nucleotide binding. This set contains residues that are not only conserved among DPCK sequences but are found in other nucleotide kinases. For example, the side chain of Arg-140 stacks with the ribose unit of ATP in the complexed H. influenzae structure in a similar way to equivalent arginine residues in related structures. A further six conserved residues (Thr-8, Asp-33, His-89, Pro-113, Leu-114, Gln-159) were identified by Obmolova et al. (2001) as key residues in their model for the binding of CoA to DPCK. Of the remaining conserved residues, Ala-36, Arg-67, Phe-75, and Leu-84 are close to the predicted CoA-binding site. Asp-31 is somewhat further away and may participate in the coordination of Mg2+ ions during catalysis. An aspartic acid residue at a qualitatively similar position interacts with Mg2+ through an intermediate water molecule, in adenylate kinase from B. stearothermophilus (Berry and Phillips 1998). Pro-90 causes a kink in helix α5, which may act as a hinge for the movement of the CoA-binding domain during catalysis. Finally, the strict conservation of Gly-55, which is located at the start of the turn region between helices α3 and α4, is likely for maintaining structural integrity.
Figure 5.
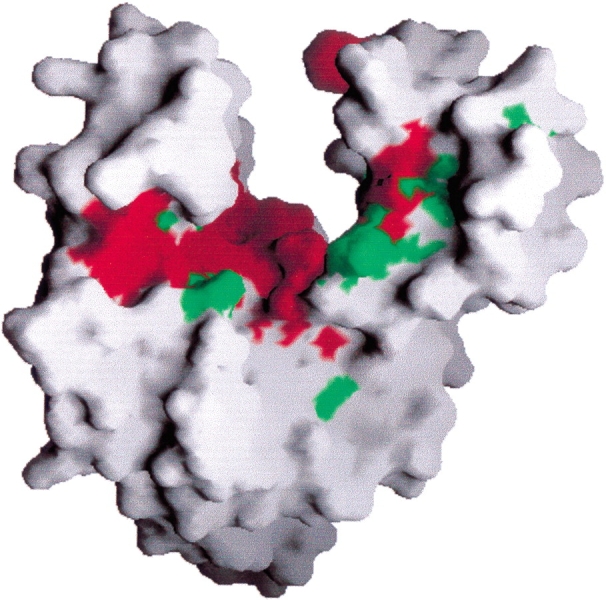
Molecular surface of the DPCK monomer, colored by sequence conservation based on sequence alignment of 25 bacterial orthologs. Red portions of the surface correspond to strictly conserved residues; green portions to conserved residues, according to the Clustal W (Thompson et al. 1994) classification. The molecule is oriented approximately as in Figure 1 ▶. This figure was produced with the program GRASP (Nicholls et al. 1991).
Orthologs of the E. coli DPCK sequence are also found in a diverse range of eukaryotic organisms (Fig. 6 ▶). Aghajanian and Worral (2002), Daugherty et al. (2002), and Zhyvoloup et al. (2002) have recently independently identified the human gene responsible for a bifunctional PPAT/DPCK enzyme and experimentally verified its function. It was demonstrated earlier by Worral and Tubbs (1983) that in mammals, PPAT and DPCK activities are carried by a single enzyme. The human PPAT/DPCK protein (Swissprot/Trembl accession no. Q8WXD4) contains a carboxy-terminal domain with 27% sequence identity to the E. coli DPCK sequence. Daugherty et al. (2002) noted another annotated human gene (Swissprot/Trembl accession no. Q8WVC6) that is homologous to E. coli DPCK, with 35% sequence identity over its entire length. This gene product was hypothesized to be a monofunctional human DPCK enzyme. The existence of monofunctional PPAT and DPCK enzymes in humans is suggested by mitochondrial subfractionation studies that show differences in PPAT and DPCK activities (Skrede and Halvorsen 1983). Interestingly, the monofunctional human DPCK sequence has lower amino acid sequence identity to the DPCK portion of bifunctional human PPAT/DPCK (32%), than to the E. coli DPCK. Orthologs of the human PPAT/DPCK are found in the pig, Drosophila melanogaster, and Caenorhabditis elegans proteomes. We have also found orthologs of monofunctional DPCK in D. melanogaster (Q9VI57) and C. elegans (P34558). Once again, the monofunctional DPCK sequence for these enzymes has a greater sequence identity to the E. coli and other bacterial DPCK sequences than to the paralogous DPCK portion of the bifunctional PPAT/DPCK sequence in each organism. Bifunctional PPAT/DPCK appears to be the result of a gene fusion in the distant evolutionary past. It is unclear at this time whether, or under what circumstances, the monofunctional DPCK genes are expressed in vivo in higher organisms. Sequence alignment of E. coli, S. cerevisiae, A. thaliana, C. elegans, D. melanogaster, and human DPCK sequences (Fig. 6 ▶) shows conservation of residues, as identified above, which are involved in catalysis for the E. coli enzyme. Most of the strictly conserved residues involved in nucleotide binding for bacterial species (e.g., the P-loop residues and the E. coli Arg-140 residue) are also conserved among the eukaryotic sequences. Note, however, that for some conserved bacterial residues suggested to be involved in CoA binding, such as Pro-113 and Leu-114, strict residue conservation among the eukaryotic sequences is absent due to mutations among the bifunctional PPAT/DPCK sequences, perhaps hinting at modified mechanisms of CoA binding in such enzymes.
Figure 6.
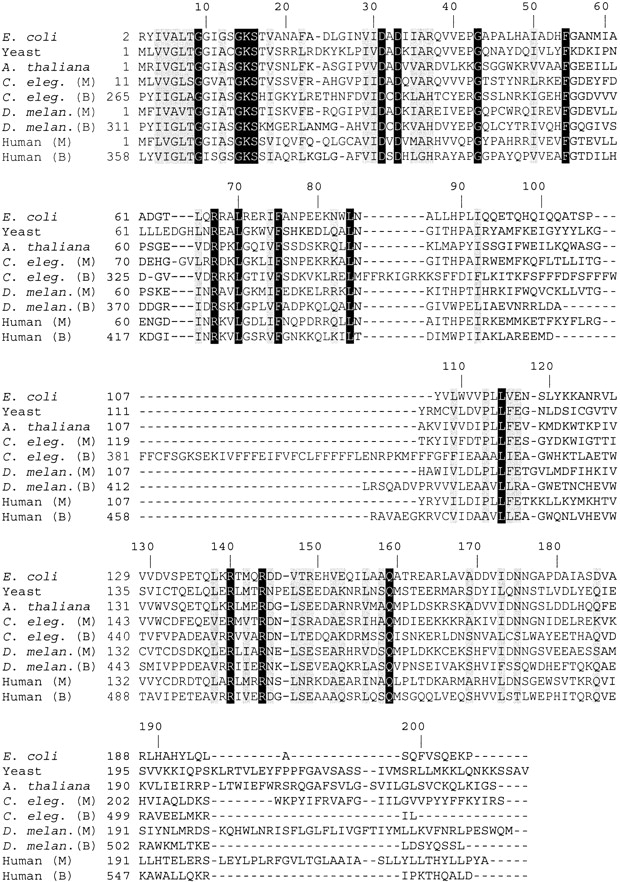
Alignment of the E. coli DPCK sequence and selected homologous sequences from eukaryotes. Sequences marked M in parentheses are putative monofunctional DPCK enzymes. Sequences marked B are putative bifunctional PPAT/DPCK enzymes. Identical residues are shown in white lettering on a black background and conserved residues are shown in black lettering on a gray background. Numbers corresponding to the position in the E. coli amino acid sequence are marked along the top of the alignment. Swiss-prot or Trembl (Bairoch and Apweiler 2000) accession numbers for these sequences are as follows: E. coli: P36679; yeast: Q03941; A. thaliana: Q9ZQH0; C. elegans (M): P34558; C. elegans (B): Q9BL56; D. melanogaster (M): Q9VI57; D. melanogaster (B): Q9VRP4; human (M): Q8WVC6; human (B): Q8WXD4. Sequence alignment was performed with the program Clustal W (Thompson et al. 1994).
Conclusions
The crystal structure of dephosphocoenzyme A kinase from E. coli has been determined at 1.8 Å resolution. The enzyme crystallizes in an unusual trimeric form that is dependent on the presence of sulfate ions in solution. Each monomer adopts a fold similar to many other P-loop-containing NTP hydrolases. The locations of residues conserved among orthologous DPCK sequences correspond to parts of the structure expected to be involved in catalysis, based on the well-characterized behavior of similar proteins. Future structural and biological analysis of this enzyme will elucidate the mechanisms of coenzyme A binding and domain closure during catalysis, as well as the biological relevance of its quaternary structure.
Materials and methods
Cloning, expression, and purification of E. coli DPCK
The coaE gene was amplified by PCR using recombinant Taq polymerase (Amersham-Pharmacia) with E. coli MC1061 genomic DNA as the template. The amplified insert was cloned into a modified pET20b vector (Novagen) using NdeI–BamH1 restriction sites. The full-length construct consists of all 206 residues of DPCK, with an additional, noncleavable His tag at the carboxyl terminus composed of 10 residues with the sequence Gly–Ser–(His)8. For SeMet protein production plasmid DNA was transformed into the strain DL41 and grown in LeMaster medium (Hendrickson et al. 1990) containing 25 mg/L l-selenomethionine. Expression of DPCK was induced in a 1-liter culture using 0.1 mM IPTG at 22°C for 20 h.
Cells were harvested by centrifugation (4000 g, 4°C, 20 min) and resuspended in 40 mL of lysis buffer (50 mM Tris-Cl at pH 7.5, 0.4 M NaCl, 1% (v/v) Triton X-100, 5% (v/v) glycerol, 10 mM beta-mercaptoethanol (BME), 10 mM imidazole) and one tablet of a protease inhibitor cocktail (Complete, Roche Diagnostics, Laval, Canada). Cells were disrupted by sonication (5 cycles, each 30 sec on, then 30 sec off, 4°C; Heat Systems Ultrasonics Inc.) and the lysate cleared by centrifugation (150,000 g, 4°C, 45 min). The protein supernatant was passed through a 3-mL DEAE-Sepharose column (Pharmacia) and the flow-through loaded on a 5-mL Ni-NTA column (Qiagen) equilibrated in lysis buffer. The column was washed with 10 bed volumes of buffer (50 mM Tris-Cl at pH 7.5, 1 M NaCl, 1% (v/v) Triton X100, 5% (v/v) glycerol, 10 mM BME, 10 mM imidazole) followed by 10 bed volumes of 50 mM Tris-Cl at pH 7.5, 0.2 M NaCl, 5% (v/v) glycerol, 10 mM BME, 20 mM imidazole to remove unbound proteins. The DPCK protein was eluted from the column using 50 mM Tris-Cl at pH 7.5, 0.2 M NaCl, 5% (v/v) glycerol, 10 mM BME, 150 mM imidazole, and ran as a single band of ∼35 kD by SDS-PAGE. Protein concentration was determined by the method of Bradford (1976) using bovine serum albumin as a standard.
Crystallization of DPCK
After purification, the buffer was changed to 20 mM Tris-Cl at pH 7.5, 0.2 M NaCl, 5% (v/v) glycerol, 10 mM dithiothreitol (DTT), and the protein concentrated by ultra-filtration using a Centriprep 10 concentrator (Amicon) to 9.3 mg/mL. Dynamic light scattering measurements (DynaPro 801, Protein Solutions) indicated that E. coli DPCK is a monomer in solution. Initial crystals were obtained by sparse-matrix screening (Hampton Research, Laguna Niguel, CA) by hanging drop vapor diffusion. The best crystals grew at 21°C from droplets containing 2 μL of protein and 2 μL of reservoir solution (20% w/v polyethylene glycol [PEG] 8K, 50 mM cacodylate at pH 6.5, 0.2 M (NH4)2SO4, 5% v/v glycerol) appearing within 4 days and grew for 2 weeks up to 0.4 × 0.5 × 0.6 mm3. The crystals were monoclinic, space group P21 with cell dimensions a = 55.4, b = 82.4, c = 76.0Å, β = 94.8° and three molecules in the asymmetric unit.
Data collection, MAD phasing, and refinement
Crystals were soaked for ∼30 sec in a cryoprotectant solution consisting of 0.1 M cacodylate buffer at pH 6.5, 0.2 M (NH4)2SO4 and 22.5% (w/v) PEG 8K and 17% (v/v) 2-methyl 4-propanediol (MPD), picked up in a rayon cryo-loop (Hampton Research) and flash-cooled in a stream of nitrogen gas at 100 K (Oxford Cryosystems, Oxford, UK). Diffraction data were collected on a Quantum-4 CCD detector (ADSC, San Diego, CA) at beamline X8C, NSLS, BNL. Data indexing, merging and scaling were performed using the HKL package (Otwinowski and Minor 1997). Data collection and processing statistics are listed in Table 1.
The MAD data from a Se-Met–labeled crystal of apo DPCK were collected to 1.75Å resolution at Se anomalous peak and inflection point wavelengths. Ice rings formed during flash-cooling resulted in incompleteness for their respective resolution shells. Eight of the nine expected Se sites in the asymmetric unit were found using SOLVE (Terwilliger and Berendzen 1999), and phases were calculated in the range of 20.0 to 1.8Å giving an overall figure of merit (FOM) of 0.37. Electron density modification was applied with the program RESOLVE (Terwilliger 2000), improving the FOM to 0.56. Using the ARP/wARP program (Perrakis et al. 1999) ∼60% of all residues were built automatically into the density-modified experimental map as glycines, alanines, serines, or valines. The remainder of the model was built manually with the program O (Jones et al. 1991) using 3Fo–2Fc electron density maps. The three molecules in the asymmetric unit are related by noncrystallographic threefold symmetry along a direction nearly parallel to the crystallographic a-axis.
Refinement of the DPCK model was performed with CNS version 1.0 (Brünger et al. 1998) in the resolution range 40–1.8 Å using the Se peak dataset. No sigma cutoff was applied to the data, and a bulk solvent correction was used. After each cycle of refinement, the model was manually fit to new 3Fo–2Fc and Fo–Fc electron density maps contoured near 1σ or ±3σ, respectively. Water molecules were added with the automatic procedure of ARP/wARP (Perrakis et al. 1999) or by visual inspection. Several strong electron density features in the difference map clearly corresponded to larger molecules and, according to their shapes and in conjunction with the high concentration in the crystallization solution, were interpreted as sulfate ions.
Coordinates
The coordinates have been deposited in the RCSB Protein Data bank (accession code 1N3B).
Gel filtration chromatography
After the structural determination gel filtration chromatography was performed on the purified DPCK enzyme with a Superdex 75 HR 10/30 column on an |f#KTA FPLC system (Amersham Pharmacia, Uppsala, Sweden). The column was equilibrated with two column volumes (CV) of the appropriate buffer below. Two hundred microliters of protein were applied to the column and eluted in 1 CV at a flow rate of 0.5 mL/min with each of the following buffers: 20mM Tris at pH 7.5, 5 mM BME, 5% glycerol, and either 0.2 M NaCl or 0.2 M (NH4)2SO4. Elution was monitored by UV adsorption at λ = 280 nm. Sample concentrations ranged from 2.3 to 4.8 mg/mL. The chromatograms were analyzed on-line with the provided software (Unicorn 3.10.11). Molecular masses were estimated by comparison with the elution profile of molecular mass standards chicken egg white lysozyme (Mr 14,300) and bovine serum albumin (Mr 67,000).
Acknowledgments
We thank Robert Larocque for cloning the coaE gene, and Dr. Joseph D. Schrag for critical comments. We also thank G. Oblomova and G. Gilliland for access to the H. influenzae DPCK coordinates before publication of their manuscript. This work was in part supported by Canadian Institutes of Health Research (CIHR) grant 200103GSP-90094-GMX-CFAA-19924 (to M.C.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0227803.
References
- Abiko, Y. 1975. Metabolism of coenzyme A. In Metabolic Pathways, vol 7 (ed. D. M. Greenberg), pp. 1–25. Academic Press, New York, NY.
- Aghajanian, S. and Worral, D.M. 2002. Identification and characterization of the gene encoding the human phosphopantetheine adenylyltransferase and dephospho-CoA kinase bifunctional enzyme (CoA synthase). Biochem. J. 365 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, M. and Phillips, G. 1998. Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+Ap5A, and Mn2+Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+. Proteins 32 276–288. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., and Pannu, N.S. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cox, S., Radzio-Andzelm, E., and Taylor, S. 1994. Domain movements in protein kinases. Curr. Opin. Struct. Biol. 4 893–901. [DOI] [PubMed] [Google Scholar]
- Daugherty, M., Polanuyer, B., Farrell, M., Scholle, M., Lykidis, A., de Crecy-Lagard, V., and Osterman, A. 2002. Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics. J. Biol. Chem. 277 21431–21439. [DOI] [PubMed] [Google Scholar]
- Geerlof, A., Lewendon, A., and Shaw, W.V. 1999. Purification and characterization of phosphopantetheine adenylyltransferase from Escherichia coli.J. Biol. Chem. 274 27105–27111. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., Schulz, G.E., and Chothia, C. 1993. Domain closure in adenylate kinase: Joints on either side of two helices close like neighbouring fingers. J. Mol Biol. 229 494–501. [DOI] [PubMed] [Google Scholar]
- Hendrickson, W.A., Horton, J.R., and LeMaster, D.M. 1990. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrick, K. and Thornton, J. 1998. PQS: A protein quaternary structure file server. Trends Biochem. Sci. 23 358–361. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20 478–480. [DOI] [PubMed] [Google Scholar]
- Izard, T. and Ellis, J. 2000. The crystal structures of chloramphenicol phosphotransferase reveal a novel inactivation mechanism. EMBO J. 19 2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izard, T. and Geerlof, A. 1999. The crystal structure of a novel bacterial adenylyltransferase reveals half of sites reactivity. EMBO J. 18 2021–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zhou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24 235–242. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Li de la Sierra, I., Munier-Lehmann, H., Gilles, A.M., B|fqrzu, O., and Delarue, M. 2001. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP and 1.95 Å resolution. J. Mol. Biol. 311 87–100. [DOI] [PubMed] [Google Scholar]
- Matte, A., Tari, L., and Delbaere, T.J. 1998. How do kinases transfer phosphoryl groups? Structure 6 413–419. [DOI] [PubMed] [Google Scholar]
- Merrit, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Mishra, P.K., Park, P.K., and Drueckhammer, D.G. 2001. Identification of yacE (coaE) as the structural gene for dephosphocoenzyme A kinase in Escherichia coli K-12. J. Bacteriology 183 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzin, A., Brenner, S., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Oblomova, G., Teplyakov, A., Bonander, N., Eisenstein, E., Howard, A., and Gilliland, G. 2001. Crystal structure of dephospho-coenzyme A kinase from Haemophilus influenzae. J. Struct. Biol. 136 119–125. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Morris, R., and Lamzin, V. S. 1999. Automated protein model building combined with iterative structure refinement. Nature Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Skrede, S. and Halvorsen, O. 1983. Mitochondrial pantetheinephosphate adenylyltransferase and dephospho-CoA kinase. Eur. J. Biochem. 131 57–63. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D 56 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein, C., Bonisch, H., Schafer, G., and Schulz, G.E. 1998. The structure of a trimeric archaeal adenylate kinase. J. Mol. Biol. 282 167–179. [DOI] [PubMed] [Google Scholar]
- Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worral, D.M. and Tubbs, P.K. 1983. A bifunctional enzyme complex in coenzyme A biosynthesis: Purification of pantetheine phosphate adenylyltransferase and dephospho-CoA kinase. Biochem. J. 215 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhyvoloup, A., Nemazanyy, I., Babich, A., Panasyuk, G., Pobigailo, N., Vudmaska, M., Naidenov, V., Kukharenko, O., Palchevskii, S., Savinska, L., et al. 2002. Molecular cloning of CoA synthase. J. Biol. Chem. 277 22107–22110. [DOI] [PubMed] [Google Scholar]