

Abstract
The turn-forming ability of a series of three-residue sequences was investigated by substituting them into a well-characterized β-hairpin peptide. The starting scaffold, bhpW, is a disulfide-cyclized 10-residue peptide that folds into a stable β-hairpin with two antiparallel strands connected by a two-residue reverse turn. Substitution of the central two residues with the three-residue test sequences leads to less stable hairpins, as judged by thiol-disulfide equilibrium measurements. However, analysis of NMR parameters indicated that each molecule retains a significant folded population, and that the type of turn adopted by the three-residue sequence is the same in all cases. The solution structure of a selected peptide with a PDG turn contained an antiparallel β-hairpin with a 3:5 type I + G1 bulge turn. Analysis of the energetic contributions of individual turn residues in the series of peptides indicates that substitution effects have significant context dependence, limiting the predictive power of individual amino acid propensities for turn formation. The most stable and least stable sequences were also substituted into a more stable disulfide-cyclized scaffold and a linear β-hairpin scaffold. The relative stabilities remained the same, suggesting that experimental measurements in the bhpW context are a useful way to evaluate turn stability for use in protein design projects. Moreover, these scaffolds are capable of displaying a diverse set of turns, which can be exploited for the mimicry of protein loops or for generating libraries of reverse turns.
Keywords: Reverse turn stability, peptide structure, turn propensity, β-hairpin
The β-hairpin conformation, two antiparallel β-strands connected by a reverse turn, is a common supersecondary structural element, typically found in multistrand β-sheets in proteins. However, certain sequences will also adopt this structure when isolated in short peptides (Blanco et al. 1994b; Munoz et al. 1997; Gellman 1998; Ramirez-Alvarado et al. 1999). Due to the higher number of stability-determining factors in β-sheets (e.g., strand propensity, strand-strand association, turn propensity), comprehensive host-guest studies are less straightforward to conduct and interpret than for α-helical systems (e.g., Chakrabartty and Baldwin 1995). Nevertheless, significant progress has been made in recent years to dissect contributions to β-sheet stability, and some design principles are beginning to emerge that may lead to reliable prediction of β-structure stability (Kortemme et al. 1998; Maynard et al. 1998; Lacroix et al. 1999). Strand positions of β-sheets have been examined systematically in the tertiary structure context of folded proteins (Kim and Berg 1993; Minor, Jr., and Kim 1994b; Smith and Regan 1995, 1997) and in model peptides (Ramirez-Alvarado et al. 1996; Blasie and Berg 1997; Russell and Cochran 2000; Cochran et al. 2001b; Syud et al. 2001).
The turn sequences used in model studies of β-hairpins are very stable and often contain a combination of D and L amino acids (Stanger and Gellman 1998; Favre et al. 1999). However, the majority of turns found in proteins are much less stable, and in many cases are insufficiently stable to form hairpins in isolated linear peptides (Blanco et al. 1994a; Zerella et al. 2000). The extent of protein destabilization by random turn sequences was examined in small β-sheet-containing proteins, which were used as scaffolds to display libraries of β-turns (Zhou et al. 1996; Gu et al. 1997). Statistical preferences for amino-acid type at each position within particular β-turn types have also been obtained by analysis of the database of protein structures (Hutchinson and Thornton 1994). Experimental determination of the relative tendency of tetrapeptides to adopt a reverse turn conformation was made through disulfide exchange equilibrium measurements (Falcomer et al. 1992). More recently, NMR-based methods were used to assess turn stability. In the isolated APGDV type II turn, any one of 12 amino acids tested could replace the i+1 Pro without detrimental effect on turn stability (Dyson et al. 1998). In the same study, i,i+3 ion pairing was shown to stabilize 2:2 turns. In a 12-residue model hairpin with the VXGK 2:2 type I′ turn, Asn was the most favored i+1 residue out of five tested residues (Ramirez-Alvarado et al. 1997). Substitutions have also led to multiple and/or nonnative conformations in hairpins derived from tendamistat (Blanco et al. 1993), ubiquitin (Searle et al. 1995), and designed peptides (de Alba et al. 1996; Ramirez-Alvarado et al. 1999; Chen et al. 2001). These changes in turn length and type indicate that controlling these factors is not straightforward.
We are interested in understanding the sequence elements responsible for stabilizing and controlling peptide structure, with the goal of developing a molecular “tool kit” that can be used to recreate a given secondary or other element of peptide structure. Such a tool kit would have utility in mimicking important regions of proteins, and in designing stable scaffolds for peptide libraries with a bias to a predefined conformational class. With a focus on β-sheets, we investigated preferences at nonhydrogen-bonding and hydrogen-bonding strand positions in β-hairpins. Host-guest studies in a disulfide-cyclized β-hairpin of the form CX8C identified tryptophan substitutions as stabilizing at nonhydrogen-bonding positions (Russell and Cochran 2000; Cochran et al. 2001b). Use of two cross-strand tryptophan pairs is capable of stabilizing linear peptides (Cochran et al. 2001a). Cross-strand substitutions at hydrogen-bonding positions can also be significantly stabilizing, although some context dependence has been noted (Russell et al. 2002).
In the present study, we examined a variety of sequences taken from protein hairpins that form 3:5 turns, the second most abundant turn type (after 2:2; Sibanda et al. 1989; Gunasekaran et al. 1997). The scaffold that we used in our earlier studies is well suited to this task. The disulfide bond and stable strand pairing produces a hairpin with defined strand register and turn location, thereby facilitating a comparison of stabilization energy between 3:5 turns and the various 2:2 turns studied previously (Cochran et al. 2001b). The solution structure of a selected peptide, PDG, was determined by NMR and compared to the NMR structure of the model peptide with a two-residue turn, bhpW. The energetic contributions of individual turn residues in a series of peptides were evaluated by quantitative stability measurements relative to the bhpW model peptide. This experimental approach allowed the relative folding energies to be evaluated and compared to statistical preferences for amino-acid type at each turn position. 1H and 13C NMR data were obtained to assess the extent and type of β-hairpin formation in each peptide. When two selected turn sequences were introduced into additional hairpin scaffolds, their relative stability relationships remained similar, supporting the generality of the results.
Results
NMR structure of PDG
It is of interest to try to design peptides with predictable structural characteristics. As part of an ongoing investigation of β-hairpins, we wanted to evaluate 3:5 turns in a well defined system. The disulfide-cyclized peptide bhpW (Ac-CTWEGNKLTC-NH2) was chosen as the starting host structure. The solution structure of bhpW showed a well folded β-hairpin (Cochran et al. 2001b), with multiple stabilizing elements in the two antiparallel β-strands. A disulfide of Cys1 and Cys10 covalently connects the two strands, while Thr2 and Thr9 are cross-strand hydrogen-bonding partners with a high propensity for extended backbone conformation. A survey of substitutions at nonhydrogen-bonding positions identified the Trp3-Leu8 cross-strand pair as significantly stabilizing the hairpin (Cochran et al. 2001b). Glu4 and Lys7 have a potential for electrostatic interaction, and would be expected to improve aqueous solubility.
We were interested in what effect changes in the turn would have on stability and conformation of this model hairpin. Three-residue substitutions were made in place of the dipeptide turn “GN” in bhpW (see Table 2), whereas the four residues in each strand were kept constant (Ac-CTWEXXXKLTC-NH2). Assuming that the strand register of bhpW is preserved, this will put the “EXXX” residues at positions i, i+1, i+2, and i+3 of a reverse turn (note that Lys9 is still the N-terminal residue of the second strand, but it is no longer in the turn i+3 position). This change in turn length can be described by the nomenclature of Thornton and colleagues (Sibanda et al. 1989) as going from a 2:2 turn to a 3:5 turn. The two digits give the number of turn residues according to two different strand residue definitions; only a single cross-strand backbone hydrogen bond is required by the first definition, whereas the second definition requires that both hydrogen bonds be present (Sibanda et al. 1989). The definition can be extended to include the presence of canonical two-residue reverse turns. Sequences that form 3:5 type I turns in peptide hairpins (de Alba et al. 1997, 1999; Santiveri et al. 2001) and in proteins (Milner-White 1987; Sibanda et al. 1989) have been described. Additionally, we performed database searches based on both sequence homology and DSSP secondary structure definitions (Kabsch and Sander 1983) to identify turns related to the known examples above.
Table 2.
Thiol-disulfide equilibrium measurements and NMR parameters of β-hairpin peptides
| Turn sequencea | Ceff (mM)c | ΔΔGEGNK-turn (kcal/mole) | Turn potentiald | J > 8.0 Hz (max = 8) | Cys δ (Hα) (ppm) | Leu9 δ (Hδ) (ppm) |
| GN | 210 ± 4 | — | 30.9e | 7 | 5.20, 5.00 | 0.58, 0.11 |
| PDG | 167 ± 10 | 0.13 | 21.1 (1.43) | 5 | 5.12, 4.97 | 0.54, 0.11 |
| SDG | 136 ± 10 | 0.25 | 9.1 (0.28) | 4 | 5.06, 4.94 | 0.58, 0.22 |
| LDG | 129 ± 5 | 0.28 | 4.2 (0.20) | 4 | 5.03, 4.92 | 0.59, 0.26 |
| RDG | 117 ± 6 | 0.34 | 5.2e (0.24) | 4 | 5.02, 4.91 | 0.59, 0.27 |
| LTG | 72 ± 7 | 0.62 | 2.4 (0.05) | 3 | 4.92, 4.82 | 0.69, 0.50 |
| SDN | 58 ± 1 | 0.75 | 4.1e (0.27) | 3 | 4.84, 4.74 | 0.74, 0.63 |
| RDN | 39 ± 1 | 0.98 | 2.4e (0.23) | 3 | 4.82, 4.77 | 0.73, 0.62 |
| PDN | 38 ± 1 | 1.0 | 9.6 (1.38) | 3 | 4.81, 4.76 | 0.74, 0.62 |
| HV-GN | 1180 ± 16 | −1.0 | 30.9f | 6 | 5.40, 5.00 | 0.43, −0.34 |
| HV-PDGb | 412 ± 14 | −0.39 | 21.1 (1.43) | 7 | 5.36, 4.99 | 0.49, −0.39 |
| HV-PDNb | 85 ± 14 | 0.53 | 9.6 (1.38) | 6 | 5.24, 4.94 | 0.54, −0.16 |
| random coil | 0 | 4.69 | 0.94, 0.90 |
a Unless otherwise noted, turns are in the bhpW framework: Ac-CTWEXXXKLTC-NH2.
b HV framework peptide sequences: Ac-CHWEXXXKLVC-NH2.
c Ceff measurements represent the mean value of at least three measurements. Values typically varied by less than 5%, giving rise to an uncertainty in ΔΔG of 0.03 kcal.mole−1 (Cochran et al. 2001b).
d Turn potentials were calculated based on positional frequencies of residues in type I turns of proteins (Hutchinson and Thornton 1994); f(i + 1)* f(i + 2)* f(i + 3). Analogous values for type II turns are indicated in parentheses.
e These values include components that are not statistically significant (Hutchinson and Thornton 1994).
f Value calculated for type II′ turn, with Lys at i + 3.
The first sequence chosen for detailed characterization was PDG. These residues form a 3:5 type I turn in several proteins (i.e., 15-lipoxygenase, 1LOX, residues 306–308; nitrous oxide reductase, 1QNI, residues 276–278; N-myristoyltransferase, 2NMT, residues 319–321), and hairpin peptides with this turn have been described (Searle et al. 1995; de Alba et al. 1997, 1999). NMR data for our PDG peptide were of sufficient quality to allow structure calculations. In addition to distance (1H NOE) and dihedral angle restraints (3JHN-Hα and 3JHα-Hβ), conservative 13C chemical shift-derived backbone dihedral angle restraints were also employed (Cornilescu et al. 1999) and significantly improved the precision of the structures. We assume that this ensemble represents the dominant, fully folded state of the peptide, although less folded conformations may also be populated transiently. The final ensemble has no NOE or dihedral angle restraint violations greater than 0.1 Å and 1.5°, respectively. Mean values of 3JHN-Hα calculated from the Karplus relationship on the basis of the final ensemble were also very close to the experimentally observed values for the strand residues (differing by less than 0.7 Hz). The φ, ξ dihedral angles lie only in the favored (91%) and allowed (9%) regions of the Ramachandran map, according to analysis with the program PROCHECK (Laskowski et al. 1993).
The peptide forms the expected antiparallel β-hairpin (Fig. 1A ▶). Comparison of the structures for bhpW (Cochran et al. 2001b) and PDG indicates that the two structures closely match in the stem region (backbone heavy atom RMSDs 0.36 Å for residues 1–4 and 8–11 of PDG) that contains the common elements of the scaffold (Fig. 1B ▶). Thus, hydrophobic contacts are observed between the “NHB” residues Cys1, Trp3, Leu9, and Cys11, whereas the “HB” pair of side chains Glu4 and Lys8 are in an appropriate orientation to form a salt bridge (disorder of these side chains suggests that this might be only transient in nature). Three of the four possible backbone-backbone hydrogen bonds between the strands are observed in all structures. That between the amide proton of Lys8 and carbonyl oxygen of Glu4 is missing because a one-residue bulge is formed at Gly7, and an additional hydrogen bond is observed from the amide hydrogen of Gly7 to the carbonyl oxygen of Glu4. Given this pattern of hydrogen bonds, and the dihedral angles of Pro5, Asp6, and Gly7, the turn in PDG can be characterized as a 3:5 type I turn (Pro5-Asp6) with Gly7 forming a G1 bulge (it is in an αL conformation with a positive φ). In contrast, bhpW features a 2:2 type II′ reverse turn.
Figure 1.
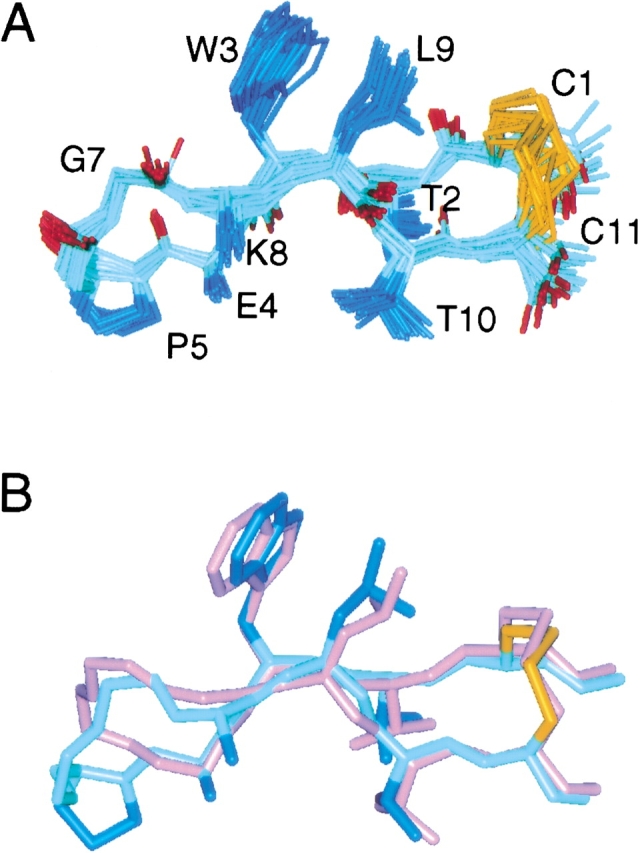
Structure of PDG and comparison to bhpW. (A) An ensemble of 20 PDG structures (mean backbone RMSD to the mean structure = 0.29 ± 0.05 Å). Side chain of N6 is omitted for clarity, and E4 and K8 are only shown to the Cβ. (B) Superposition of the minimized mean structures of PDG (blue) onto bhpW (magenta). Side chains between residues 4 and 8 are omitted for clarity. Backbone heavy atom RMSD of the strands (residues 1–3, 9–11) is 0.36 Å.
A prominent feature of the bhpW and the tryptophan zipper scaffolds is a considerable right-handed twist of the β-strands (Cochran et al. 2001a,b). The majority of the twist occurs N-terminal to the hydrophobic NHB residues (Russell et al. 2002). The strands of PDG are also twisted preceding Trp3 and Leu9 with an enhanced twist preceding Glu4 relative to bhpW (Table 1). As a result of the twist, the φ angles of Trp3 and Leu9 are significantly more positive than the value of −120° observed in regular, extended residues (Table 1). As observed previously (Cochran et al. 2001a,b), this local deviation from an idealized extended structure is apparent from the low 3JHN-Hα values for these residues (7.3 and 7.9 Hz for Trp3 and Leu9 in PDG, respectively, compared to > 8 Hz for an extended conformation). Thus, the observed twist is a general feature of the scaffold, and is not attributable to the influence of any particular turn sequence or turn type.
Table 1.
Measures of strand twisting in β-hairpin peptides
| Parametera | bhpW | PDG |
| ξThr2 + φTrp3 | 59 ± 14° | 56 ± 9° |
| ξTrp3 + φGlu4 | 10 ± 15° | 41 ± 8° |
| ξLys8 + φLeu9 | 52 ± 15° | 49 ± 6° |
| φTrp3 | −94 ± 14° | −85 ± 17° |
| φLeu9 | −98 ± 17° | −83 ± 7° |
a ξi + φi+1 is zero for an untwisted strand and generally >50° for the most twisted part of highly-twisted hairpins; the twisting in part occurs due to a less negative value for φ (last two rows) (Russell et al. 2002).
Also in line with the above observations are the deviations of Hα and Cα chemical shifts from random coil values (“secondary chemical shifts”) for strand residues in PDG (Fig. 2A ▶). 1Hα and 13Cα effects are positive and negative, respectively, in regular β secondary structures (Spera and Bax 1991). However, Trp3, Glu4, and Leu9 have secondary chemical shifts of low magnitude that do not fit the pattern expected for residues in β-strands. Low-magnitude secondary chemical shifts are also observed for the NHB Trp-Leu pair in bhpW and for a Tyr-Leu NHB pair in a linear β-hairpin peptide reported recently (Stanger et al. 2001). In the latter case, the anomalous shifts were attributed to aromatic ring current effects. Calculations of 1Hα and 13Cα chemical shifts on the basis of the bhpW ensemble were performed using the program SHIFTS (v4.1; Xu and Case 2001). These calculations indicate that ring-current effects from Trp3 are indeed responsible for decreasing the 1Hα secondary chemical shift of Trp3 and Leu8. However, the low-magnitude 13Cα secondary chemical shifts of Trp3 and Leu8 can be explained on the basis of the twisted backbone conformation only (data not shown). Thus, the anomalous backbone 13C secondary chemical shifts in PDG are consistent with an extended but twisted backbone in the vicinity of Trp3, Glu4, and Leu9.
Figure 2.
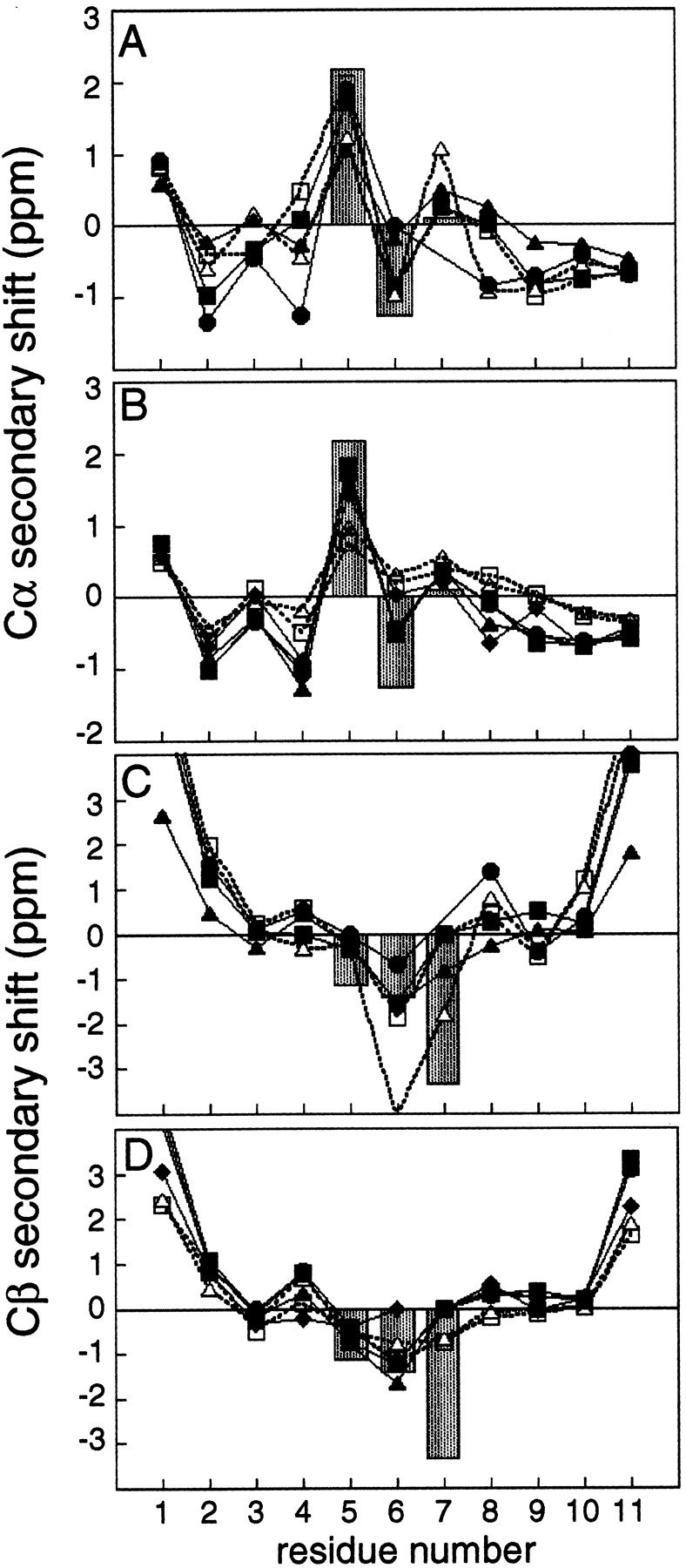
13C secondary chemical shift profiles. Gray bars represent secondary chemical shifts in 3:5 type I + G1 bulge turns in proteins (Santiveri et al. 2001). (A) Cα secondary chemical shifts for PDG (▪, solid line) PDN (▴, solid line), bhpW (•, solid line), HV-PDG (□, dashed line), and HV-PDN (▵, dashed line). A one-residue gap was inserted into the bhpW sequence at residue position 7, to obtain appropriate alignment of the data for the second strand. (B) Cα secondary chemical shifts for SDG (▪, solid line), RDG (▴, solid line), SDN (□, dashed line), RDN (▵, dashed line), LDG (•, solid line), and LTG (♦, solid line). (C) Cβ secondary chemical shifts for peptides in panel A. The gray bars indicate a large expected secondary chemical shift at the i+3 position; however in most examples it is irrelevant because the i+3 residue is typically Gly with no Cβ. (D) Cβ secondary chemical shifts for peptides in panel B.
The stability of PDG relative to bhpW was evaluated using thiol-disulfide equilibrium measurements in a redox buffer (Lin and Kim 1989; Stroup and Gierasch 1990). The effective concentration (Ceff) of disulfide obtained in this assay monitors the appropriate positioning of the two Cys side chains for disulfide bond formation, and therefore the hairpin-forming tendency of a given sequence (Cochran et al. 2001b). Table 2 shows that the PDG sequence that forms a hairpin with a 3:5 turn is slightly less stable than bhpW (ΔΔG= 0.13 kcal/mole).
3:5 turn substitution series
Seven additional turn sequences were selected to examine the general applicability of the scaffold to display 3:5 turns (Table 2). Three of these selected sequences are represented in 3:5 type I + G1 bulge turns of proteins with known structure, and were identified by the strategy used for PDG. RDG is found in cytochrome Cd1 nitrite reductase (1AOQ residues 216–218), LTG occurs in the N-terminal hairpin of ubiquitin (1UBQ residues 8–10), and SDN is found in ovomucoid (1OVO residues 26–28). The four other sequences listed in Table 2 were chosen to test the effects that single-residue substitutions have on turn stability. Such single-residue changes allow the factors that determine turn preference to be probed, and also allow comparison with the turn residue preferences inferred from statistical analyses of protein structures (Hutchinson and Thornton 1994).
The Ceff of dithiol in the thiol-disulfide equilibrium was determined for each 3:5 turn peptide (Table 2), and in every case it indicates a reduced stability relative to bhpW. The most stable of the series is the PDG turn. Changing the Pro in the PDG sequence to Ser, Leu, or Arg leads to a slight loss of stability (∼0.12–0.21 kcal/mole). At the i+2 position, the Asp to Thr substitution in LXG affects a larger destabilization of the hairpin (0.34 kcal/mole). A context dependence of the effects of individual residue changes on hairpin stability is a general feature of the data in Table 2. For example, Gly is always more stabilizing at i+3 than Asn, which agrees well with the observation that Gly is the predominant residue at this position within 3:5 turns observed in proteins (Milner-White 1987; Sibanda et al. 1989; Hutchinson and Thornton 1994). However, the extent of this effect is influenced significantly by context, with the Gly to Asn substitution destabilizing by 0.50 kcal/mole in the case of SDX, but 0.87 kcal/mole in the PDX context. Accordingly, Pro at i+1 gives the most stable hairpin with an XDG turn, but Ser at i+1 gives the most stable hairpin in the context of XDN turns. This context dependence (or nonadditivity) implies that associating a turn propensity score to amino-acid types at specific turn positions will not be meaningful (Hutchinson and Thornton 1994). This parallels observations of context dependence in β-strands, both in statistical analyses of protein structures (Minor, Jr., and Kim 1994a; Wouters and Curmi 1995) and experimental studies of β-sheet protein model systems (Otzen and Fersht 1995; Zaremba and Gregoret 1999).
All peptides in the series were examined by NMR. Structures were not calculated for these peptides; instead, a number of NMR parameters sensitive to conformation were evaluated and compared to equivalent data from PDG described above. Several indicators of the extent of hairpin folding correlate well with the ΔΔG values determined from the thiol-disulfide equilibrium measurements (Table 2); a similar correlation was noted previously (Cochran et al. 2001b). The number of 3JHN-Hα coupling constants above 8 Hz in the strand regions of the molecules ranges from 5 to 3, suggesting less-extended or dynamically averaging backbone torsion angles for the less stable peptides. Downfield shifts of Hα resonance positions are also expected for β-structure (Dalgarno et al. 1983), and indeed such effects are observed for the two Cys residues, with the magnitude of downfield shift correlating with the stability of the hairpin (Table 2). A similar trend is noted for the upfield shifts of the methyl protons of Leu9 (Table 2), reflecting the fraction of time spent in the shielding environment of the indole ring system of the cross-strand partner Trp3.
A large number of cross-strand NOEs were observed in each of the four peptides with the XDG turn sequence, that is, those in the top half of the stability ranking. Although fewer NOEs are observed for the rest of the peptides, significant intensity Hα–Hα NOEs were observed between Trp3 and Leu9 in all peptides (including PDN, the least folded example of the series) and between Cys1 and Cys11 for all peptides except RDN and PDN, where degeneracy precludes observation. These data indicate that in all cases the strands are engaged in an antiparallel interaction. Several NOEs in the turn region were also observed in each peptide. An intense HN–HN i+1 to i+2 NOE indicates the presence of a type I or I′ turn but not a type II or II′ turn (Wüthrich et al. 1984). The intense NOE between the HNs of the i+2 and i+3 residues is expected for a type I turn but not for a type I′ turn (Wüthrich et al. 1984). Both of these interactions are strong in every peptide where resonance overlap or the absence of HN in Pro does not preclude observation, lending additional support to the presence of a type I turn.
The secondary chemical shifts in the strands of our hairpin peptides are small in general (0.1–1 ppm; Fig. 2 ▶); nevertheless, they follow the expected pattern (Spera and Bax 1991). Secondary chemical shift profiles specific for particular turn types were recently described (Santiveri et al. 2001). For hairpins with a 3:5 type I + G1 bulge turn, the 13Cα secondary chemical shift of the i+1 turn residue is usually positive, and the 13Cβ secondary chemical shifts of the i+1 and i+2 residue are both negative (Santiveri et al. 2001). Figure 2 ▶ shows that the hairpin sequences examined in the present study fit this profile. The secondary chemical shifts in this study are about half or less than the average perturbations in protein hairpins; however, they more closely match the observed values for an isolated β-hairpin of this turn type, which had an estimated 60% folded population (Santiveri et al. 2001). In addition to partial folding, strand twisting may also reduce the secondary chemical shifts in peptides relative to the less twisted backbone typically observed in protein β-sheets (see above). The characteristic shifts at the indicated turn positions have the expected polarity and are large (0.8–2 ppm) relative to the secondary chemical shifts of the strand residues in each peptide.
One turn sequence, LTG, which is the native 3:5 type I turn of ubiquitin, does not have the expected negative secondary chemical shift for Thr6 13Cβ, the i+2 residue of the turn. The possibility that this sequence forms a turn with a different conformation is unlikely, based on several observations. As indicated above, an intense NOE is present between the amide protons of Leu5 and Thr6, which is a distinct characteristic of the type I turn (Wüthrich et al. 1984). 3JHN-Hα coupling constants and other NOE intensities in the turn region are also similar between LTG and each of the four XDG turn peptides (data not shown). As threonine has a tendency to adopt an extended conformation even in the absence of secondary structure, the random coil value may not be relevant for this residue, thereby skewing the calculated secondary chemical shift.
The 3JHN-Hα coupling constants, NOE intensity pattern, and 13Cα and 13Cβ secondary chemical shift profiles in the turn region are very similar to those of PDG for each of the examined sequences. These data indicate that the predominant turn conformation present in all of these peptides is a 3:5 type I with G1 bulge, supporting the utility of the bhpW scaffold to predispose various turn sequences to adopt such a conformation. However, other less well folded conformations may also be sampled (if only transiently), especially in those peptides with low Ceff values that have NMR parameters less indicative of a fully folded state (Table 2).
Other scaffolds
Two of the eight turn sequences were selected for further investigation. The PDG and PDN turns, from the most and least stable peptides of the series, were studied in two other contexts. First, the hydrogen bonding residues adjacent to the cysteine residues were replaced with histidine and valine (HV; Ac-CHWEXXXKLVC-NH2), a modification that confers a considerable stabilization to the bhpW reference hairpin (Russell et al. 2002). The PDG and PDN turn peptides are stabilized by ∼0.5 kcal/mole by these substitutions (Table 2). With respect to the parent HV peptide with the GN turn sequence, introduction of either the PDG or the PDN turn is again destabilizing. However, the amount of destabilization in going from the two- to the three-residue turn is larger in the HV context (0.62 kcal/mole and 1.54 kcal/mole for HV-PDG and HV-PDN, respectively), than in the bhpW context (0.13 kcal/mole and 1.0 kcal/mole for PDG and PDN, respectively). NMR evaluation of the secondary structure reveals that the HV peptides are considerably more folded than their counterparts in the bhpW framework, with HV-PDG being more highly folded than HV-PDN (Table 2).
We showed previously that short linear peptides containing two pairs of tryptophan residues at adjacent nonhydrogen-bonding sites (the tryptophan zipper motif) fold readily into remarkably stable monomeric β-hairpins in aqueous solution (Cochran et al. 2001a). PDG and PDN turns were therefore introduced into the tryptophan zipper scaffold (TZ; SWTWEPDXKWTWK-NH2) and compared to trpzip1 of Cochran et al. (2001a); trpzip1 contains the same two-residue GN turn as bhpW in the disulfide-linked series. Both TZ-PDG and TZ-PDN exhibit cross-strand Hα–Hα NOEs between the two pairs of Trp residues, although they are more intense in the former peptide. Compared to the reference molecule trpzip1 (Cochran et al. 2001a), the backbone secondary chemical shifts are smaller for these two peptides, with values for TZ-PDN being less extreme than for TZ-PDG.
Due to the compact arrangement of aromatic rings in the tryptophan zipper, there are a number of extreme secondary chemical shifts present in the fully folded state. For example, the ɛ3 protons of Trp4 and Trp11 of trpzip1 resonate at 5.49 and 5.31 ppm, respectively, −2.16 and −2.31 ppm up-field of the random coil shift of 7.65 ppm for this proton (Wüthrich 1986). Deviations from these extreme values provide a sensitive measure of the degree of folding in less stable peptides. For TZ-PDG, a broad resonance is observed for Hɛ3 of Trp12 at 6.21 ppm at 298 K (−1.44 ppm secondary chemical shift; Trp12 of TZ-PDG is equivalent to Trp11 of trpzip1). The signal for this proton is sharper and less perturbed relative to its random coil chemical shift at higher temperatures (dδ/dT = 45 ppb/K), and is broadened beyond detection at temperatures lower than 298 K. Hɛ3 of Trp4 is not observed in our experimental temperature range (278 to 308 K), presumably as a result of severe line broadening. These observations suggest that TZ-PDG is in fast to intermediate exchange between a folded and an unfolded state. Similarly for TZ-PDN, the Hɛ3 resonances of both Trp4 and Trp12 are broad and undetectable below 298 K. Their secondary chemical shifts are only −0.51 ppm (Trp4) and −0.45 (Trp12) at 298 K, and both shift further down-field at higher temperatures (dδ/dT ∼15 ppb/K). Thus, for both TZ-PDG and TZ-PDN, the chemical shift data indicate incomplete folding at 298 K, with TZ-PDN being less stable. Given that trpzip1 is ∼80% folded at 298K (Cochran et al. 2001a) and assuming that Hɛ3 chemical shifts vary linearly with the extent of folding, we can estimate from these chemical shifts that TZ-PDG and TZ-PDN are ∼50% and ∼20% folded, respectively, at 298 K.
The CD spectra of TZ-PDG and TZ-PDN are qualitatively similar to those reported for other tryptophan zipper peptides (Cochran et al. 2001a). The spectra are dominated by a positive and a negative band at 229 nm and 215 nm, respectively. These can be attributed to the strong exciton couplets of interacting aromatic ring systems (Grishina and Woody 1994). Compared to the spectrum of trpzip1, these bands for TZ-PDG and TZ-PDN are approximately one-half and one-third the intensity, respectively (Fig. 3A ▶), consistent with a reduced β-hairpin content relative to trpzip1. Although the reduced CD signal intensity could result from a significantly different orientation of the indole rings in the presence of a three-residue turn, the temperature dependence of the CD signal at 229 nm suggests that the reduced intensity is a result of partial folding at 298 K (Fig. 3B ▶). Analysis of the temperature dependence of the CD signal identifies the thermal denaturation midpoints at 304 K and 292 K for TZ-PDG and TZ-PDN, respectively. These melting temperatures are in line with the stability relationships observed by other experimental approaches, and are both much lower than the value of 323 K measured previously for trpzip1 (Cochran et al. 2001b). Due to incomplete folding even at the lowest temperatures studied, extraction of thermodynamic parameters from the thermal melts of TZ-PDG and TZ-PDN has not been attempted.
Figure 3.

CD spectra and thermal unfolding of trpzip peptides. (A) CD spectra of trpzip-1 (triangles; Cochran et al. 2001a), tz-pdg (circles), and tzpdn (crosses). (B) Thermal unfolding of trpzip-1 (triangles, upper; Cochran et al. 2001a), tz-pdg (circles, middle), and tzpdn (crosses, lower), followed by CD at the 229 nm maximum of the prominent aromatic band in panel A.
Discussion
We investigated the effects of several turn substitutions on the stability of a β-hairpin model system. Eight homologous 11-residue peptides were designed, each with a different three-residue turn region in the center of the sequence. NMR data indicate that each molecule forms an antiparallel hairpin, and that the turn in each belongs to the 3:5 type I + G1 bulge turn category. Because the strand and disulfide residues are kept constant, differences in stability of the β-hairpin obtained from thiol-disulfide exchange measurements can be correlated with substitutions made in the reverse turn. This approach is quantitative, and requires no assumptions about the NMR parameters associated with the fully folded or unfolded state, as do NOE-based or chemical shift perturbation-based comparative stability evaluation methods (Gellman 1998; Ramirez-Alvarado et al. 1999; Santiveri et al. 2001). Conformation-dependent NMR parameters for each peptide are in general agreement with the thermodynamic stability differences. Both the NMR and thiol-disulfide equilibrium measurements indicate that the introduction of three residues in the turn leads to some loss of hairpin stability, with the degree of destabilization depending on turn sequence.
The frequencies of amino-acid types at positions within particular β-turn types were obtained by analysis of the database of protein structures. These frequencies were used to derive turn potentials that estimate the stability of each amino-acid type at each position within different types of reverse turn (Hutchinson and Thornton 1994). One way that these turn potentials can be used is to predict turn type. The combined turn potential for the four residues in each reverse turn studied in the present work was calculated (Table 2); in all cases the potential is higher for type I turn than for type II turn, in accord with the experimental observations based on the NMR data. The turn potentials can also be a basis for predicting relative turn stability. In this case, there is only a limited agreement between the turn potential-derived relative propensity and the relative stability determined from Ceff. For example, the PDG sequence has the highest calculated turn potential, in agreement with experiment, but PDN is predicted to have the second highest turn stability, when in fact it is the least stable. There may be several reasons for such discrepancies. The number of examples of particular turns in the protein structure database may not be large enough for accurate determination of potentials; this is underscored by the fact that the turn potentials for some residues are not statistically significant. Furthermore, some of the turns in proteins have a biological function that does not necessarily maximize turn stability (Kwong et al. 1998). Tertiary and quaternary structure formation may be able to order turn sequences, which will also eliminate the need for the most stable turns to be the most common (Zhou et al. 1996). Perhaps most importantly, context is ignored in individual residue positional frequencies. As seen from our experimental results, contributions of individual residues to turn stability can be context-dependent. Pro at i+1 in conjunction with Gly at i+3 was the most stable, whereas Ser was best at i+1 when Asn was present at i+3, revealing a context dependence. This context dependence may result from specific interactions between certain side chains, or from the subtle interplay between the backbone conformation enforced by one residue, and the side chain at another.
There is now a growing body of data suggesting that turns can play a crucial role in protein folding as nucleation sites for local structure formation (see, e.g., Freund et al. 1996); amino acid changes that disrupt turn formation can also affect the rate of protein folding and unfolding (Serrano et al. 1992; Gu et al. 1997). Thus, attempting to understand these folding processes in more detail requires detailed and accurate determination of relative turn stability, for example, in model peptide systems. Although a number of experimental and statistical methods have been used to roughly quantitate turn relative stability (see introductory text), these often suffer from having a poor dynamic range or requiring assumptions about the behavior of the fully folded or unfolded states. The measurement of disulfide equilibria represents the only published method that circumvents these problems. Our previous studies indicated that in disulfide-linked hairpins containing a two-residue turn, substitutions in the strands and turn are energetically independent (Russell and Cochran 2000; Cochran et al. 2001b). Comparison of PDG versus PDN turns in the two disulfide-cyclized scaffolds used in the present study also indicate a scaffold-independent stability difference of ∼ 0.9 kcal/mo1 (Table 2). Although the exact stability of PDG or PDN turns is difficult to assess in the tryptophan zipper scaffold, the latter is clearly less stable (Fig. 3 ▶), indicating that the relative energies of the three-residue turn sequences are preserved in several different scaffolds. Thus, the relative energies listed in Table 2 are probably relevant to a wide variety of hairpin or protein scaffolds, and may be used to predict the relative stability of de novo designed peptides or proteins that contain these turns.
The three-residue turn sequences described in this study were initially selected from loops in proteins, where a type I turn followed by a G1 bulge conformation is observed. In the case of PDG, structures determined on the basis of NMR restraints indicate that this conformation is preserved in the peptide. Superpositions of hairpin segments from 15 protein loops with central 3:5 type I + G1 bulge turns on nine residues of PDG yielded a 1.29 ± 0.65 Å RMSD over backbone heavy atoms (Fig. 4 ▶). In some cases, these protein hairpins contain noncanonical strand pairing (extra bulges or some other tertiary context-induced difference) that contribute to a high RMSD, even though the turn region in each is very similar to the conformation of the turn residues in the PDG structure. The hairpin from 15-lipoxygenase has the closest structural similarity to PDG (0.43 Å RMSD, Fig. 4 ▶) and extends away from a multistranded antiparallel β-sheet with a pair of leucine residues in the NHB site adjacent to the turn and significant surface exposure around the turn. Presumably, these features allow this lipoxygenase hairpin to adopt a highly twisted strand conformation that is very similar to the PDG peptide. However, there are nonbonded contacts to a helix that is roughly perpendicular to the hairpin, which may also encourage the strand twisting. At the backbone level, the structure of PDG is also quite similar to the LTG turn in the N-terminal β-hairpin of ubiquitin (0.46 Å RMSD over backbone heavy atoms of nine residues). Previous studies of this N-terminal hairpin have shown that a change of turn from LTG to LDG is very stabilizing (Zerella et al. 2000). This effect was attributed to an ion pairing between Asp16 in the turn and Lys18 following the bulge. In the bhpW context, such a change is also stabilizing (LDG and LTG in Table 2; 0.34 kcal/mole), with the possibility of an analogous ion pair (Asp6–Lys8) explaining the stabilization. We observe no NOE between these side chains. However, if the two residues form an ion pair, the χ1 orientation of Asp7 may actually place the Hβ atoms beyond NOE range for side chain H atoms of Lys9. Therefore, such a stabilizing interaction cannot be excluded.
Figure 4.
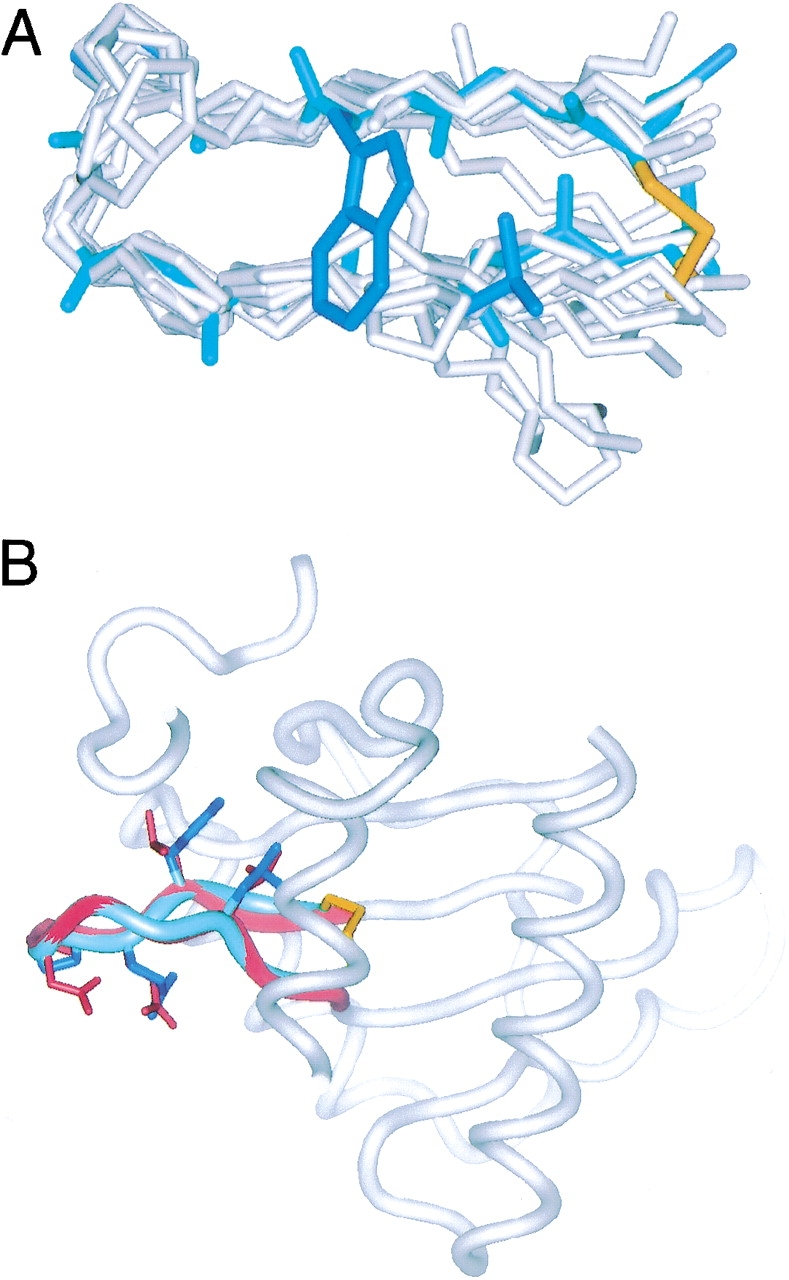
Comparison of PDG and protein hairpins. (A) Superposition of PDG (shown as in Fig. 1 ▶) on 15 protein hairpins shown in gray, with only backbone and proline side chains shown. The backbone heavy atom RMSD = 1.29 ± 0.65 Å. (B) Superposition of PDG (blue ribbon) onto a hairpin (highlighted in red) in lipoxygenase (1LOX). Surrounding backbone segments of the crystal structure of the protein are shown as gray ribbon. Side chains are shown only for C1, W3, E4, P5, N6, L9, and C11 in PDG and the corresponding residues in lipoxygenase. The backbone heavy atom RMSD of 1LOX residues 305–313 (KLQPDGKLM) and PDG residues 2–10 is 0.43 Å.
A detailed analysis of 1H and 13C secondary chemical shifts suggests that the turn conformation observed in PDG is retained in all of the peptides studied, even those of low relative stability. In previous studies, several linear hairpin peptides displayed some strand-register ambiguity, with designed two-residue turns forming three-residue bulged turns (Searle et al. 1995) or designed three-residue turns containing a mixture of turn types (de Alba et al. 1996). Not only does this lead to a final conformation different from what was intended, but it also confounds a detailed energetic comparison of different turn sequences, because the stability of the peptide depends upon the relative stabilities of a large number of alternative strand pairings and turn propensities. The defined nature of the conformations in our disulfide-linked peptides suggests that it is a more robust scaffold for the mimicry of loops in proteins, a property that might be useful in the display of protein-binding epitopes present in hairpin loops of proteins.
Materials and methods
Peptide synthesis
Peptides were synthesized as described (Cochran et al. 2001b) by standard solid-phase Fmoc chemistry on a Perseptive Biosystems Pioneer 8000 synthesizer, except for LTG, which was synthesized by Boc chemistry on an ABI 430A synthesizer. All peptides were made on a 0.2 mmole scale, cleaved from the resin in 95% TFA, 5% triisopropylsilane, and purified by reverse-phase HPLC. Intramolecular disulfides were formed by dropwise addition of a saturated solution of I2 in acetic acid, followed by a second HPLC chromatography step and lyophilization. Purity was assessed by analytical HPLC, and identity was confirmed by electrospray mass spectrometry.
Thiol-disulfide equilibrium measurements
Thiol-disulfide equilibria were established by mixing 0.5–0.05 mM peptide and a redox buffer containing 18.75 mM reduced and 3.125 mM oxidized glutathione, respectively, in 200 mM Tris pH 8.0, 1 mM EDTA. Solutions were deoxygenated and stirred under argon at 20°C. Samples were taken after 1.5, 2.5, and 3.5 h, quenched in 30 mM HCl, and analyzed by HPLC. Ceff values were calculated from the molar ratios of oxidized and reduced peptide and glutathione (Cochran et al. 2001b): Ceff = ([peptideox]/[peptidered])([GSH]2/[GSSG]).
NMR data collection and analysis
One- and two-dimensional (2QF-COSY, TOCSY, ROESY, COSY-35) 1H NMR spectra of each peptide were acquired on a Bruker DRX500 spectrometer at temperatures between 5 and 30°C in 90% H2O/10% D2O and 100% D2O. Samples contained 1–5 mM solution of peptide at pH 5.0. Data on TZ-PDG and TZ-PDN were acquired on a Bruker DRX600 spectrometer. Spectra were processed and analyzed with FELIX (Accelrys). Resonance assignments were made by standard stepwise spin-system identification and sequential through-space interaction methods (Wüthrich 1986). 3JHN-Hα were obtained by fitting Lorentzian lines to the antiphase doublets of HN–Hα peaks in 2QF-COSY spectra processed to high digital resolution in F2. 3JHα-Hβ were extracted from the COSY-35 spectra collected in D2O. 13C resonance assignments were obtained from 13C HMQC and HMQC-TOCSY spectra collected on a Bruker DRX600 in D2O under the same solution conditions and temperatures as the 1H data collection.
Structure calculations
Interproton distance restraints were generated from ROEs observed in 2D-ROESY spectra collected in both H2O and D2O. φ and χ1 dihedral angle restraints were derived from 3JHN-Hα and 3JHα-Hβ scalar coupling constants, respectively. Additional φ and ξ dihedral angle restraints were obtained from the Hα, Cα, and Cβ chemical shift-based predictions using the program TALOS (Cornilescu et al. 1999). Calculation steps were as described (Skelton et al. 1994). One hundred structures were calculated using DGII (CVFF force field parameters), and 80 structures were refined using DISCOVER (AMBER all-atom force field parameters; Accelrys). The 20 conformations of lowest restraint violation energy were used to represent the structure and were evaluated using the program PROCHECK (Laskowski et al. 1993). 1H and 13C NMR chemical shifts were calculated on the basis of this ensemble of structures and also on the basis of the ensemble determined previously for bhpW (Cochran et al. 2001b) using the program SHIFTS, v4.0 (Xu and Case 2001). This program reports on the individual components that contribute to a particular secondary chemical shift. The coordinates for the PDG ensemble and the minimized mean structure have been deposited with the Protein Data Bank (accession codes 1N0A).
CD spectroscopy and analysis of thermal denaturation curves
Spectra were acquired with an Aviv Instruments model 202 spectrophotometer. Peptide concentrations were determined spectrophotometrically as described (Gill and von Hippel 1989). Melting curves were acquired at 229 nm with 1.5-min equilibration at each temperature and an averaging time of 15 sec. Thermal denaturation was reversible, as judged by recovery of CD signal (> 95%) upon cooling. The midpoint of the thermal folding transition was identified from polynomial fitting to a first-derivative curve; due to incomplete folding even at the lowest temperatures, the values reported in the text should be taken as estimates only.
Motif searching
A nonredundant database of 462 protein structures was searched for defined structure motifs, using the program WHATIF (Vriend 1990). Queries that yielded useful information included a seven-residue sequence search restricting position 5 to glycine combined with DSSP secondary structure definitions of strand for 1–2 and 6–7 and helix/turn/coil for 3–5 (26 hits); and a seven-residue DSSP secondary structure definition of strand for 1–2 and 6–7 and turn for 3–5 (65 hits). Hits were visually filtered to assure that the strands were forming the appropriate hydrogen bonds and the turns were the desired turn type, a step that left 14 of 87 examples. A BLAST search was also performed on the PDB using the sequence EPDGK, yielding three additional examples of hairpins with this turn type after visual inspection of hits.
Electronic supplementary material
NMR resonance assignments and coupling constant data for PDG, PDN, HV-PDG, HV-PDN, TZ-PDG, and TZ-PDN (Supplementary Tables 1–6). Summary of input restraints and structural statistics for PDG (Supplementary Table 7).
Acknowledgments
We thank Dr. David Case and Dr. Wayne Fairbrother for assistance with the chemical shift calculations; Melissa Starovasnik and Stephen Russell for many helpful discussions; and Jeff Tom for the Boc synthesis of the LTG turn peptide.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
2QF-COSY, double quantum filtered correlation spectroscopy
CD, circular dichroism spectroscopy
Ceff, effective concentration
COSY-35, correlation spectroscopy with 35° read pulse
DSSP, dictionary of secondary structure in proteins
Fmoc, 9-fluorenyl-methyloxycarbonyl
GSH, reduced glutathione
GSSG, oxidized glutathione
HB, hydrogen bonding
HMQC, heteronuclear multiple quantum spectroscopy
HPLC, high performance liquid chromatography
NHB, nonhydrogen bonding
NMR, nuclear magnetic resonance spectroscopy
NOE, nuclear Overhauser effect
RMSD, root mean squared deviation
ROESY, rotating frame Overhauser spectroscopy
Boc, tert-butyloxycarbonyl
TOCSY, total correlation spectroscopy
Tris, tris-hydroxymethyl-aminomethane
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0228603.
Supplemental material: See www.proteinscience.org.
References
- Blanco, F.J., Jimenez, M.A., Herranz, J., Rico, M., Santoro, J., and Nieto, J.L. 1993. NMR evidence of a short linear peptide that folds into a β-hairpin in aqueous solution. J. Am. Chem. Soc. 115 5887–5888. [Google Scholar]
- Blanco, F.J., Jimenez, M.A., Pineda, A., Rico, M., Santoro, J., and Nieto, J.L. 1994a. NMR solution structure of the isolated N-terminal fragment of protein-G B1 domain. Evidence of trifluoroethanol induced native-like β-hairpin formation. Biochemistry 33 6004–6014. [DOI] [PubMed] [Google Scholar]
- Blanco, F.J., Rivas, G., and Serrano, L. 1994b. A short linear peptide that folds into a native stable β-hairpin in aqueous solution. Nat. Struct. Biol. 1 584–590. [DOI] [PubMed] [Google Scholar]
- Blasie, C.A. and Berg, J.M. 1997. Electrostatic interactions across a β-sheet. Biochemistry 36 6218–6222. [DOI] [PubMed] [Google Scholar]
- Chakrabartty, A. and Baldwin, R.L. 1995. Stability of α-helices. Adv. Protein Chem. 46 141–176. [PubMed] [Google Scholar]
- Chen, P.Y., Lin, C.K., Lee, C.T., Jan, H., and Chan, S.I. 2001. Effects of turn residues in directing the formation of the β-sheet and in the stability of the β-sheet. Protein Sci. 10 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, A.G., Skelton, N.J., and Starovasnik, M.A. 2001a. Tryptophan zippers: Stable, monomeric β-hairpins. Proc. Natl. Acad. Sci. 98 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, A.G., Tong, R.T., Starovasnik, M.A., Park, E.J., McDowell, R.S., Theaker, J.E., and Skelton, N.J. 2001b. A minimal peptide scaffold for β-turn display: Optimizing a strand position in disulfide-cyclized β-hairpins. J. Am. Chem. Soc. 123 625–632. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Dalgarno, D.C., Levine, B.A., and Williams, R.J. 1983. Structural information from NMR secondary chemical shifts of peptide α C-H protons in proteins. Biosci. Rep. 3 443–452. [DOI] [PubMed] [Google Scholar]
- de Alba, E., Jimenez, M.A., Rico, M., and Nieto, J.L. 1996. Conformational investigation of designed short linear peptides able to fold into β-hairpin structures in aqueous solution. Fold. Des. 1 133–144. [DOI] [PubMed] [Google Scholar]
- de Alba, E., Rico, M., and Jimenez, M.A. 1997. Cross-strand side-chain interactions versus turn conformation in β-hairpins. Protein Sci. 6 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1999. The turn sequence directs β-strand alignment in designed β-hairpins. Protein Sci. 8 2234–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, H.J., Bolinger, L., Feher, V.A., Osterhout, Jr., J.J., Yao, J., and Wright, P.E. 1998. Sequence requirements for stabilization of a peptide reverse turn in water solution—Proline is not essential for stability. Eur. J. Biochem. 255 462–471. [DOI] [PubMed] [Google Scholar]
- Falcomer, C.M., Meinwald, Y.C., Choudhary, I., Talluri, S., Milburn, P.J., Clardy, J., and Scheraga, H.A. 1992. Chain reversals in model peptides: Studies of cystine-containing cyclic peptides. 3. Conformational free energies of cyclization of tetrapeptides of sequence. J. Am. Chem. Soc. 114 4036–4042. [Google Scholar]
- Favre, M., Moehle, K., Jiang, L., Pfeiffer, B., and Robinson, J.A. 1999. Structural mimicry of canonical conformations in antibody hypervariable loops using cyclic peptides containing a heterochiral diproline template. J. Am. Chem. Soc. 121 2679–2685. [Google Scholar]
- Freund, S.M., Wong, K.B., and Fersht, A.R. 1996. Initiation sites of protein folding by NMR analysis. Proc. Natl. Acad. Sci. 93 10600–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman, S.H. 1998. Minimal model systems for β sheet secondary structure in proteins. Curr. Opin. Chem. Biol. 2 717–725. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Grishina, I.B. and Woody, R.W. 1994. Contributions of tryptophan side chains to the circular dichroism of globular proteins: Exciton couplets and coupled oscillators. Faraday Discuss. 99 245–262. [DOI] [PubMed] [Google Scholar]
- Gu, H., Kim, D., and Baker, D. 1997. Contrasting roles for symmetrically disposed β-turns in the folding of a small protein. J. Mol. Biol. 274 588–596. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Ramakrishnan, C., and Balaram, P. 1997. β-hairpins in proteins revisited: Lessons for de novo design. Protein Eng. 10 1131–1141. [DOI] [PubMed] [Google Scholar]
- Hutchinson, E.G. and Thornton, J.M. 1994. A revised set of potentials for β-turn formation in proteins. Protein Sci. 3 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kim, C.A. and Berg, J.M. 1993. Thermodynamic β-sheet propensities measured using a zinc-finger host peptide. Nature 362 267–270. [DOI] [PubMed] [Google Scholar]
- Kortemme, T., Ramirez-Alvarado, M., and Serrano, L. 1998. Design of a 20-amino acid, three-stranded β-sheet protein. Science 281 253–256. [DOI] [PubMed] [Google Scholar]
- Kwong, P.D., Wyatt, R., Robinson, J., Sweet, R.W., Sodroski, J., and Hendrickson, W.A. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, E., Kortemme, T., Lopez de la Paz, M., and Serrano, L. 1999. The design of linear peptides that fold as monomeric β-sheet structures. Curr. Opin. Struct. Biol. 9 487–493. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lin, T.Y. and Kim, P.S. 1989. Urea dependence of thiol-disulfide equilibria in thioredoxin: Confirmation of the linkage relationship and a sensitive assay for structure. Biochemistry 28 5282–5287. [DOI] [PubMed] [Google Scholar]
- Maynard, A., Sharman, G., and Searle, M. 1998. Origin of β-hairpin stability in solution—Structural and thermodynamic analysis of the folding of model peptide supports hydrophobic stabilization in water solution. J. Am. Chem. Soc. 120 1996–2007. [Google Scholar]
- Milner-White, E.J. 1987. β-bulges within loops as recurring features of protein structure. Biochim. Biophys. Acta 911 261–265. [DOI] [PubMed] [Google Scholar]
- Minor, Jr., D.L., and Kim, P.S. 1994a. Context is a major determinant of β-sheet propensity. Nature 371 264–267. [DOI] [PubMed] [Google Scholar]
- ———. 1994b. Measurement of the β-sheet-forming propensities of amino acids. Nature 367 660–663. [DOI] [PubMed] [Google Scholar]
- Munoz, V., Thompson, P.A., Hofrichter, J., and Eaton, W.A. 1997. Folding dynamics and mechanism of β-hairpin formation. Nature 390 196–199. [DOI] [PubMed] [Google Scholar]
- Otzen, D.E. and Fersht, A.R. 1995. Side-chain determinants of β-sheet stability. Biochemistry 34 5718–5724. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado, M., Blanco, F.J., and Serrano, L. 1996. De novo design and structural analysis of a model β-hairpin peptide system. Nat. Struct. Biol. 3 604–612. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado, M., Blanco, F.J., Niemann, H., and Serrano, L. 1997. Role of β-turn residues in β-hairpin formation and stability in designed peptides. J. Mol. Biol. 273 898–912. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado, M., Kortemme, T., Blanco, F.J., and Serrano, L. 1999. β-hairpin and β-sheet formation in designed linear peptides. Bioorg. Med. Chem. 7 93–103. [DOI] [PubMed] [Google Scholar]
- Russell, S.J. and Cochran, A.G. 2000. Designing stable β-hairpins: Energetic contributions from cross-strand residues. J. Am. Chem. Soc. 122 12600–12601. [Google Scholar]
- Russell, S.J., Blandl, T., Skelton, N.J., and Cochran, A.G. 2002. Stability of cyclic β-hairpins: Energetic contributions from hydrogen-bonded cross-strand pairs. J. Am. Chem. Soc. 124: (in press). [DOI] [PubMed]
- Santiveri, C.M., Rico, M., and Jimenez, M.A. 2001. 13C(α) and 13C(β) chemical shifts as a tool to delineate β-hairpin structures in peptides. J. Biomol. NMR 19 331–345. [DOI] [PubMed] [Google Scholar]
- Searle, M.S., Williams, D.H., and Packman, L.C. 1995. A short linear peptide derived from the N-terminal sequence of ubiquitin folds into a water-stable non-native β-hairpin. Nat. Struct. Biol. 2 999–1006. [DOI] [PubMed] [Google Scholar]
- Serrano, L., Matouschek, A., and Fersht, A.R. 1992. The folding of an enzyme. III. Structure of the transition state for unfolding of barnase analysed by a protein engineering procedure. J. Mol. Biol. 224 805–818. [DOI] [PubMed] [Google Scholar]
- Sibanda, B.L., Blundell, T.L., and Thornton, J.M. 1989. Conformation of β-hairpins in protein structures. A systematic classification with applications to modeling by homology, electron density fitting and protein engineering. J. Mol. Biol. 206 759–777. [DOI] [PubMed] [Google Scholar]
- Skelton, N.J., Garcia, K.C., Goeddel, D.V., Quan, C., and Burnier, J.P. 1994. Determination of the solution structure of the peptide hormone guanylin: Observation of a novel form of topological stereoisomerism. Biochemistry 33 13581–13592. [DOI] [PubMed] [Google Scholar]
- Smith, C.K. and Regan, L. 1995. Guidelines for protein design: The energetics of β sheet side chain interactions. Science 270 980–982. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Construction and design of β-sheets. Acc. Chem. Res. 30 153–161. [Google Scholar]
- Spera, S. and Bax, A. 1991. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113 5490–5492. [Google Scholar]
- Stanger, H.E. and Gellman, S.H. 1998. Rules for antiparallel β-sheet design—D-pro-gly is superior to L-asn-gly for β-hairpin nucleation. J. Am. Chem. Soc. 120 4236–4237. [Google Scholar]
- Stanger, H.E., Syud, F.A., Espinosa, J.F., Giriat, I., Muir, T., and Gellman, S.H. 2001. Length-dependent stability and strand length limits in antiparallel β-sheet secondary structure. Proc. Natl. Acad. Sci. 98 12015–12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroup, A.N. and Gierasch, L. 1990. Reduced tendancy to form a β-turn in peptides from the P22 tailspike protein correlates with a temperature-sensitive folding defect. Biochemistry 29 9765–9771. [DOI] [PubMed] [Google Scholar]
- Syud, F.A., Stanger, H.E., and Gellman, S.H. 2001. Interstrand side chain-side chain interactions in a designed β-hairpin: Significance of both lateral and diagonal pairings. J. Am. Chem. Soc. 123 8667–8677. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Wouters, M.A. and Curmi, P.M. 1995. An analysis of side chain interactions and pair correlations within antiparallel β-sheets: The differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins 22 119–131. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids./ITL J. Wiley, New York.
- Wüthrich, K., Billeter, M., and Braun, W. 1984. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton–proton distances. J. Mol. Biol. 180 715–740. [DOI] [PubMed] [Google Scholar]
- Xu, X.P. and Case, D.A. 2001. Automated prediction of 15N, 13Cα, 13Cβ and 13C′ chemical shifts in proteins using a density functional database. J. Biomol. NMR 21 321–333. [DOI] [PubMed] [Google Scholar]
- Zaremba, S.M. and Gregoret, L.M. 1999. Context-dependence of amino acid residue pairing in antiparallel β-sheets. J. Mol. Biol. 291 463–479. [DOI] [PubMed] [Google Scholar]
- Zerella, R., Chen, P.Y., Evans, P.A., Raine, A., and Williams, D.H. 2000. Structural characterization of a mutant peptide derived from ubiquitin: Implications for protein folding. Protein Sci. 9 2142–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H.X., Hoess, R.H., and DeGrado, W.F. 1996. In vitro evolution of thermodynamically stable turns. Nat. Struct. Biol. 3 446–451. [DOI] [PubMed] [Google Scholar]