

Abstract
The aim of this study has been to develop a strategy for purifying correctly oxidized denatured major histocompability complex class I (MHC-I) heavy-chain molecules, which on dilution, fold efficiently and become functional. Expression of heavy-chain molecules in bacteria results in the formation of insoluble cellular inclusion bodies, which must be solubilized under denaturing conditions. Their subsequent purification and refolding is complicated by the fact that (1) correct folding can only take place in combined presence of β2-microglobulin and a binding peptide; and (2) optimal in vitro conditions for disulfide bond formation (∼pH 8) and peptide binding (∼pH 6.6) are far from complementary. Here we present a two-step strategy, which relies on uncoupling the events of disulfide bond formation and peptide binding. In the first phase, heavy-chain molecules with correct disulfide bonding are formed under non-reducing denaturing conditions and separated from scrambled disulfide bond forms by hydrophobic interaction chromatography. In the second step, rapid refolding of the oxidized heavy chains is afforded by disulfide bond–assisted folding in the presence of β2-microglobulin and a specific peptide. Under conditions optimized for peptide binding, refolding and simultaneous peptide binding of the correctly oxidized heavy chain was much more efficient than that of the fully reduced molecule.
Keywords: MHC class I, protein folding, disulfide bond formation, inclusion bodies, hydrophobic interaction chromatography
Major histocompatibility complex class I (MHC-I) molecules are expressed on the surface of almost all cells in the human body. These molecules are ternary complexes consisting of three components: (1) a glycosylated heavy chain (44 kD), containing two disulfide bonds; (2) a noncovalently associated light chain, β2-microglobulin (12 kD), containing a single disulfide bond; and (3) a tightly bound peptide (Springer et al. 1977). Their function is to sample endogenously derived peptides, transport them to the cell surface, and present them to cytotoxic T cells, which continuously scan cell surfaces for peptide–MHC-I complexes. Peptides presented in context with MHC-I molecules originate from the digestion of intracellular proteins, normal ones and those of pathogens. MHC-I molecules therefore serve as a link between the intracellular compartment, which is inaccessible to the cells of the immune system, and the extracellular compartment, in which the immune cells reside. This mechanism is crucial in the immune defense against intracellular pathogens, such as viruses.
The availability of highly pure and functional MHC-I molecules is important for analytical, as well as preparative, purposes. Conventional purification of soluble peptide–MHC-I complexes from human cell lines, using monoclonal antibodies, is a cumbersome and expensive process resulting in low protein yields. Expression of MHC-I molecules in bacterial hosts offers significant advantages, including ease of genetic manipulation and reasonably simple growth and induction methodologies. However, high levels of expression frequently lead to the formation of insoluble inclusion bodies (Marston 1986), and this phenomenon has been shown to apply to the expression of the heavy chain and β2m in Escherichia coli (Garboczi et al. 1992). To acquire the biological activity inherent in these molecules, extraction into denaturing buffer followed by in vitro protein refolding is necessary. The refolding process is complicated by the fact that the heavy chain cannot fold to its native state in the absence of β2m and peptide. Thus far, it has been impossible to generate empty MHC-I molecules, as they are highly unstable and prone to aggregate. Refolding of the heavy-chain molecule from the completely reduced configuration must be performed under mildly alkaline conditions, typically around pH 8.0, so as to promote disulfide bond formation (Rudolph and Lilie 1996). However, we have previously shown that peptide binding, which is necessary for stable complex formation, is optimal at slightly acidic pH, around pH 6.6 (Stryhn et al. 1996; Pedersen et al. 2001). This poses a serious problem, as disulfide bond formation and peptide binding cannot operate under optimized conditions simultaneously.
A possible solution to the above conundrum lies in uncoupling these two events from one another so that each of them can be optimized independently. Although the E. coli cytosol is generally considered a reducing environment, we have previously observed the presence of oxidized species of MHC-I heavy chains in extracts of inclusion bodies (Pedersen et al. 2001). A large part of these oxidized species have apparently obtained the correct disulfide bond configuration and were able to undergo efficient refolding even at pH 6.6, whereas heavy chains with nonnative configurations misfolded or aggregated under the same conditions.
Here we demonstrate that disulfide bond isomers of the heavy-chain molecules can be separated by hydrophobic interaction chromatography under non-reducing, denaturing conditions, and that pure preparations of oxidized MHC-I heavy-chain molecules can be obtained. The active isomer is subsequently identified and shown to undergo essentially complete disulfide-assisted refolding under conditions optimized for peptide binding.
Results
Expression of MHC-I heavy chains in E. coli
Membrane-truncated recombinant HLA-A*0201 (rA2), HLA-A*1101 (rA11), and H2-Kk (rKk [des cys]) were produced as insoluble inclusion bodies in E. coli. Expression was initiated with IPTG and allowed to continue for 3 h at 42 °C. Low levels of expression were reached ∼1 h after addition of IPTG for both rA2 and rKk (des cys) (Fig. 1A,B ▶). In contrast, the expression of rA11 reached much higher levels, which were attained after 3 h of induction (Fig. 1C ▶). The observed difference in the expression levels are probably because the gene encoding rA11 was optimized for E. coli codon usage, which was not the case for rA2 and rKk (des cys).
Figure 1.

Reducing SDS-PAGE analysis of expression levels of MHC-I heavy chains in Escherichia coli. (A) Expression levels of rA2. (B) Expression levels of rKk (des cys). (C) Expression levels of rA11. Fermentor samples (15 μL) were withdrawn before and every hour after induction. The bacterial cell pellet was resuspended in 50 μL MgCl2/SDS lysis buffer to release and solubilize heavy-chain inclusion bodies as described by Chen and Christen (1997). After centrifugation at 20,000g for 2 min, 15 μL of the supernatant was loaded directly on the gel. Lane 1, protein marker; lane 2, before induction; and lanes 3–5, samples taken 1, 2, and 3 h after induction with IPTG, respectively. Positions of heavy-chain monomers are shown with arrows.
Isolation and solubilization of inclusion bodies
Inclusion bodies were released from harvested E. coli cells by enzymatic disruption with lysozyme, and co-released DNA/RNA was subsequently digested with DNase I and RNase A to reduce the viscosity. Centrifugally collected inclusion bodies were then washed free of loosely adsorbed and entrained contaminants in several washing steps, before solubilization in 8 M urea under non-reducing conditions. A prerequisite for the chosen strategy is that inclusion body solubilization must be performed under non-reducing conditions so as to preserve the oxidation state of the heavy-chain molecules. The initial purities were estimated to be 20% to 30% by densitometric analysis of non-reducing SDS–polyacrylamide gels, and total inclusion body protein recovery was ∼0.3 to 1.5 g/L bacterial culture. Final optical density O.D.600 values of the fermentor culture prior to harvest were in the range of 30 to 60.
SDS-PAGE analysis of inclusion body-derived MHC-I heavy chains
Reducing SDS-PAGE analysis of solubilized inclusion body preparations revealed a monomeric band of high intensity with an apparent molecular mass of 32 kD, which corresponds well with the theoretical molecular masses of the truncated recombinant heavy-chain monomers. However, non-reducing SDS-PAGE analysis revealed a number of monomeric heavy-chain isomers, which could only differ from the reduced isomer in their disulfide bond configuration (Fig. 2 ▶), in a similar manner to that which we have previously reported for rA2 (Pedersen et al. 2001). The band with the lowest mobility is the fully reduced heavy-chain isomer (designated 0 in Fig. 2 ▶), whereas partially/fully oxidized species have a higher mobility (designated 1,2 in Fig. 2 ▶). We expect that only one of these oxidized forms have attained the correct disulfide bond configuration. In the case of rA2 and rA11 only oxidized species were observed, whereas for rKk (des cys), the reduced form was also found. Isomers 1 and 2 were present in equimolar amounts in the rA2 preparation, but in all other preparations, isomer 1 was the dominant form. Thus, >90% of the observed rKk (des cys) heavy-chain forms were accounted for by isomer 1, indicating that this isomer is the most stable one.
Figure 2.

Reducing and non-reducing SDS-PAGE analysis of solubilized inclusion body preparations. (A) Solubilized inclusion bodies containing rA2. (B) Solubilized inclusion bodies containing rA11. The intensity of the heavy-chain monomer band increased on reduction, owing to the release of disulfide bond cross-linked monomers, and made the distinction of the isomers difficult. To improve the visualization of heavy-chain isomers, reducing samples of rA2 and rA11 were diluted twice as much in sample buffer as the non-reducing counterparts. The positions of heavy-chain isomers 0, 1, and 2 are indicated on the figure. NR indicates non-reducing; R indicates reducing.
Separation of oxidized species of MHC-I heavy-chain monomers
In order to determine the most effective refolding candidate among the observed oxidized heavy-chain isomers, we screened several chromatographic separation techniques (i.e., hydrophobic interaction, anion-exchange, cation-exchange chromatography) and media for their ability to resolve the different isomers. Although the anion-exchanger Q Sepharose Fast Flow was able to partly separate isomers 1 and 2 in the rA2 preparation (data not shown), the separation was much better when using hydrophobic interaction chromatography (HIC). Several HIC media were tested, including butyl and octyl Sepharose Fast Flow and three different versions of the phenyl Sepharose media. In all cases, however, the best resolution was achieved on phenyl Sepharose High Performance. Figures 3 and 4 ▶ ▶, respectively, show chromatograms corresponding to the fractionation of rA2 and rA11 on this media and the non-reducing SDS-PAGE analysis of selected fractions. In the case of rA11, isomer 1 eluted first, whereas the order was reversed for rA2. An additional isomer, not discovered during the initial analysis, appeared during the fractionation of rA11 and was designated isomer 3 (Fig. 4 ▶). The band representing isomer 3 is more diffuse than bands 1 and 2 and could conceivably represent more than one band, corresponding to different disulfide bond isomers of rA11. Isomer 3 co-eluted with the rA11 isomer 1, and it was not possible to resolve the two with any of the chromatographic techniques tested in this study.
Figure 3.
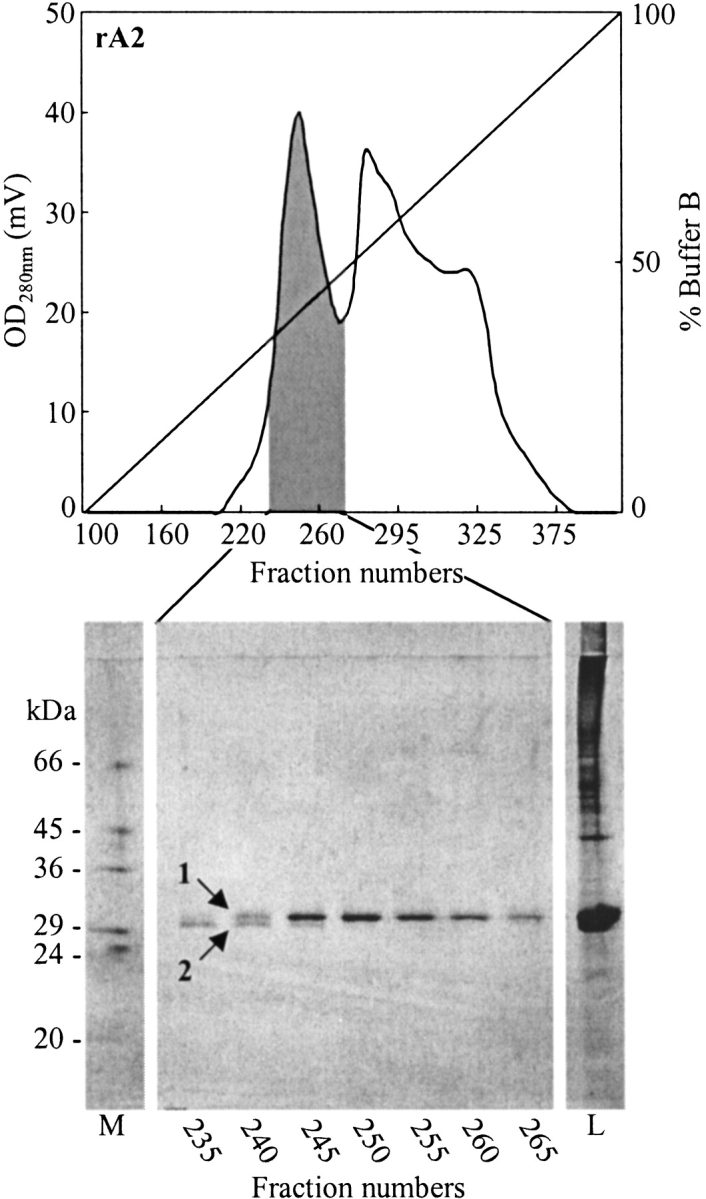
Separation of rA2 heavy-chain isomers by hydrophobic interaction chromatography on phenyl Sepharose High Performance under non-reducing denaturing conditions. Aliquots (7.5 μL) of fractions collected in the shaded area of the chromatogram were analyzed by non-reducing SDS-PAGE, and the result is shown below. Analyzed fractions are indicated on the figure, as well as the positions of heavy-chain isomers. Lane M, protein marker; lane L, sample applied on the hydrophobic interaction chromatography column.
Figure 4.
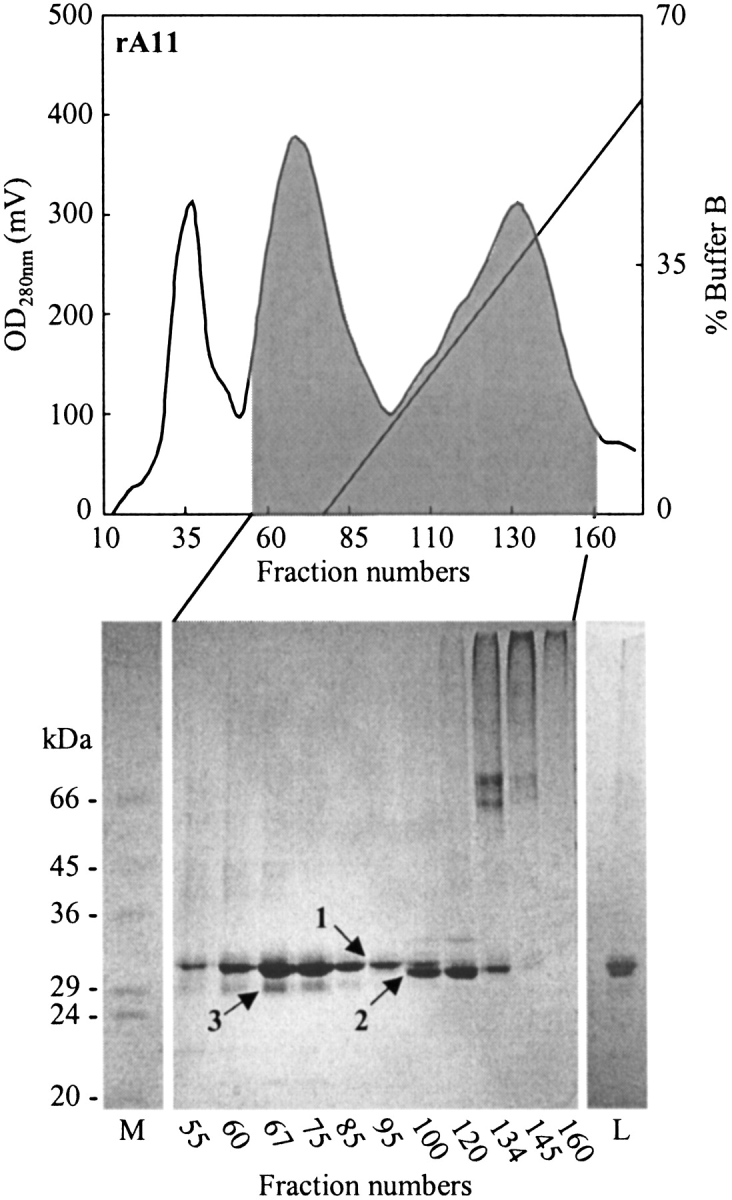
Separation of rA11 heavy-chain isomers by hydrophobic interaction chromatography on phenyl Sepharose High Performance under non-reducing denaturing conditions. Aliquots (12 μL) of fractions collected in the shaded area of the chromatogram were analyzed by non-reducing SDS-PAGE, and the result is shown below. Analyzed fractions are indicated on the figure, as well as the positions of heavy-chain isomers. Lanes M, protein marker; lane L, sample applied on the hydrophobic interaction chromatography column.
High-molecular-weight contaminants and multimerized forms of the heavy-chain monomer were removed by size exclusion chromatography (SEC) on Sephacryl 200-HR (rA2 & rA11) and Sephacryl 400-HR (rKk [des cys]). By combining anion-exchange on Q Sepharose Fast Flow with HIC on phenyl Sepharose High Performance and SEC, the final purities of heavy-chain preparations were ∼95% as estimated by densitometric analysis of non-reducing SDS–polyacrylamide gels, and yields were ∼20 to 30 mg/L bacterial culture. Table 1 shows a summary of the purification steps for all three heavy chains.
Table 1.
Summary of purification steps for rA2, rA11, and rKk (des cys)
| rA2 | rA11 | rKk (des cys) | ||||||||||
| Purification step | Pool volume (mL) | Total protein (mg) | Purity of isomer 1a (%) | Yield (%) | Pool volume (mL) | Total protein (mg) | Purity of isomer 1a (%) | Yield (%) | Pool volume (mL) | Total protein (mg) | Purity of isomer 1a (%) | Yield (%) |
| Solubilized IB | 180 | 624 | 25 | 100 | 200 | 2265 | 34 | 100 | 175 | 1940 | 22 | 100 |
| Q Sepharose Fast Flow | 150 | 392 | 36 | 90 | 180 | 652 | 39 | 33b | 235 | 832 | 41 | 78 |
| (NH4)2SO4 precipitation | 160 | 42 | 43 | 46 | 190 | 218 | 37 | 10 | 245 | 71 | 87 | 14 |
| Phenyl Sepharose HPc | 200 | 38 | 92 | 23 | 200 | 72 | 90 | 8 | 280 | 71 | 87 | 14 |
| Sephacryl 200-HRd | 30 | 34 | >99 | 22 | 40 | 57 | 92 | 7 | 50 | 58 | 91 | 13 |
a The purity of isomer 1 is calculated from the total amount of protein combined with densitometric analysis of non-reducing Coomassie-stained SDS–polyacrylamide gels.
b The Q Sepharose Fast Flow media was heavily overloaded with protein, resulting in a significant loss of rA11 in the flow through.
c Collected pools were concentrated to ∼15 mL final volume prior to loading on the following SEC column (see Materials and Methods).
d SEC for purification of rKk (des cys) was performed on Sephacryl 400-HR instead of Sephacryl 200-HR.
Analysis of peptide binding to fractionated oxidized heavy-chain isomers
The ability of fractionated heavy chains to undergo productive refolding in the presence of β2m, and a specific radiolabeled peptide at pH 6.6 was examined with a biochemical-binding assay (see Materials and Methods). In short, samples were withdrawn from collected fractions containing heavy-chain monomer and assayed for peptide binding. To avoid ligand depletion, which would lead to peptide binding affinity being underestimated, each sample was diluted to obtain a final heavy-chain concentration, resulting in approximately 25% or less of the ligand being bound.
Figure 5 ▶ shows the peptide binding analysis performed on fractions collected during purification of rA2 and rA11 on phenyl Sepharose High Performance. From Figure 5 ▶, it is evident that the peptide-binding signal coincides with the protein tracing for isomer 1 and not for isomer 2 for both rA2 (Fig. 5A ▶) and rA11 (Fig. 5B ▶). A similar profile, although not shown, was observed during purification of rKk (des cys). The peptide binding observed in fractions enriched in isomer 2 is most likely the result of low amounts of isomer 1 present in these fractions (Figs. 4, 5B ▶ ▶; cf. the analysis of fraction 55 and 120).
Figure 5.
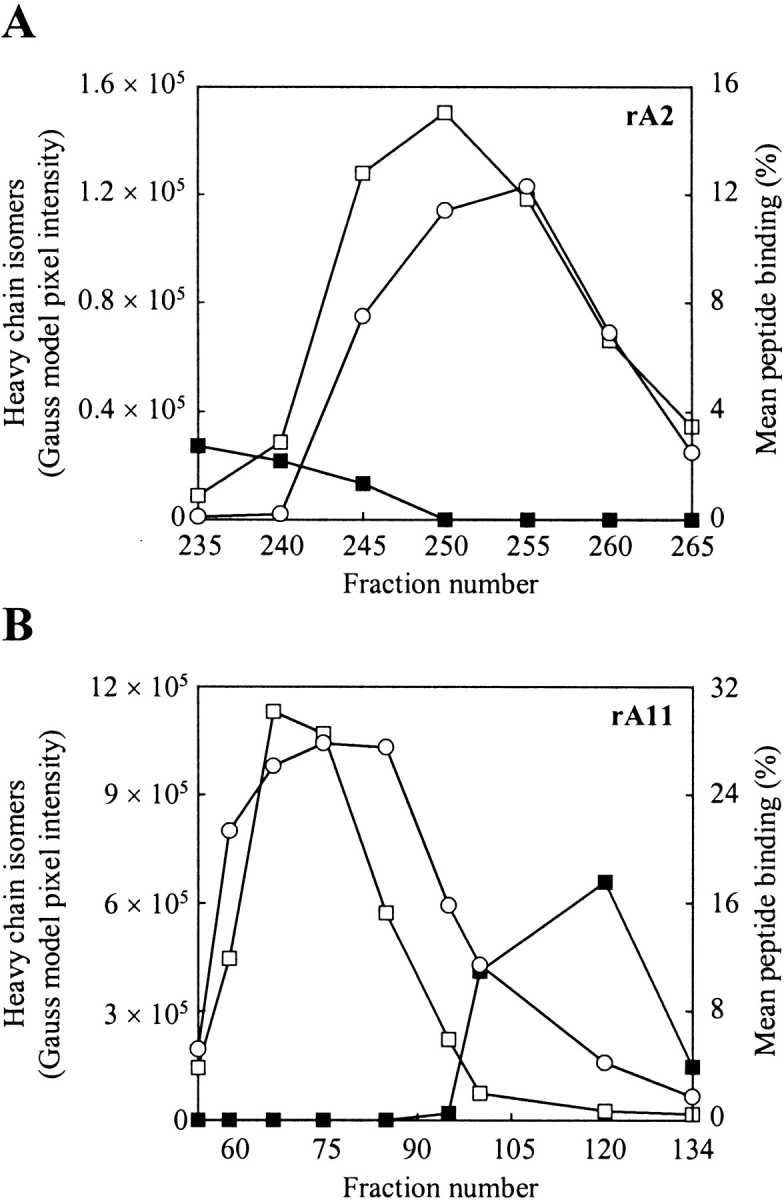
Refolding and peptide binding analysis of fractionated MHC-I heavy chain isomers. (A) Analysis of fractions collected during purification of rA2 on phenyl Sepharose High Performance (see Fig. 3 ▶). (B) Analysis of fractions collected during purification of rA11 on phenyl Sepharose High Performance (see Fig. 4 ▶). Folding was initiated by diluting aliquots (1 μL) from selected fractions 100-fold in 100 mM Tris-maleate (pH 6.6) buffer, containing human β2m (3 μM) and a radiolabeled peptide (15,000 cpm). The pixels intensities of heavy-chain isomers 1 and 2 were calculated from a densitometric analysis of SDS–polyacrylamide gels shown in Figure 3 and Figure 4 ▶ ▶, respectively. Fraction numbers are shown on the figure. Empty squares indicate isomer 1 protein tracing; solid squares, isomer 2 protein tracing; and circles, mean peptide binding. The standard deviation of duplicate peptide binding measurements was typically within 5%.
Evaluation of refolding efficiency with a quantitative and conformationally sensitive ELISA
In the case of rA11, isomer 1 co-eluted with isomer 3, making it difficult to determine whether only one or both of them were contributing to the observed peptide-binding signal (Fig. 5 ▶). Based on the results presented in Figure 5 ▶, isomer 1 is likely to be refolding properly and binding peptide as one fraction enriched in this isomer, but essentially free of isomer 3, gave rise to a peptide-binding signal of 15% (Figs. 4, 5B ▶ ▶; fraction 95).
To further pursue the question of refolding efficiency and peptide-binding abilities of rA11 heavy-chain isomers 1 and 3, a newly developed quantitative ELISA was exploited (Sylvester-Hvid et al. 2002). When β2m and peptide are present at saturating conditions, this ELISA assay allows the determination of the concentration of heavy-chain molecules, which can fold into complexes.
The refolding reaction was performed with a purified batch of rA11, enriched in isomer 1, but also containing low amounts of isomer 3. Figure 6 ▶ shows a non-reducing SDS-PAGE analysis of the tested rA11 preparation as well as the processed results from the ELISA analysis. A linear fit to the ELISA data (y = 0.96× − 4.14, R2 = 0.99) estimates the amount of heavy chain available for complex formation to ∼96% of the total amount. By comparison the densitometric analysis of the non-reducing SDS–polyacrylamide gel in Figure 6 ▶ gives the following distribution of isomers 1 and 3: 92% and 8%, respectively. This strongly indicates that isomer 1 is the active form of the two isomers.
Figure 6.

Determination of the amount of oxidized rA11 heavy-chain monomer that refolds properly into the matured state with a quantitative and conformationally sensitive ELISA. The double log plot shows the amount of heavy-chain monomer offered to the folding reaction that was detected in the fully matured MHC-I complex. A non-reducing SDS-PAGE analysis of the purified rA11 sample is shown in the insert. Graded concentrations of denatured rA11 were diluted in a 100 mM Tris-maleate (pH 6.6) refolding buffer containing an excess of human β2m (3 μM) and a specific peptide (10,000 nM). Detection of properly folded complexes and conversion of measured O.D.450 values to picomolar complex was performed as described in Materials and methods. Lane 1, rA11 sample; lane 2, protein marker. Positions of rA11 isomers 1 and 3 are shown with arrows. Standard deviations of triplicate measurements are indicated on the figure.
Comparison of refolding efficiency between the correctly oxidized (isomer 1) and fully reduced (isomer 0) rA11 heavy-chain monomer
To evaluate the performance of the chosen strategy the refolding of the apparently correctly oxidized heavy-chain monomer (isomer 1) was compared with that of the fully reduced monomer (isomer 0). An inclusion body preparation of rA11 was solubilized under non-reducing conditions and purified on phenyl Sepharose High Performance as described in Materials and Methods. Fractions enriched in isomer 1 and completely free from isomer 2 were pooled and then divided into two equal aliquots. One of them was reduced by an overnight treatment with 4 mM DTT at 4°C.
Subsequently, both were purified on Sephacryl 400-HR to remove high-molecular-weight contaminants. For the reduced heavy-chain preparation, the buffer was supplemented with 2 mM DTT to prevent reoxidation during the size exclusion chromatography. The resulting heavy-chain preparation only contained reduced species as ascertained by non-reducing SDS-PAGE mobility analysis (data not shown).
Fractions containing highly purified rA11 monomer were pooled, and the ability of the pooled material to undergo productive refolding at acidic pH in the presence of β2m and a radiolabeled peptide was examined (Fig. 7 ▶). Binding of 10% of the offered peptide was reached at a concentration of only 30 nM of isomer 1, whereas 150 nM of reduced heavy chain was required to reach the same degree of binding, demonstrating that isomer 1 undergoes refolding with considerably higher efficiency than the fully reduced heavy chain (isomer 0).
Figure 7.

Dose-response curve for purified rA11 isomer 0 (reduced) and isomer 1 (oxidized). Graded concentrations of purified A11 isomer 0 and 1 were diluted 100-fold into 100 mM Tris-maleate (pH 6.6) buffer containing human β2m (3 μM) and a specific radiolabeled peptide (15,000 cpm) and 1 mg/mL pluriol. The mean peptide binding values were calculated as described in Materials and Methods. Empty squares indicate rA11 isomer 1; solid squares, rA11 isomer 0. The standard deviation of duplicate peptide binding measurements was typically within 5%.
Discussion
Denatured MHC class-I heavy-chain molecules are particularly difficult proteins to fold. In vivo, the complicated nature of the MHC-I folding pathway is emphasized by the number of endoplasmic reticulum chaperones and disulfide bond isomerases, which are involved in folding and peptide loading (Cresswell 2000). In vitro, the heavy chain only exists as part of a ternary complex involving the MHC-I light chain (β2m) and a specific peptide (Garboczi et al. 1992). In the absence of either of the latter two components, denatured MHC-I heavy chain misfolds when diluted into a refolding buffer. Even in their presence, denatured and reduced MHC-I heavy chain folds only slowly and incompletely. In contrast, we have recently observed that denatured MHC-I heavy chains, if they are oxidized, can fold and generate the ternary complex rapidly and efficiently (Pedersen et al. 2001). Similar disulfide bond–assisted folding has been observed for other oxidized molecules with native disulfide bonds (van den Berg et al. 1999). In this report, we demonstrate that oxidized preparations of denatured MHC-I heavy-chain molecules can be purified to virtual homogeneity under denaturing conditions, yielding subspecies of oxidized denatured molecules, which become fully active when appropriately diluted. This indicates a new strategy in protein folding whereby the overall process is divided into two parts; one of purifying denatured protein species with native disulfide bonds, and another of folding these species. The advantage of such an approach is that the two parts can be optimized independently. Recombinant generation of MHC-I is an example of the potential benefits.
The primary structure of the MHC-I heavy chain has four conserved cysteine residues, constituting two disulfide bonds in the correctly folded MHC-I molecule (Springer et al. 1977). One is located in the α2 domain (Cys101 to Cys164), which is part of the peptide-binding groove and the other is located in the membrane proximal α3 domain (Cys203 to Cys259). With four cysteine residues, 10 different conformations are possible, namely, one completely reduced, six partially oxidized (with one disulfide bond), and three completely oxidized (with two disulfide bonds). Although the distribution of these isomers during protein expression and extraction of the inclusion bodies is not known, we have previously noted that only two major bands are apparent in non-reducing SDS–polyacrylamide gels of inclusion body extracts (Pedersen et al. 2001).
Our approach is based on bacterial protein expression followed by extraction into denaturing buffer. Very low amounts of the fully reduced heavy-chain monomer are found, indicating that oxidation has occurred either within the bacteria (Teilum et al. 1999) or as a result of air-oxidation during the extraction process (Schoemaker et al. 1985). We have not attempted to distinguish between these two possibilities, but it seems reasonable to assume that all disulfide bond configurations are possible and that our preparations contain at least some scrambled versions. During extraction and purification in the current work we identified different bands by SDS-PAGE, which were designated 0, 1, 2, and 3 (Figs. 2, 4 ▶ ▶). Non-reducing and reducing SDS-PAGE analysis indicated that band 0 is the fully reduced heavy chain, whereas bands 1, 2, and 3 are either partially or fully oxidized species of the heavy-chain monomer (Figs. 2, 4 ▶ ▶). Tector and coworkers (1997) analyzed the distribution of natural MHC-I heavy chain isomers in extracts from Daudi cells and suggested, on the grounds of electrophoretic mobility, that isomers 0, 1, and 2 represent the fully reduced, partially oxidized, and fully oxidized forms, respectively. It is tempting to speculate that this distribution is identical to that one found in our recombinant preparations.
Isomers 1 and 2 were partially resolved on the anion exchanger Q Sepharose Fast Flow and almost complete separation was achieved by hydrophobic interaction chromatography on phenyl Sepharose High Performance media (Figs. 3, 4 ▶ ▶). It is interesting to note that isomer 1 of rA2 elutes after isomer 2, whereas isomer 1 of rA11 elutes before isomer 2. HLA-A*0201 has a preference for hydrophobic peptides (Falk et al. 1991), whereas HLA-A*1101 have a preference for hydrophilic peptides (Zhang et al. 1993; Kubo et al. 1994). The structural nature of the bound peptides is complementary to the structure of the peptide-binding groove in which the major differences between the HLA molecules are located. We speculate that the observed shift in the elution profile during hydrophobic interaction chromatography reflects the hydrophobicity of the peptide-binding groove exposed under the denaturing conditions.
Peptide–MHC-I binding experiments conducted on selected fractions during fractionation on phenyl Sepharose High Performance combined with a quantitative ELISA analysis, indicated that only isomer 1 was capable of efficient refolding and simultaneous peptide binding in the presence of β2m and a specific peptide (Figs. 5, 6 ▶ ▶). Isomer 1 most likely assumes the native disulfide bond configuration, locking the molecule in a favorable conformation for subsequent folding, whereas isomer 2 probably adopts an incorrectly oxidized form, which prevents it from reaching the 3-dimensional structure required for peptide binding. Compared with the fully reduced heavy-chain monomer, isomer 1 is considerably more efficient at refolding and simultaneous peptide binding (Fig. 7 ▶). The weak binding observed when using fully reduced MHC-I molecules probably reflects the inefficient formation of correct disulfide bonds by air-oxidation followed by rapid peptide binding, rather than inefficient peptide binding to reduced MHC-I. This is consistent with previous evidence indicating that disulfide bond formation precedes peptide loading into the peptide-binding groove (Smith et al. 1995). However, recent studies indicate that isomerization of the disulfide bond in the α2 domain may play a role in the peptide binding process and the final maturation of the MHC-I receptor complex (Dick et al. 2002).
In summary, we have shown that oxidized species of heavy-chain monomers can be separated by hydrophobic interaction chromatography under non-reducing and denaturing conditions, and that one of these isomers is able to undergo efficient refolding and simultaneously peptide binding under acidic conditions. The feasibility of the suggested production and purification process was demonstrated for both murine and human MHC-I molecules, and we believe that it can be extended to include all MHC-I molecules. Such recombinant MHC class I molecules are ideally suited for quantitative peptide–MHC-I binding studies (Buus 1999) and for the production of tetrameric peptide–MHC-I complexes (Altman et al. 1996). Refolding of other immunoglobulin-like proteins containing multiple disulfide bonds might also benefit from this strategy.
Materials and methods
Cloning of human and murine MHC-I heavy chains
All recombinant MHC-I heavy chains were expressed without the transmembrane sequence. E. coli strain XA90 transformed with pHN1+ containing an HLA-A*0201 (rA2) heavy-chain (1–275) cDNA insert was a kind gift from Drs. Wiley and Garboczi (Harvard University). cDNA segments encoding HLA-A*1101 (rA11) heavy chain (1–275) was PCR amplified and inserted into the pET28a+ vector (Novagen). The rA11 sequence was optimized for E. coli codon usage, by using the QuickChange kit (Stratagene) and appropriate primers. The plasmid was subsequently transformed into E. coli strain BL21-CodonPlus(DE3)-RP (Stratagene). Clones, which produced protein on induction with IPTG, were identified and the inserted sequence was verified by DNA sequencing (ABI310, Perkin Elmer). Murine H2-Kk (des cys) cDNA containing four cysteines was inserted into the pET28a+ vector and transformed into BL21(DE3) (Pedersen et al. 2001). Briefly, cysteine 121, which is not involved in disulfide bond formation, was exchanged for arginine as found in other murine MHC-I molecules.
Expression of rA2, rA11, rKk (des cys) in E. coli
Large-scale production was done in a 2-L Labfors fermentor (Infors AG), according to the manufacturer’s instructions. All fermentations were run with ECPM 1 media (Bernard and Payton 2002). Primatone (20 %, w/v; Rode and Rode) was used instead of casamino acids, and 0.3 mL antifoam 289 (Sigma) was added to prevent foaming. The fermentor, including media, was autoclaved for 1 h, and appropriate sterile antibiotics were added after autoclavation. Inoculation cultures were set up in 200 mL YT×2 medium containing 10 mM MgCl2 and 0.5% (v/v) glucose plus the respective antibiotics. An inoculum, corresponding to 10 mL of an O.D.600 = 1 culture, was transferred to the fermentor, and expansion was done over night at 25°C to 28°C with atmospheric air as oxygen source. The growth rates were followed by measuring the O.D.600 of samples withdrawn every hour. As soon as the atmospheric oxygen became limited, the air source was changed to 100% oxygen. When the O.D.600 reached 25, induction was initiated with 1 mM IPTG. At the same time, the temperature was increased to 42°C, and additional sugar (87% glycerol) was added in a fed batch mode. After 3 h of induction, the fermentor was harvested. Samples taken just before and during induction were kept for SDS-PAGE analysis.
Isolation and solubilization of inclusion bodies
The isolation and solubilization procedure was performed essentially according to Sambrook et al. (1989). Cells lysis was done with lyzosyme (Sigma), and liberated DNA/RNA was digested with DNase I (Sigma) and RNAse A (Sigma). Inclusion bodies were collected by centrifugation at 17,000g for 10 min at 4°C and washed in PBS supplemented with 0.5 % (v/v) NP-40 (Sigma) and 0.1 % (w/v) deoxycholic acid (Sigma) followed by washing in 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 100 mM NaCl. Washed inclusion bodies were solubilized in 20 mM Tris-HCl (pH 8.0) and 8 M urea (200 mL for each 100 g of wet cell paste). Insoluble material was removed by centrifugation at 17,000g for 15 min at 4°C. Supernatants were pooled and successively filtered through 8-, 3-, 1.2-, and 0.45-μm filters and stored at -20°C until further processing.
Purification of MHC-I heavy chains
Chromatographic separations were performed at 12°C on a ÄKTA prime workstation (Amersham Biosciences), using a flow rate of 60 mL/h. All chromatographic media and columns were from Amersham Biosciences, Sweden, including 1 mL HiTrap columns for screening experiments. The purification was monitored by SDS-PAGE and bicinchoninic acid (BCA) assay (Pierce). Urea decomposition leads to the generation of cyanate. To minimize the concentration of cyanate in urea-containing buffers, all solution were made freshly and used immediately, and the primary amine, Tris, was included as cyanate scavenger (Wingfield 2002). A preparation of solubilized inclusion bodies was applied to a Q Sepharose Fast Flow column (2.6 × 65 cm) equilibrated with 20 mM Tris-HCl (pH 8.0) and 8 M urea (Merck; loading buffer). Unbound material was washed off with 2 column volumes (CV) of loading buffer. Bound proteins were eluted with a linear 0 to 1 M NaCl gradient (5 CV). Fractions containing functional MHC-I heavy chain were pooled. Functional pools from Q Sepharose Fast Flow fractionation were adjusted to 100 mM Tris-HCl (pH 8.0) and 8 M urea, and ammonium sulfate (Sigma) was added to 20% (w/v) saturation. The solution was stirred at room temperature for 30 min, and insoluble material was removed by centrifugation at 17,000g for 10 min at 4°C. The supernatant was filtered through a 0.45-μm filter and applied to a phenyl Sepharose High Performance column (2.6 × 70 cm), equilibrated with 100 mM Tris-HCl (pH 8.0), 8 M urea, and 20% ammonium sulfate. Unbound proteins were washed off with 1.5 CV of the same buffer. Bound proteins were eluted from the column with a linear 20% to 0% ammonium sulfate gradient (5 CV). Prior to the subsequent size exclusion chromatography step, fractions were pooled and concentrated on a 10-kD nominal-molecular-weight-limit filter (Millipore) in a stirred nitrogen pressure cell (Amicon) to a final volume of 15 mL. Size exclusion chromatography was done on two Sephacryl 200-HR or two Sephacryl 400-HR columns (2.6 × 100 cm) connected in series. Columns were equilibrated with 20 mM Tris-HCl (pH 8.0), 8 M urea, or 20 mM Tris-HCl (pH 8.0), 6 M guanidine hydrochloride. Purified heavy chains were pooled, aliquoted, and stored at −20°C until further analysis.
Peptide radioiodination
Peptides were purchased from Schaefer-N, purified to homogeneity by reverse-phase HPLC chromatography, lyophilized, and stored at −20°C. All preparations were quantified by using the BCA assay. Radiolabeling was done with 125Iodine (Amersham Biosciences). Peptides used for refolding of MHC-I heavy chains, and biochemical binding assays had the following sequences (in single letter code): FLPSDYFPSV for HLA-A*0201, KLFPPLYLR for HLA-A*1101, and SDYEGRLI (Influenza NP peptide50–57) for H2-Kk (des cys).
Biochemical peptide binding assay
Refolding conditions reported by Pedersen et al. (2001) for MHC-I heavy chain were used. Purified MHC-I heavy chain samples were refolded by 100-fold dilution in the presence of excess human β2m (3 μM) and radiolabeled peptide (1 to 3 nM, 15,000 cpm/sample) for ∼24 h at 18°C in a total reaction volume of 100 μL per sample. The refolding buffer was 100 mM Tris-maleate buffer (pH 6.6) in PBS supplemented with 1 mg/mL pluronic copolymer Lutrol F-68 (BASF). The final concentration of urea after dilution was 80 mM. The recombinant human β2m was produced in our laboratory from E. coli fermentations (Pedersen et al. 2001). Binding of peptide to MHC-I heavy chains were measured by Sephadex G-50 spun column chromatography (Buus et al. 1995). The radioactivity of the excluded "void" volume, containing formed MHC-I complexes, and of the retained volume, containing unbound peptide, was measured by gamma spectrometry (Packard Instruments). Peptide binding values were calculated by dividing excluded radioactivity with the total amount of radioactivity offered. Mean peptide binding values were obtained from duplicate spun column chromatography runs and expressed in percent.
Quantitative ELISA assay measuring peptide–MHC-I complex formation
Quantitative detection of peptide–MHC-I complex formation was performed as described by Sylvester-Hvid et al. (2002). In summary, peptide–MHC-I complex formation was measured with a sandwich ELISA by using the conformationally sensitive monoclonal antibody W6/32 for capturing complexes and horseradish peroxidase-conjugated, polyclonal rabbit anti-human β2m antibody (DAKO) for detection of correctly folded complexes. The ELISA was developed with 3,3′ 5,5′ -tetramethylbenzidine hydrogen peroxide (TMB-one, Kem-En-Tec) for 30 min at room temperature and the color reaction was read at 450 nm on a Victor2Multilabel ELISA counter (Wallac). A standard curve was constructed by plotting the measured O.D.450 response against the logarithm of an MHC standard with known protein concentration. The curve was optimally fitted to a sigmoid curve (Prism™ 3.0, GraphPad), thereby allowing O.D.450 of any sample to be converted to the concentration of MHC-I complexes in the sample.
Electrophoresis
One-dimensional SDS-PAGE was performed as described by Laemmli (1970), by using 1-mm-thick mini gels containing 12% polyacrylamide resolving gels (w/v) and 5% (w/v) stacking gels. SDS-PAGE analysis of samples taken during fermentation was done according to the procedure described by Chen and Christen (1997). Protein bands were visualized with Coomassie brilliant blue. Protein standards (SDS-7) were from Sigma. Densitometric analysis was performed on non-reducing, Coomassie blue–stained gels, using an one-dimensional image analysis program (Kodak).
Acknowledgments
We thank Annette Hauge Byrde and Rannvá Skála for performing the peptide binding experiments. This work was supported in part by the Danish MRC (grant 9601615) and THOR, the fifth Framework Program of the European Commission (grant QLRT-1999-00173), and the NIH (grant AI49213-02). Henrik Ferré is a recipient of a Ph.D. stipend from the Technical University of Denmark.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
β2m, β2-microglobulin
CV, column volumes
HIC, hydrophobic interaction chromatography
HLA, human leucocyte antigen
IB, inclusion bodies
MHC-I, major histocompability complex class I
SEC, size exclusion chromatography
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0233003.
References
- Altman, J.D., Moss, P.A., Goulder, P.J., Barouch, D.H., McHeyzer-Williams, M.G., Bell, J.I., McMichael, A.J., and Davis, M.M. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science 274 94–106. [DOI] [PubMed] [Google Scholar]
- Bernard, A. and Payton, M. 2002. Fermentation and growth of Escherichia coli for optimal protein production. In Current protocols of protein science (eds. J.E. Coligan et al.), unit 5.3. John Wiley & Sons, New York. [DOI] [PubMed]
- Buus, S. 1999. Description and prediction of peptide-MHC binding: The "human MHC project." Curr. Opin. Immunol. 11 209–213. [DOI] [PubMed] [Google Scholar]
- Buus, S., Stryhn, A., Winther, K., Kirkby, N., and Pedersen, L.Ø. 1995. Receptor-ligand interactions measured by an improved spun column chromatography technique: A high efficiency and high throughput size separation method. Biochim. Biophys. Acta. 1243 453–460. [DOI] [PubMed] [Google Scholar]
- Chen, M., and Christen, P. 1997. Removal of chromosomal DNA by Mg2+ in the lysis buffer: An improved lysis protocol for preparing Escherichia coli whole-cell lysates for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 246 263–264. [DOI] [PubMed] [Google Scholar]
- Cresswell, P. 2000. Intracellular surveillance: Controlling the assembly of MHC class I-peptide complexes. Traffic 1 301–305. [DOI] [PubMed] [Google Scholar]
- Dick, T.P., Bangia, N., Peaper, D.R., and Cresswell, P. 2002. Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity 16 87–98. [DOI] [PubMed] [Google Scholar]
- Falk, K., Rotzschke, O., Stevanovic, S., Jung, G., and Rammensee, H.G. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351 290–296. [DOI] [PubMed] [Google Scholar]
- Garboczi, D.N., Hung, D.T., and Wiley, D.C. 1992. HLA-A2-peptide complexes: Refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. 89 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, R.T., Sette, A., Grey, H.M., Apella, E., Sakaguchi, K., Zhu, N-Z., Arnott, D., Sherman, N., Shabanowitz, J., Michel, H., et al. 1994. Definition of specific motifs for four major HLA-A alleles. J.Immunol. 152 3913–3924. [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Marston, F.A. 1986. The purification of eukaryotic polypeptides synthesized in Escherichia coli. Biochem J. 240 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, L.Ø., Nissen, M.H., Hansen, N.J., Nielsen, L.L., Lauemøller, S.L., Blicher, T., Nansen, A., Sylvester-Hvid, C., Thomsen, A.R., and Buus, S. 2001. Efficient assembly of recombinant major histocompatibility complex class I molecules with preformed disulfide bonds. Eur. J. Immunol. 31 2986–2996. [DOI] [PubMed] [Google Scholar]
- Rudolph, R. and Lilie, H. 1996. In vitro folding of inclusion body proteins. FASEB J. 10 49–56. [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schoemaker, J. M., Brasnett, A.H., and Marston, F.A. 1985. Examination of calf prochymosin accumulation in Escherichia coli: Disulphide linkages are a structural component of prochymosin-containing inclusion bodies. EMBO J. 4 775–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J.D., Solheim, J.C., Carreno, B.M., and Hansen, T.H. 1995. Characterization of class I MHC folding intermediates and their disparate interactions with peptide and β2-microglobulin. Mol. Immunol. 32 531–540. [DOI] [PubMed] [Google Scholar]
- Springer, T.A., Robb, R.J., Terhorst, C., and Strominger, J.L. 1977. Subunit and disulfide structure of monomeric and dimeric forms of detergent-soluble HLA antigens. J. Biol. Chem. 252 4694–4700. [PubMed] [Google Scholar]
- Stryhn, A., Pedersen, L.Ø., Romme, T., Olsen, A.C., Nissen, M.H., Thorpe, C.J., and Buus, S. 1996. pH dependence of MHC class I–restricted peptide presentation. J. Immunol. 156 4191–4197. [PubMed] [Google Scholar]
- Sylvester-Hvid, C., Kristensen, N.S., Blicher, T., Ferré, H., Lauemøller, S.L., Wolf, X.A., Lamberth, K., Nissen, M.H., Pedersen, L.Ø., and Buus, S. 2002. Measuring peptide–MHC class I affinity by a quantitative ELISA. Tissue Antigens 59 251–258. [DOI] [PubMed] [Google Scholar]
- Tector, M., Zhang, Q., and Salter, R.D. 1997. β2-Microglobulin and calnexin can independently promote folding and disulfide bond formation in class I histocompatibility proteins. Mol. Immunol. 34 401–408. [DOI] [PubMed] [Google Scholar]
- Teilum, K., Ostergaard, L., and Welinder, K.G. 1999. Disulfide bond formation and folding of plant peroxidases expressed as inclusion body protein in Escherichia coli thioredoxin reductase negative strains. Protein Expr. Purif. 15 77–82. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., Ellis, R.J., and Dobson, C.M. 1999. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield, P.T. 2002. Use of protein folding reagents. In Current protocols of protein science (eds. J.E. Coligan et al.), appendix 3A. John Wiley & Sons, New York. [DOI] [PMC free article] [PubMed]
- Zhang, Q-J., Gavioli, R., Klain, G., and Masucci, M.G. 1993: An HLA-A11-specific motif in nonamer peptides derived from viral and cellular proteins. Proc. Natl. Acad. Sci. 90 2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]