

Abstract
Polyproline II (PPII) is reported to be a dominant conformation in the unfolded state of peptides, even when no prolines are present in the sequence. Here we use isothermal titration calorimetry (ITC) to investigate the PPII bias in the unfolded state by studying the binding of the SH3 domain of SEM-5 to variants of its putative PPII peptide ligand, Sos. The experimental system is unique in that it provides direct access to the conformational entropy change of the substituted amino acids. Results indicate that the denatured ensemble can be characterized by at least two thermodynamically distinct states, the PPII conformation and an unfolded state conforming to the previously held idea of the denatured state as a random collection of conformations determined largely by hard-sphere collision. The probability of the PPII conformation in the denatured states for Ala and Gly were found to be significant, ∼30% and ∼10%, respectively, resulting in a dramatic reduction in the conformational entropy of folding.
Keywords: Sem-5, SH3, conformational entropy, Ala and Gly mutants, protein folding, thermodynamics, polyproline II helix, ITC
Polyproline II (PPII) has been implicated as an important conformation in the ensemble of disordered states of proteins and peptides (Tiffany and Krimm 1968,Tiffany and Krimm 1972; Krimm and Tiffany 1974; Drake et al. 1988; Dukor and Keiderling 1991; Woody 1992; Wilson et al. 1996; Park et al. 1997). Recent experimental studies indicate that the number of available conformations accessible to denatured proteins and peptides may be much lower than originally estimated, with the peptide backbone occupying polyproline II conformations a significant fraction of the time (Rucker and Creamer 2002; Shi et al. 2002). This result could have a profound impact on research efforts focused on protein fold prediction, macromolecular interactions, and most notably protein stability.
Up to now, it has been largely held that restrictions in the available φ and ξ angles for different amino acids are determined primarily by side-chain and backbone hard-sphere collisions, herein referred to as the hard-sphere collision (HSC) model. One of the most compelling arguments in support of the HSC view of the denatured state has been the ability of this model to quantitatively predict the ΔΔG of unfolding associated with changing a surface-exposed alanine (Ala) to a glycine (Gly). The observed decrease in stability (∼0.73 kcal/mole) of the Gly variant (Lee et al. 1994; D’Aquino et al. 1996) is consistent with a 3.4-fold increase in the number of available conformations for the Gly variant in the denatured (i.e., unfolded) state, an approximation of which can be ascertained by straightforward inspection of the Ramachandran map of each amino acid (Ramachandran and Sasisekharan 1968). It should be noted however, that these experimental results provide access only to the difference in the available conformational space for each amino acid. As such, one cannot discriminate between the HSC model, where all of φ and ξ space is available, and models wherein the unfolded states of the Gly and the Ala variants are both significantly constrained.
Here we test whether the unfolded ensembles of peptides are biased toward PPII conformations by studying the binding of the C-terminal SH3 domain (C-SH3) from SEM-5 to a series of designed peptides. We show that because the bound state of the peptide is PPII, experimentally observed differences in binding affinity among proline (Pro), alanine (Ala), and glycine (Gly) substitutions at solvent-exposed sites in the peptide provide direct access to the degree to which the unfolded ensembles are biased toward PPII at the substituted position. In addition, the experimental setup provides quantitative estimates of the conformational free energy of Ala and Gly residues in the unfolded states.
Experimental model
Figure 1 ▶ shows the structure of the SEM-5 C-SH3:Sos peptide complex (PDB entry 1SEM) used in this study. The binding of the wild-type and single-amino-acid variants of the Sos peptide to a series of SEM-5 mutants was measured by isothermal titration calorimetry (ITC). Amino acid substitutions of the peptide were made at the surface-exposed residues Pro 3 and Pro 6 (see Table 1). These residues were targeted for substitution because the side chains are not involved in binding, and thus differences in Pro to Ala or Pro to Gly at these positions are caused by differences in the conformational ensemble of the peptides in the unbound state.
Figure 1.

The experimental model system investigated in this study: Sem-5 C-SH3 domain (ribbon) complexed with the polyproline peptide (Ac-VPPPVPPRRRY-amide, CPK model). The green highlighted residues are located in the solvent-exposed site of the peptide and do not interact in the binding interface. These residues were mutated to Ala and Gly residues for the purpose of this study.
Table 1.
Summary of the solvent-exposed Ala/Gly mutants generated for both the Sem-5 C-SH3 domain and the Sos peptide
| Nomenclature | Sem-5 C-SH3 domain mutations | Location |
| G171 | Cys 209 → Ala (C209A; pseudo-WT) | |
| S170A | C209A, Ser 170 → Ala | RT loop |
| S170G | C209A, Ser 170 → Gly | RT loop |
| Nomenclature | Sos peptide variants |
| SosY | Ac-V0P1P2P3V4P5P6R7R8R9Y10-amide (wild type) |
| SosY-P3A | Ac-VPPAVPPRRRY-amide (Pro 3 → Ala) |
| Sosy-P3G | Ac-VPPGVPPRRRY-amide (Pro 3 → Gly) |
| SosY-P6A | Ac-VPPPVPARRRY-amide (Pro 6 → Ala) |
| SosY-P6G | Ac-VPPPVPGRRRY-amide (Pro 6 → Gly) |
Results
Free energy of binding of SEM-5 C-SH3 to Pro, Ala, and Gly peptide variants
Binding constants from ITC measurements were obtained for all protein–peptide complexes shown in Table 1. Figure 2 ▶ shows a typical ITC binding isotherm. Uncertainties in the binding constants are ∼2% as determined from multiple experiments. Although ΔHbinding was obtained from the integral of the titration peaks (see Materials and Methods), Table 2 reveals that the differences in enthalpy among different peptides were found not to be statistically significant. Figure 3 ▶ shows the binding constant profile of the S170G C-SH3 domain to each peptide (Pro, Ala, and Gly mutations at the third and sixth positions). As indicated in Tables 3A, 3B, and 3C, the binding (association) constant profiles for all SH3 mutants are similar, showing the following rank order of binding for amino acid substitutions at the third and 6th positions: Pro > Ala > Gly.
Figure 2.

A typical binding isotherm acquired using Isothermal Titration Calorimetry (ITC). All experiments were performed in 20 mM NaPhos, 200 mM NaCl (pH 7.5) at 25°C.
Table 2.
Average and standard deviation for the enthalpy of binding between the SosY peptide variants and the different C-SH3 domain mutants from Table 1
| Sos peptide | ΔHbinding, SEM5/Sos (cal/mole) |
| WT | −7767 ± 151 |
| P3A | −8781 ± 176 |
| P3G | −8353 ± 523 |
| P6A | −8413 ± 117 |
| P6G | −8500 ± 174 |
Figure 3.

Binding constants of Sem-5 C-SH3 S170G domain with five different Sos peptides. Error bars are shown and obtained from the fitting error of ITC measurements.
Table 3A.
Summary of the differences in binding free energy measurements (ΔΔ Gbinding) between Pro/Ala SosY peptide mutants bound with different Sem-5 C-SH3 domain mutants
| Sem-5 C-SH3 | ΔΔGbinding, SosY-Pro3 → Ala (cal/mole) | ΔΔGbinding, SosY-Pro6 → Ala (cal/mole) |
| S170A | −552 | −650 |
| S170G | −550 | −613 |
| G171 | −545 | −653 |
| Average | −549 ± 27 | −639 ± 27 |
Table 3B.
Summary of the differences in binding free energy measurements (ΔΔ Gbinding) between Pro/Gly SosY peptide mutants bound with different Sem-5 C-SH3 domain mutants
| Sem-5 C-SH3 | ΔΔGbinding, SosY-Pro3 → Gly (cal/mole) | ΔΔGbinding, SosY-Pro6 → Gly (cal/mole) |
| S170A | −1115 | −1181 |
| S170G | −1189 | −1145 |
| G171 | −1201 | −1179 |
| Average | −1168 ± 46 | −1168 ± 40 |
Table 3C.
Summary of the differences in binding free energy measurements (ΔΔ Gbinding) between Ala/Gly SosY peptide mutants bound with different Sem-5 C-SH3 domain mutants
| Sem-5 C-SH3 | ΔΔGbinding, SosY-P3Ala → Gly (cal/mole) | ΔΔGbinding, SosY-P6Ala → Gly (cal/mole) |
| S170A | −565 | −533 |
| S170G | −642 | −534 |
| G171 | −658 | −528 |
| Average | −622 ± 48 | −532 ± 43 |
As indicated in Table 3A, the average and standard deviation of ΔΔGbinding from Pro to Ala at the third position for all three C-SH3 mutants is −549 ± 27 cal/mole. For the sixth position, the difference is −639 ± 27 cal/mole. Table 3B lists the ΔΔGbinding of the Pro-to-Gly mutation in the peptide at both the third and the sixth positions. The average and standard deviation of ΔΔGbinding from Pro to Gly at the third position for all three C-SH3 mutants is −1168 ± 46 cal/mole. For the sixth position, the difference is −1168 ± 40 cal/mole. Table 3C lists the ΔΔGbinding of the Ala-to-Gly mutation in the peptide at both the third and the sixth positions. The average and standard deviation of ΔΔGbinding from Ala to Gly at the third position for all three C-SH3 mutants is −622 ± 48 cal/mole. For the sixth position, the difference is −532 ± 43 cal/mole. The fact that duplicate experiments performed on multiple protein variants at two different peptide positions produced similar results indicates that the same effects are being monitored for each type of substitution (i.e., Pro to Ala or Pro to Gly). The significance of the observed differences is discussed below.
Chemical shift perturbation
To attribute the binding free energy changes to conformational free energy differences between the different peptides, it is important to show that the mutations do not affect the binding interface. The mutations made in the Sos peptide are located in solvent-exposed sites (see Fig. 1 ▶), and have no surface area interacting with the SH3 domain, as determined from accessible surface-area calculations of the free and bound peptide (data not shown). In such cases, major structural effects are rarely seen (Blaber et al. 1994). To ensure no major structural rearrangements in the SEM-5:Sos complex upon changing the third and sixth positions in the peptide, 1H–15N HSQC spectra of the Sem-5 C-SH3 domain were obtained with the different peptide variants (see Materials and Methods).
Pro to Ala at third position
Figure 4A ▶ shows the 1H–15N HSQC overlay spectra of the Sem-5:Sos(Ala3) complex and the Sem-5:Sos(Pro3) complex. Residues in SEM-5 involved in the binding interface and that have accessible surface area (ASA) buried upon binding are the following: N190, W191, P204, N206, Y207, F163, D164, F165, N166, Q168, E169, and E172. The almost perfect overlay between the two spectra is clear. The only slight, but significant, chemical shift differences are for the backbone and side-chain amides of N206. As evident from Figure 5 ▶, N206 is proximal to Pro 3 of the Sos peptide in the complex. Indeed, N206 H2Nδ forms an H-bond with the O of the backbone Pro 3 of the peptide (Lim et al. 1994). The observation that chemical shift changes were isolated to a single residue that is in direct contact with the substituted position in the peptide indicates that substitution from Pro to Ala at this position does not significantly affect the structure of the complex.
Figure 4.
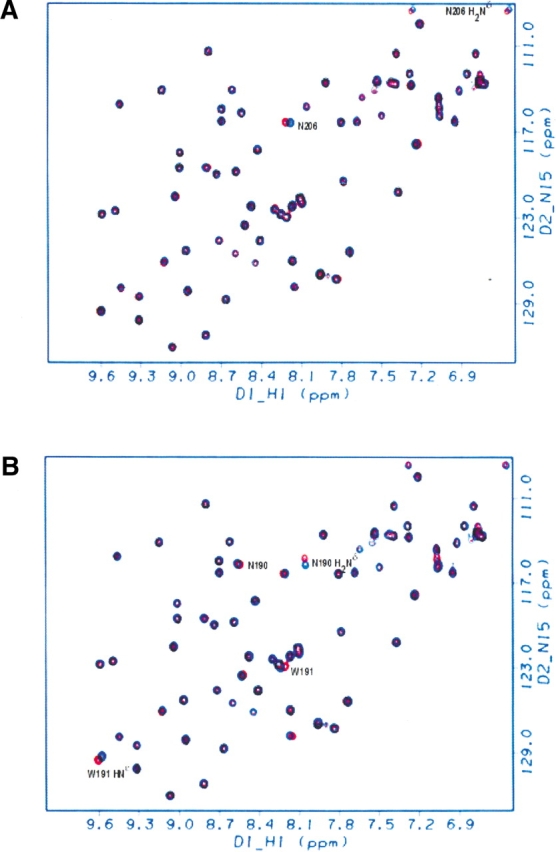

Structural changes associated with the mutations for the Pro-to Ala-residues: HSQC spectra of the complexed Sem-5 C-SH3 domain with different Sos peptides. (A) Overlay of complex with the Ala peptide mutant at the third position (Ala 3, in blue) onto the complex with the wild-type peptide (Pro 3, in red). (B) Overlay of complex with the Ala peptide mutant at the sixth position (Ala 6, in blue) onto the complex with the wild-type peptide (Pro 6, in red). Almost all peaks have identical chemical shifts. Residues with significant differences are labeled. (C) Structural changes associated with the mutations for the Ala-to-Gly residues: HSQC spectra of the complexed Sem-5 C-SH3 domain with different Sos peptides. Overlay of complex with the Gly peptide mutant at the third position (Gly 3, in blue) onto the complex with the Ala peptide (Ala 3, in red). (D) Overlay of complex with the Gly peptide mutant at the sixth position (Gly 6, in blue) onto the complex with the Ala peptide (Ala 6, in red). Almost all peaks have identical chemical shifts. Residues with significant differences are labeled.
Figure 5.

Binding interface of the Sem-5 C-SH3 domain (ribbon) with the Sos peptide (gold thick line). Solvent-exposed Pro residues at the third and sixth positions that are mutated in this study to Ala and Gly residues are labeled. Residues highlighted in green (F163, Y207, N206, and W191) form intermolecular H-bonds with certain Os (red) in the backbone of the Sos peptide in the crystal structure. Some residues with noticeable chemical shift changes are in the RT loop (Q168 and E169, in blue).
Pro to Ala at sixth position
Figure 4B ▶ shows the 1H–15N HSQC overlay spectra of the Sem-5:Sos(ALA6) complex and the Sem-5:Sos(PRO6) complex. Similar to Figure 4A ▶, the Pro-to-Ala change in the sixth position results in chemical shift differences in the immediate vicinity of the substituted residue. Specifically, differences are seen in the backbone and side-chain amide of N190 as well as the backbone and side-chain amide of the neighboring W191 (Fig. 5 ▶). Indeed, as with N206 in the previous comparison (Fig. 4A ▶), W191 HNɛ forms an H-bond with the O of the backbone Pro 6.
Ala to Gly at third and sixth positions
Figure 4 ▶, C and D, shows the 1H–15N HSQC overlay spectra of the Sem-5:Sos(GLY3) complex and the Sem-5:Sos(ALA3) complex and the Sem-5:Sos(GLY6) complex and the Sem-5:Sos(ALA6) complex, respectively. Interestingly, Ala-to-Gly mutations at positions 3 and 6 caused more differences in the chemical shifts compared with Pro and Ala (Fig. 4A,B ▶). Also, many of the differences observed for the Ala-to-Gly change at both sites are found in residues that are distal to the substituted residues (Fig. 5 ▶; i.e., Q168, E169, F163, and F165).
Although the differences observed for Ala-to-Gly changes at each position are generally more extensive than those seen for Pro to Ala, two important features indicate that the differences are not indicative of significant structural changes. First, most binding-site residues do not show chemical shift differences. Second, HSQC spectra are extremely sensitive to small changes in the electronic environment (Jameson 1996). The average change observed here, which is <0.1 ppm, is significantly less than what is observed for binding-site residues as a result of peptide titration (>0.5 ppm in the 15N dimension; data not shown). The fact that the observed thermodynamic differences between Ala and Gly variants are similar at each position further supports the idea that the changes do not significantly affect the structure of the complex.
Direct experimental access to the conformational free energy of unstructured alanine and glycine
Inspection of Table 1 reveals that the substituted positions in the Sos peptide (positions 3 and 6) contain Pro residues, which themselves are immediately preceded in the sequence by other Pro residues. Such a system provides a unique opportunity to directly access the conformational free energy of any substituted amino acid. As discussed previously (MacArthur and Thornton 1991; Creamer 1998), a Pro residue followed by another Pro residue is restricted by hard-sphere collisions to PPII. Thus, in the wild-type peptide, which has Pro at the third and sixth positions, Pro 2 and Pro 5 will experience essentially no conformational entropy change upon binding to SEM-5, as they are already prefolded to PPII in the unbound state. Pro 3 and Pro 6, however, are not restricted to PPII and can occupy two conformations, PPII or α-helix, although it is not clear what the true distribution for these two conformations will be at either position. Such an uncertainty would ordinarily render interpretation of the Ala and Gly substitution data problematic. However, by mutating residues 3 and 6 to Ala (or Gly), the conformational entropy contributions for each residue in the peptide change according to the following scheme:
Conformational Entropy Change:
where SP, SA, and SX are the conformational entropy contributions for Pro, Ala, and any flanking residue X, respectively. The overall difference in conformational entropy (ΔStot) between each peptide is simply the sum of the site-specific differences:
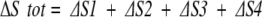 |
which in this case becomes SA (= [Sx − Sx] + [SP − 0] + [SA − SP] + [Sx − Sx]). The important feature is that, provided there are no position-specific differences for Pro (i.e., SP in position 2 is equivalent to SP in position 3), the precise value of SP need not be known, as it will cancel in the calculation. Consequently, the overall measured energy difference will, as shown below, provide access to the conformational entropy of the substituted residue.
As the HSQC spectra indicate that no significant structural differences result from the Pro-to-Ala change at either position in the Sos peptide, the observed ΔΔGbinding at each site (Table 3A) corresponds to the apparent conformational free energy of Ala in the denatured state. We note that the values obtained at position 3 (549 cal/mole = −1.8 e.u.) and position 6 (639 cal/mole = −2.2 e.u.) are strikingly different from the 1222 cal/mole (= −4.1 e.u.) that has previously been reported for the conformational entropy for Ala (D’Aquino et al. 1996; Fig. 6 ▶). Interestingly, the results for the Pro-to-Gly and Ala-to-Gly changes also significantly underestimate the previously published values. It will be shown that the underestimation in all three cases is indicative of a bias in the unliganded peptide ensemble toward the PPII conformation.
Figure 6.

Conformational entropy changes (ΔΔS) from (A,B) Pro to Ala, (C,D) Pro to Gly, and (E,F) Ala to Gly at the third and sixth positions, respectively. The values labeled HSC come from D’Aquino et al. (1996) and are similar in magnitude to the differences in available φ/ξ space obtained from simple hard-sphere collisions. The data shown represent the experimental values obtained for the difference in binding between each peptide pair and the different C-SH3 protein mutants (S170A, S170G, G171).
The effect of proline cis–trans isomerization on the binding energetics
For the Pro-to-Ala and the Pro-to-Gly substitutions, it is necessary to account for the effect of changes in the number of isomerizing Pro residues. As Table 1 indicates, Pro 3 and Pro 6 in the wild-type peptide are immediately preceded by Pro residues. These preceding Pro residues affect the cis–trans isomerization equilibrium as described previously (Reimer et al. 1998; Huyghues-Despointes et al. 1999). For the present case, the Pro residues at positions 3 and 6 will occupy the cis configuration ∼6% of the time in the unbound state. As a consequence, the Pro-containing peptides have an additional, albeit minimal (0.037 kcal/mole), energetic penalty for binding that the Ala- and Gly-substituted peptides do not. In the subsequent analysis, this energetic correction is considered explicitly.
The relationship between conformational free energy and conformational entropy in the SEM-5:Sos system
The three sets of binding results (i.e., monitoring the ΔΔG of binding for Pro-to-Ala, Pro-to-Gly, and Ala-to-Gly substituted peptides) provide access to a quantitative estimate of the probability that the substituted amino acid is occupying a PPII conformation. Figure 7 ▶ is a schematic representation of the binding of SEM-5 to the Sos peptide. The overall binding energy can be divided into three contributions:
Figure 7.

Schematic representation of the binding of SEM-5 C-SH3 to the polyproline helix. For simplicity the bound state is represented as a single conformation, and the unbound states are represented as an ensemble. In reality, the bound and free forms are both ensembles, but the conformational ensemble of bound form is significantly reduced. Changing Pro to Ala and Ala to Gly on surface-exposed noninteracting sites on the peptide should change only the conformational free energy of the peptide (ΔGconf,P). See Appendix for details.
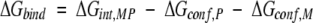 |
(1) |
where ΔGint,MP is the free energy of interaction at the binding interface, ΔGconf,P (= −RT ln[1 + Kconf,P]) is the conformational free energy change for the peptide, and ΔGconf,M (= −RT ln[1 + Kconf,M]) is the conformational free energy change for the protein. In essence, the latter two terms are the energy associated with redistributing the conformational ensemble of both the protein and the peptide to the binding-competent species.
For the case described here, wherein the protein is unchanged and the structure of the complex is the same when different substituted peptides are used (see Appendix), the difference in binding free energy between, for example, the Ala and the Gly variant will be
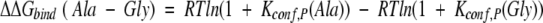 |
(2) |
where the Kconf,P(Ala) and Kconf,P(Gly) are the conformational equilibrium constants between the binding competent conformations (i.e., PPII) of the Ala- and Gly-substituted peptide, and the binding incompetent conformations (i.e., unstructured states). The important feature of this relationship is that if the conformational ensemble of the substituted residue is significantly biased toward PPII in the unbound state, that bias will be manifested in the magnitude of the observed ΔΔGbinding as determined by the linkage relationship in equation 2. In other words, the expression
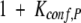 |
is the conformational partition function for the unbound peptide, wherein the statistical weights of the PPII conformation and the unfolded state are 1 and Kconf,P, respectively. In equation 2, if Kconf,P(Ala) ≫ 1 and
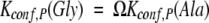 |
where Ω is the difference in degeneracy of the unfolded conformational ensembles between Ala and Gly (i.e., ΔΔSconf(Gly–Ala) = R ln Ω), the observed difference in binding affinity will correspond approximately to the difference in conformational free energy between the Ala- and Gly-containing peptides in the unstructured state (i.e., ΔΔGbinding ∼ −TΔΔSconf). If, on the other hand, Kconf,P ∼ 1, the observed difference in binding affinity will be less than the conformational free energy difference in the unstructured states. Because ΔΔGbinding(Pro–Ala), ΔΔGbinding(Pro–Gly), and ΔΔGbinding(Ala–Gly) are known from experiment (Tables 3A, 3B, and 3C), it is possible to work backwards and determine the magnitude of Kconf,P and thus the PPII bias (i.e., the ratio 1/[1 + Kconf,P]) with either Ala or Gly at the substituted positions.
For Ala-to-Gly substitutions, Kconf,P(Ala) can be obtained from equation 2 by recognizing that the denatured conformational ensemble of a Gly-substituted peptide is according to previous reports (D’Aquino et al. 1996; Fig. 6 ▶) 3.4 times larger than the denatured ensemble of an Ala-substituted peptide. As such, Kconf,P(Gly) = 3.4Kconf,P(Ala). Using this substitution, the calculated equilibrium between binding-competent and binding-incompetent species with an Ala in the substituted position—Kconf,P(Ala)—is ∼2.2, meaning that of the total unbound peptide molecules at any instant, ∼30% (i.e., 1/[1 + 2.2]) of the ensemble has the Ala residue prefolded in the PPII conformation. The conformation of the Ala residue in the remaining 70% is unfolded or randomly distributed.
Of course, the fraction of PPII calculated above is dependent on the validity of the assumption that once the bias for PPII in the unfolded state is considered explicitly (as in equation 2), there is no other major conformational preference, and the differences in the conformational ensembles of Ala and Gly are random and determined primarily from hard-sphere collisions. Although it is not possible to assess the validity of this assumption directly, it is useful to apply the identical assumption to the other substitutions and determine the agreement between prediction and experiment. This is done for the Pro-to-Ala and Pro-to-Gly substitutions.
As shown in Scheme I ▶, the ΔΔGbinding associated with changing the Pro to any other amino acid within the context of the present experimental system should correspond to the contribution of the substituted residue. The values for ΔS for Ala and Gly in Figure 6 ▶ (4.1 e.u. and 6.5 e.u., respectively) correspond to a 7.87- and 26.7-fold increase in size of the conformational ensembles over the Pro-containing peptide. Substituting the equalities Kconf,P(Pro) = Kconf,P(Ala)/7.87 and Kconf,P(Pro) = Kconf,P(Gly)/26.7 into the respective equations provides additional estimates of the PPII bias (see Tables 4A and 4B).
Scheme 1.

Table 4A.
Percentage PPII ( PPPII)aat substituted position with Ala
| Mutation | Position 3 | Position 6 | Averagea |
| P → Ab | 28% | 22% | 25% |
| A → Gc | 22% | 39% | 31% |
| P → Gd | 27% | 26% | 27% |
aPPPII = 1/[1 + Kconf,P(Ala)].
bKconf,P(Ala) = (1 − xz)/(xz/Ω − 1), where x is the ratio Kbind(Pro)/ Kbind(Ala), Ω is the ratio Kconf,P(Ala)/Kconf,P(Pro) = 7.67 (see text), and z is the contribution of proline cis/trans isomerization (1 + 0.064, see text).
cKconf,P(Ala) = (1 − x)/(x − Ω), where x is the ratio Kbind(Ala)/Kbind(Gly) and Ω is the ratio Kconf,P(Ala)/KKconf,P(Gly) = 3.4 (see text).
dKconf,P(Ala) = Kconf,P(Gly)e/Ω, where Ω is the ratio Kconf,P(Ala)/ Kconf,P(Gly) = 3.4 (see text).
e Determined as described in Table 4B.
Table 4B.
Percentage PPII ( PPPII)aat substituted position with Gly
| Mutation | Position 3 | Position 6 | Averagea |
| P → Gb | 9.6% | 9.5% | 9.6% |
| A → Gc | 8% | 16% | 12% |
| P → Ad | 10.1% | 7.5% | 8.8% |
aPPPII = 1/[1 + Kconf,P(Gly)].
bKconf,P(Gly) = (1 − xz)/(xz/Ω − 1), where x is the ratio Kbind(Pro)/Kbind(Gly), Ω is the ratio Kconf,P(Gly)/Kconf,P(Pro) = 26.72 (see text), and z is the contribution of proline cis/trans isomerization (1 + 0.064; see text).
cKconf,P(Gly) = (1 − x)/(x/Ω − 1), where x is the ratio Kbind(Ala)/ Kbind(Gly) and Ω is the ratio Kconf,P(Ala)/Kconf,P(Gly) = 3.4 (see text).
dKconf,P(Gly) = Ω Kconf,P(Ala)e, where Ω is the ratio Kconf,P(Ala)/ Kconf,P(Gly) = 3.4 (see text).
e Determined as described in Table 4A.
The agreement between the PPII conformational bias for Ala and Gly, calculated from Pro-, Ala-, and Gly-substituted peptides, is remarkable and provides unprecedented insight into the thermodynamic nature of the denatured state ensemble. The results indicate that the denatured ensemble for non-proline amino acids is significantly biased toward PPII (∼30% for Ala and ∼10% for Gly), and that the non-PPII conformations appear to conform to the previously held idea of the denatured state as a random collection of states, which are determined largely by hard-sphere collisions.
Discussion
The structure and energetics of the denatured state have long been enigmatic, primarily because of the difficulty associated with spectroscopically resolving the different denatured conformations. Recent NMR and theoretical studies, however, point to the importance of the PPII conformation, even for non-proline residues. The implication is that an understanding of the denatured state is predicated on determining not only residue-specific PPII biases, but also the energetic attributes of the non-PPII conformations. Here we address both issues by studying a peptide that binds in a PPII conformation. Such a situation allows us to explicitly account for the energetic contribution of PPII through the linkage relationships. Because the bound conformation is PPII, which is also one of the thermodynamically distinct states in the unbound ensemble, the thermodynamic dissection of the binding reaction is straightforward. In contrast, studies based on folding from the denatured state to other types of regular structure, such as a helix or sheet, will be complicated by the heterogeneity in the denatured state. In this respect, the binding of SH3 domains to PPII helices represents an ideal venue for a residue-specific investigation of PPII structure and energetics in the denatured state.
It is important to point out that the experimental setup is such that the results provide access to the position-specific PPII bias, not the PPII bias of the peptide as a whole. Because of the high proline content, it is indeed the case that certain positions of the peptide will occupy PPII a significant fraction of the time, especially at proline positions that are followed by other prolines. The influence of prolines on the PPII preference of other residues, however, is very locally driven (Creamer 1998), and does not bias the interpretation of the results presented here. Our analysis considers explicitly the effect of neighboring prolines on the observed results. In fact, it is the unique conformational properties of proline that facilitate direct access to the conformational free energy of the substituted residue, as described in Scheme I ▶.
It should also be noted that the PPII estimates for Ala reported here differ considerably from those determined for a polyalanine peptide using NMR (Shi et al. 2002). In that study it was estimated that PPII is occupied >80% of the time for Ala at 0°C, with very little temperature dependence. Although we have no unambiguous explanation for this effect, it is plausible that the difference arises from the different techniques used in each study. In the present analysis, the definition of PPII is restricted to those conformations that are binding-competent, regardless of φ and ξ angle. Although the unbound peptide may experience fluctuations around the canonical φ/ξ values such that a structural probe like NMR may register it as PPII, it is likely that the more extreme fluctuations will be excluded from binding. As such, only a subset of the structurally defined PPII conformations will likely correspond to the thermodynamically (or functionally) defined PPII. In this respect, the thermodynamically defined PPII may represent a lower limit from a structural perspective.
The preference for PPII in denatured peptides has potentially broad reaching implications, particularly with respect to modeling the denatured state or understanding the determinants of protein stability. For instance, the conformational entropy of the denatured state of a 100-amino-acid protein with no PPII bias would be ∼200 cal/(mole × K) greater than for a denatured state with a PPII bias of ∼30% at every position. This corresponds to a denatured state that is >1040 times larger than the PPII-biased denatured state, and indicates that the search space accessible to unfolded peptides and proteins is significantly smaller than previously estimated. More importantly, the search space is restricted to a unique region, a result that may have a significant impact on ab initio fold prediction approaches.
Finally, previous studies (Sreerama and Woody 1999; Kelly et al. 2001; Pappu and Rose 2002; Rucker and Creamer 2002) point toward preferential backbone solvation in the PPII conformation as a major component determining the conformational bias. Although it is not possible with the present experimental system to ascertain the molecular origins of the observed bias, careful determination of the temperature dependence of the PPII bias may prove useful in determining the enthalpic and entropic contributions to the observed free energy differences. Such a dissection should prove invaluable in the construction of a more realistic and experimentally verifiable model for the denatured state ensemble.
Materials and methods
Preparation of Sem-5 SH3 domain mutants
The cloning, overexpression, and purification of the C terminus of the Sem-5 SH3 domain was as described previously (Lim et al. 1994). The Sem-5 C-SH3 used throughout the study was the pseudo-wild type wherein Cys 209 was mutated to alanine to prevent possible oxidation and intermolecular cross-linking. Other mutants were also generated by the same method. Table 1 lists the mutants used in this study.
Preparation of SosY peptides
The original Sos peptide has the sequence (Ac-PPPVPPRRR-NH2). As this sequence presents the problem of accurate peptide concentration determination, we adopted the peptide sequence (Ac-VPPPVPPRRRY-NH2) used by Wittekind et al. (1994, 1997), with an additional valine residue at the N terminus and tyrosine residue at the C terminus, which we term throughout the paper as the SosY peptide. The peptides were synthesized and purified by the Peptide Synthesis Laboratory (UTMB). The purity of the peptide (>95%) was checked by mass spectrometry (LSU) and reverse phase high performance liquid chromatography.
Isothermal Titration Calorimetry (ITC)
All titration experiments were performed using the Microcal ITC system at 25°C as described elsewhere (Wiseman et al. 1989; Gómez and Freire 1995). The protein was extensively dialyzed against each buffer condition. Peptides were lyophilized from water and dissolved with the buffer from the final dialysis. Proteins and peptides were then centrifuged to remove particulates, degassed, and pH-adjusted to their final desired pH. Protein and peptide concentrations were measured using UV absorbance spectroscopy and the Edelhoch method (Edelhoch 1967; Gill and von Hippel 1989) with the protein and peptide in unfolded states (6 M Gdm-Cl) and using the extinction coefficients determined in Edelhoch (1967). For the Sem-5 C-SH3 domain, we used 13,940 M−1 cm−1, whereas for the SosY peptide, we used 1280 M−1 cm−1. Protein concentrations ranged from 0.3 to 1.2 mM, with the final c values ranging from 1 to 17. Peptide concentrations were usually from 8 to 10 times that of the protein concentration. The 1.3-mL sample cell was washed first with the dialyzed buffer before the protein was injected, making sure bubbles were not introduced, and then the corresponding peptide was loaded into the injector, with 10-μL injections made for a total of 30–31 injections and a 6-min equilibration time between injections. To avoid anomalies associated with the initial titration point, an initial injection of 5 μL was used. The heat of dilution for the peptide was measured using a control experiment of peptide titrated into buffer (without protein). The heats of dilution were small (∼70 cal/mole) compared with the heat of reaction. Data were then collected during the titration and fitted using a nonlinear least squares routine using a single site binding model in Origin for ITC v. 5.0 (Microcal), varying the stoichiometry (N), binding constant (Kb), and the change in the binding enthalpy (ΔH°). The Kb determined from the best-fit curve was then used to determine the free energy of the interaction (ΔG°) and then the entropy (ΔS°) involved in binding by using the following thermodynamic relation:
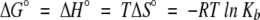 |
(3) |
where R is the gas constant, T is the absolute temperature, ΔG°, ΔH°, and ΔS° are the standard free energy, enthalpy, and entropy for the binding, respectively.
NMR spectroscopy
All 1H–15N-HSQC NMR spectra were collected at 25°C on a Varian UnityPlus 750-MHz instrument using a triple resonance probe equipped with a pulsed field z gradient. The HN and 15N chemical shifts for the unliganded form (pH 4.8) were assigned using various 3D and 2D heteronuclear NMR experiments (data not shown). Assignments at a higher pH (pH 7.5) were obtained by monitoring chemical shift changes as a function of pH titration (data not shown). For the complexed SH3 domain, unlabeled SosY peptide (Ac-PPPVPPRRRY-amide) was titrated to the 15N-labeled SH3 domain and chemical shift changes were followed. The final peptide concentration was almost 7 times that of the protein concentration, to completely ensure saturation. All spectra were processed using the FELIX v. 98.0 software (MSI, Inc.) on a Silicon Graphics Indy workstation. The spectra were apodigized with 90° shifted sinebell window function and zero-filled to a 256 × 1024 matrix.
Acknowledgments
We thank George Rose, Rohit Pappu, and Wayne Bolen for helpful discussions, and Trevor Creamer for critical evaluation of this manuscript. This work was funded by the NIH R01-GM13747, by NSF MCB-9875689, and by the Welch Foundation H-1461.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
SH3, src-homology domain 3
C-SH3, C-terminal SH3 domain
NMR, nuclear magnetic resonance
2D, two-dimensional
HSQC, heteronuclear single quantum coherence
ITC, Isothermal Titration Calorimetry
PPII, polyproline II helix
Ala, Alanine
Pro, Proline
Gly, Glycine
PDB, Protein Data Bank
Appendix
For the coupled equilibrium shown in Figure 7 ▶, the intrinsic binding equilibrium and the conformational equilibrium between binding-competent and binding-incompetent conformations of the protein and peptide can be written as
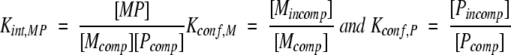 |
(A1) |
The overall linked equilibrium can thus be written as
 |
(A2) |
Combining expressions one obtains
 |
(A3) |
which when written in terms of free energy gives
 |
(A4) |
The ΔΔGbinding between any two substituted peptides (i.e. Ala & Gly) is given by:
 |
(A5) |
where
 |
(A6) |
 |
In the case that the structures of the complex are the same and the protein is not changed, the ΔΔGbinding (Ala-Gly) becomes
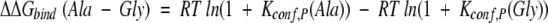 |
(A7) |
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0237803.
References
- Blaber, M., Zhang, X.J., Lindstrom, J.L., Pepiot, S.D., Baase, W.A., and Matthews, B.W. 1994. Determination of α-helix propensity within the context of a folded protein, sites 44 and 131 in bacteriophage T4 lysozyme. J. Mol. Biol. 235 600–624. [DOI] [PubMed] [Google Scholar]
- Creamer, T.P. 1998. Left-handed polyproline II helix formation is (very) locally driven. Proteins 33 218–226. [PubMed] [Google Scholar]
- D’Aquino, J.A., Gomez, J., Hilser, V.J., Lee, K.H., Amzel, L.M., and Freire, E. 1996. The magnitude of the backbone conformational entropy change in protein folding. Proteins 25 143–156. [DOI] [PubMed] [Google Scholar]
- Drake, A.F., Siligardi, G., and Gibbons, W.A. 1988. Reassessment of the electronic circular dichroism criteria for random coil conformations of poly(L-lysine) and the implications for protein folding and denaturation studies. Biophys. Chem. 31 143–146. [DOI] [PubMed] [Google Scholar]
- Dukor, R.K. and Keiderling, T.A. 1991. Reassessment of the random coil conformation: Vibrational CD study of proline oligopeptides and related polypeptides. Biopolymers 31 1747–1761. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gómez, J. and Freire, E. 1995. Thermodynamic mapping of the inhibitor site of the aspartic protease endothiapepsin. J. Mol. Biol. 252 337–350. [DOI] [PubMed] [Google Scholar]
- Huyghues-Despointes, B.M.P., Scholtz, J.M., and Pace, C.N. 1999. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat. Struct. Biol. 6 910–912. [DOI] [PubMed] [Google Scholar]
- Jameson, C.J. 1996. Understanding NMR chemical shifts. Annu. Rev. Phys. Chem. 47 135–169. [Google Scholar]
- Kelly, M., Chellgren, B.W., Rucker, A.L., Troutman, J.M., Fried, M.G., Miller, A.-F., and Creamer, T.P. 2001. Host–guest study of left-handed polyproline II helix formation. Biochemistry 40 14376–14383. [DOI] [PubMed] [Google Scholar]
- Krimm, S. and Tiffany, M.L. 1974. The circular dichroism spectrum and structure of unordered polypeptides and proteins. Israel J. Chem. 12 189–200. [Google Scholar]
- Lee, K.H., Xie, D., Freire, E., and Amzel, L.M. 1994. Estimation of changes in side chain configurational entropy in binding and folding: General methods and application to helix formation. Proteins Struct. Funct. Genet. 20 68–84. [DOI] [PubMed] [Google Scholar]
- Lim, W.A., Richards, F.M., and Fox, R.O. 1994. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372 375–379. [DOI] [PubMed] [Google Scholar]
- MacArthur, M.W. and Thornton, J.M. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218 397–412. [DOI] [PubMed] [Google Scholar]
- Pappu, R.V. and Rose, G.D. 2002. A simple model for polyproline II structure in unfolded states of alanine-based peptides. Protein Sci. 11 2437–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.H., Shalongo, W., and Stellwagen, E. 1997. The role of PII conformations in the calculation of peptide fractional helix content. Protein Sci. 6 1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, G.N. and Sasisekharan, V. 1968. Conformation in polypeptides and proteins. Adv. Protein Chem. 23 283–437. [DOI] [PubMed] [Google Scholar]
- Reimer, U., Scherer, G., Drewello, M., Kruber, S., Schutkowski, M., and Fischer, G. 1998. Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J. Mol. Biol. 279 449–460. [DOI] [PubMed] [Google Scholar]
- Rucker, A.L. and Creamer, T.P. 2002. Polyproline II helical structure in protein unfolded states: Lysine peptides revisited. Protein Sci. 11 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Z., Olson, C.A., Rose, G.D., Baldwin, R.L., and Kallenbach, N.R. 2002. Polyproline II structure in a sequence of seven alanine residues. Proc. Natl. Acad. Sci. 99 9190–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama, N. and Woody, R.W. 1999. Molecular dynamics simulations of polypeptide conformations in water: A comparison of a, b, and poly(Pro)II conformations. Proteins 36 400–406. [PubMed] [Google Scholar]
- Tiffany, M.L. and Krimm, S. 1968. New chain conformations of poly(glutamic acid) and polylysine. Biopolymers 6 1379–1382. [DOI] [PubMed] [Google Scholar]
- ———. 1972. Effect of temperature on the circular dichroism spectra of polypeptides in the extended states. Biopolymers 11 2309–2316. [DOI] [PubMed] [Google Scholar]
- Wilson, G., Hecht, L., and Barron, L.D. 1996. Residual structure in unfolded proteins revealed by Raman optical activity. Biochemistry 35 12518– 12525. [DOI] [PubMed] [Google Scholar]
- Wiseman, T., Williston, S., Brandts, J.F., and Lin, L. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179 131–137. [DOI] [PubMed] [Google Scholar]
- Wittekind, M., Mapelli, C., Farmer II, B.T., Suen, K., Goldfarb, V., Tsao, J., Lavoie, T., Barbacid, M., Meyers, C.A., and Mueller, L. 1994. Orientation of peptide fragments from Sos proteins bound to the N-terminal SH3 domain of Grb2 determined by NMR spectroscopy. Biochemistry 33 13531–13539. [DOI] [PubMed] [Google Scholar]
- Wittekind, M., Mapelli, C., Lee, V., Goldfarb, V., Friedrichs, M.S., Meyers, C.A., and Mueller, L. 1997. Solution structure of the Grb2 N-terminal SH3 domain complexed with a ten-residue peptide derived from SOS: Direct refinement against NOEs, J-couplings and 1H and 13C chemical shifts. J. Mol. Biol. 267 933–952. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1992. Circular dichroism and conformation of unordered polypeptides. Adv. Biophys. Chem. 2 37–79. [Google Scholar]