

Abstract
We previously reported that under certain experimental conditions, many variants of the B1 domain of IgG-binding protein G from Streptococcus form fibrils reproducibly. The variant I6T53 was the focus of the present study because the lag phase in the kinetics of fibril formation by this variant is significantly longer than that of other variants. This lag phase is distinguished by changes in both intrinsic fluorescence intensity and in light scattering of the protein. NMR diffusion measurements suggest that the soluble protein during the lag phase is monomeric. The kinetic profiles of fibril formation are found to depend on experimental conditions. The first kinetic phase diminishes almost completely when the reaction is seeded with preformed amyloid fibrils.
Keywords: Protein G B1 domain, amyloid, amyloid kinetics, sodium sulfate, fluorescence, NMR, light scattering
The amyloidoses are a group of diseases characterized by the deposition of protein in an insoluble form. It has been proposed that these are diseases of protein misfolding and that the protein deposition causes cellular toxicity. To date, more than 20 proteins have been found to form amyloid deposits associated with disease, and many other proteins can be induced to form amyloid fibrils in vitro (Kelly 1996,Kelly 1998; Serpell et al. 1997).
Proteins involved in amyloidogenic diseases have no obvious sequence similarity, nor do their native folds resemble each other. Despite the fact that the amyloidogenic proteins are biochemically and structurally diverse, there is considerable evidence that amyloid fibrils from different protein sources share a common global structure of long, nonbranched fibrils about 120 Å in diameter, with an underlying cross-β-sheet structure (Blake and Serpell 1996; Sunde et al. 1997; Jimenez et al. 1999). The molecular basis of the similarity of fibril structure has not been determined. One explanation may be that the similarity among the amyloidogenic proteins develops not at the level of their natively folded structure, but arises from the intermediate conformations whose existence is usually transient (Kelly and Lansbury 1994). This hypothesis is supported by an increasing number of reports in which mutations resulting in subtle conformational changes have been shown to be sufficient for amyloidogenic conversion of, for example, the human proteins λ or κ Ig light chain (Hurle et al. 1994), transthyretin (Lai et al. 1996), and lysozyme (Booth et al. 1997).
The development of in vitro model systems is of critical importance to our understanding of the physicochemical basis of amyloidosis. The formation of amyloid fibrils by a number of proteins unlinked to disease has been recently described, suggesting that the ability to form fibrils may be a common property of many if not all proteins under the appropriate conditions (Guijarro et al. 1998; Litvinovich et al. 1998; Chiti et al. 1999; Gross et al. 1999; Konno et al. 1999; West et al. 1999; Krebs et al. 2000; Ohnishi et al. 2000; Ramírez-Alvarado et al. 2000; Villegas et al. 2000; Yutani et al. 2000; Fandrich et al. 2001; Bucciantini et al. 2002). We previously reported that under certain experimental conditions, certain variants of the B1 domain of IgG-binding protein G from Streptococcus (β1) form fibrils reproducibly (Ramírez-Alvarado et al. 2000). β1 is a variant of the B1 domain of protein G with the second residue mutated (T2Q) to ensure homogeneity of N-terminal processing (Smith et al. 1994). By controlling the stability of the protein (either by introducing mutations or by changing experimental conditions), we are able to modulate the ability of the protein to form fibrils. For all variants, the key requirement for fibril formation is to choose conditions under which the population of intermediate states is maximized.
For a number of other proteins, it has been shown that a range of experimental conditions (such as changes in temperature, pH, presence of organic solvents, bivalent cations, or mutations) is effective in promoting the formation of amyloid fibrils. As in the case of the β1 variants, it appears that the effect of these agents or mutations is not to change the fold of the native state of the protein but rather to promote the formation, or enhance the stability, of amyloidogenic intermediates.
In transthyretin (TTR), for example, the point mutations that are associated with familial amyloid polyneuropathy (FAP) do not significantly change the tertiary or quaternary structure of native tetrameric TTR (Hamilton et al. 1993; Terry et al. 1993; Damas et al. 1996; Sebastiao et al. 1998); instead, they appear to function by destabilizing the TTR tetramer in favor of a monomeric amyloidogenic intermediate (Lai et al. 1996). It is of interest that sites of pathogenic changes are distributed throughout the molecule, with a particularly high number in the β-sheet comprising the most stable part of the core, supporting the idea that general destabilization of the folded states contributes to amyloidogenicity (Liu et al. 2000).
In another example, it was recently found that copper (II) plays a role destabilizing the native state of β2-microglobulin and in promoting fibril formation (Morgan et al. 2001). Acidic conditions also induce β2-microglobulin to form amyloid fibrils. Unfolding studies of β2-microglobulin suggest the presence of two partially structured species (Chiti et al. 2001), the more highly structured of which appears similar to the amyloidogenic form of β2-microglobulin (McParland et al. 2000). β2-microglobulin amyloid precursor is a noncooperatively stabilized ensemble that retains stable structure in five of the seven β-strands that comprise the native fold (McParland et al. 2002).
In the case of human lysozyme, specific point mutations are associated with autosomal dominant hereditary amyloidosis. The mutant proteins behave differently in solution compared to wild-type lysozyme, even though the crystal structures are all quite similar. One effect of the amyloidogenic mutations is to make the unfolding transition noncooperative (10). In addition, both the unfolding rates and hydrogen/deuterium exchange at equilibrium are much faster for the amyloidogenic mutants than for the wild type (Canet et al. 1999, 2002). A common feature of these mutant proteins appears to be an increase in the dynamics of the native state, and thus the conversion of intermediate into amyloid is facilitated by destabilizing the native state (Takano et al. 2001).
Similar observations have been made with mutations in human Ig light chains—it seems that again the propensity of different mutants to form fibrils correlates with a decrease in thermodynamic stability and an increase in the population of intermediates (Hurle et al. 1994; Wall et al. 1999).
The B1 domain of IgG-binding protein G is an extremely well characterized protein, both structurally and thermodynamically (Gronenborn et al. 1991; Alexander et al. 1992a,b; Gallagher et al. 1994; Smith et al. 1994; Smith and Regan 1995; Park et al. 1997, 1999; Merkel and Regan 1998; Merkel et al. 1999). In an earlier report, we investigated amyloid formation in a series of β1 mutants, differing only in the identity of two cross-parallel-strand amino acids (Ramírez-Alvarado et al. 2000).
Amyloid fibril formation by most of this series of β1 variants shows a short lag phase followed by a second single kinetic phase. However, for the I6T53 variant (Fig. 1 ▶), the lag phase in the kinetics of amyloid fibril formation is significantly longer.
Figure 1.
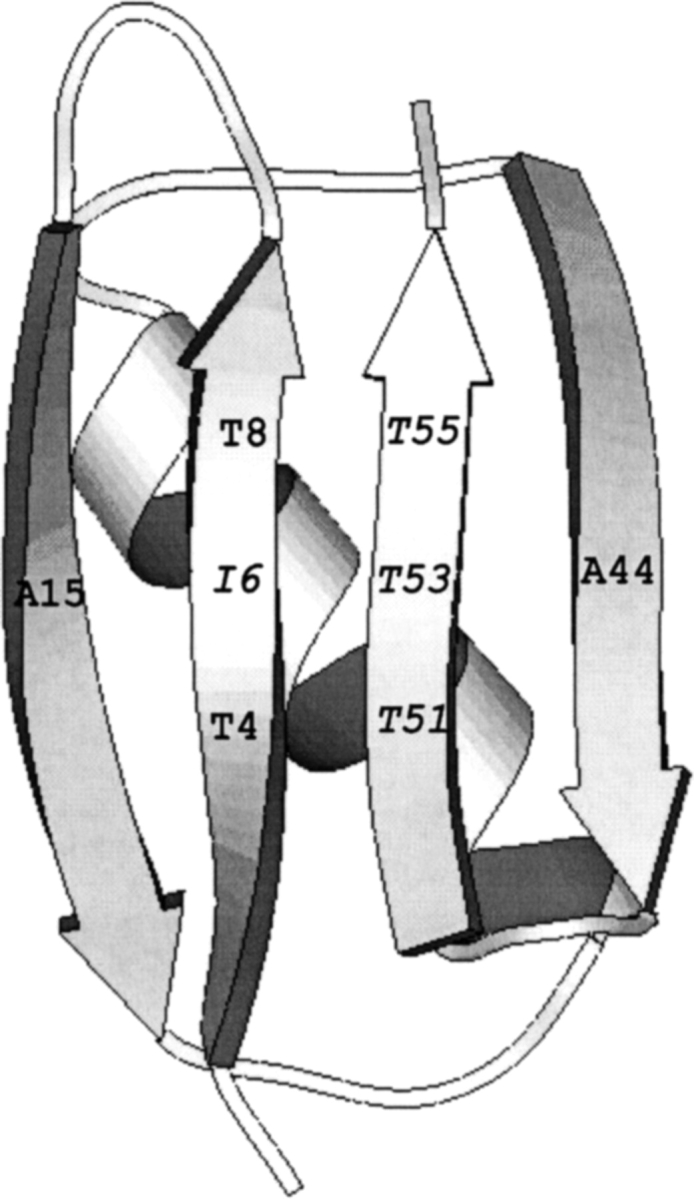
Schematic representation of I6T53 variant, showing the position numbers where the host-site mutations were introduced (35). Isoleucine (I) and threonine (T) are the wild-type residues in positions 6 and 53.
The aim of the work presented here was to further characterize the structural changes that occur during amyloid fibril formation by I6T53.
Results
The variant I6T53 was the focus of this study because of its longer lag phase in the kinetics of fibril formation (Fig. 1 ▶). I6T53 is the most stable variant of the family of mutants described previously with a melting temperature of 72°C and a ΔGunf = 4.7 kcal/mole (Ramirez-Alvarado et al. 2000). We analyzed the kinetics of amyloid fibril formation under different solvent conditions. All the experiments were performed at the midpoint unfolding transition:
100 μM I6T53 in 50 mM sodium acetate, pH 5.2, 72°C.
100 μM I6T53 in 10mM borate/citrate/acetate pH 9.0, 54°C.
100 μM I6T53, 1 M sodium sulfate, in 10mM borate/citrate/acetate pH 9.0 and 74°C.
100 μM I6T53 in 50 mM sodium acetate, pH 5.2, 72°C in the presence of 1/100 (v/v) of preformed fibrils.
Fluorescence and scattering of I6T53 at pH 5.2, 72°C
Figure 2A ▶ shows the kinetic profile of fibril formation by I6T53 at pH 5.2 and 72°C followed by the changes in the ratio of tryptophan fluorescence emission intensity at 310 and 350 nm (denoted as 310/350; Ramírez-Alvarado et al. 2000). In the initial phase, the intensity of the emission remains unchanged for about 2 h. After that initial phase, the emission intensity increases and then levels off to a constant final value around 3 h. These observations suggest that the tryptophan environment remains unchanged during the first phase of the kinetics. In the second phase, the fluorescence intensity increases, possibly due to the more hydrophobic environment that the amyloid fibril provides to the tryptophan.
Figure 2.


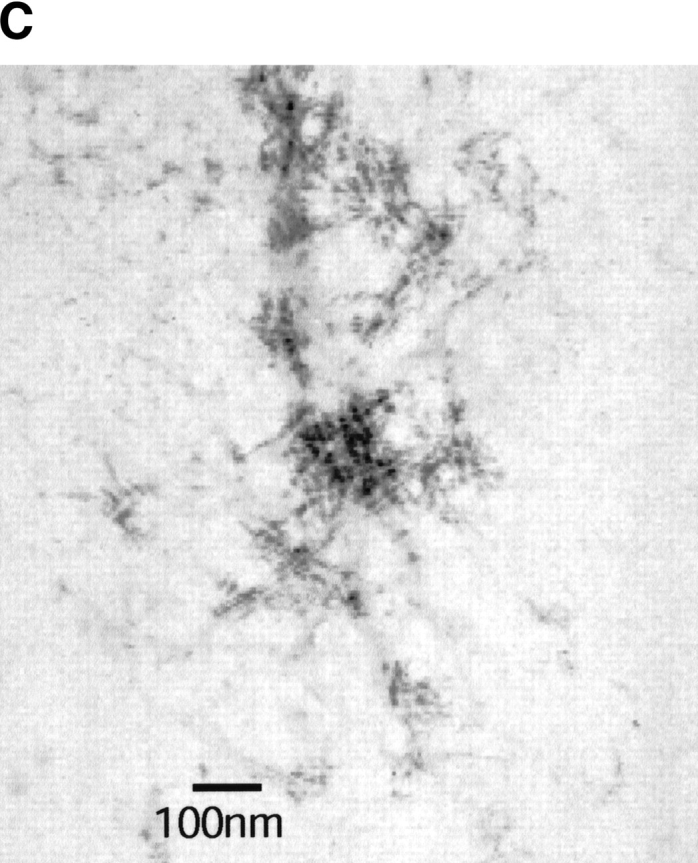
Kinetics of fibril formation of I6T53 at pH 5.2. (A) Normalized intrinsic tryptophan fluorescence intensity as a function of time (excitation 294 nm, ratio of emission intensity at 310 nm and 350 nm). (B) Normalized light scattering of amyloid formation as a function of time (excitation 340 nm, emission at 340 nm). Experimental conditions: 100μM I6T53, 50mM sodium acetate buffer, pH 5.2, 72°C. (C) Electron micrographs of a sample at the end of the kinetics experiment.
We note that although we typically monitor tryptophan fluorescence at two wavelengths, we have also monitored the kinetics of fibril formation by recording complete emission spectra (excitation at 294 nm, emission spectra from 300–400 nm). The same kinetic profile is observed. In order to rule out the possibility of an influence of scattering on the fluorescence at 310/350, we changed the excitation wavelength to 274 nm and recorded an emission scan from 300 to 400 nm with a soluble protein and a fibril sample. There was no observed fluorescence emission fluorescence in any of the samples, confirming that we were observing changes in the fluorescence as a consequence of changes in the environment of the tryptophan and not artifacts due to scattering.
Figure 2B ▶ shows the light scattering over the course of the same reaction. The kinetic profile closely parallels that of the fluorescence emission. During the first 2.0 h, the scattering is very similar to the monomeric initial population; after that, the scattering increases and reaches a plateau around 2.8 h. This indicates that the population present during the first phase of kinetics is close to monomeric size. The half-times for the fluorescence emission change and scattering change are both 2.6 h. The presence of amyloid fibrils was confirmed at the endpoint by electron microscopy (Fig. 2C ▶). We estimated that typically about 50% of the soluble protein has converted to amyloid fibrils at the end of the reaction (Ramírez-Alvarado et al. 2000).
NMR characterization of I6T53 at pH 5.2
NMR spectra were collected before, during, and after fibril formation under similar conditions to that of (1) above, at 100- and 200-μM protein concentrations. To visualize the protein throughout the thermal unfolding transition, we collected 1D spectra at increasing temperatures during the lag phase of fibril formation. Figure 3 ▶ shows the aromatic regions of 1D NMR spectra of 200 μM I6T53 at 25°C, 70°C, and 79°C in D2O. The spectrum of I6T53 at the midpoint of thermal denaturation is a weighted sum of folded and unfolded forms, demonstrating that the two states are in slow exchange even at this high temperature.
Figure 3.
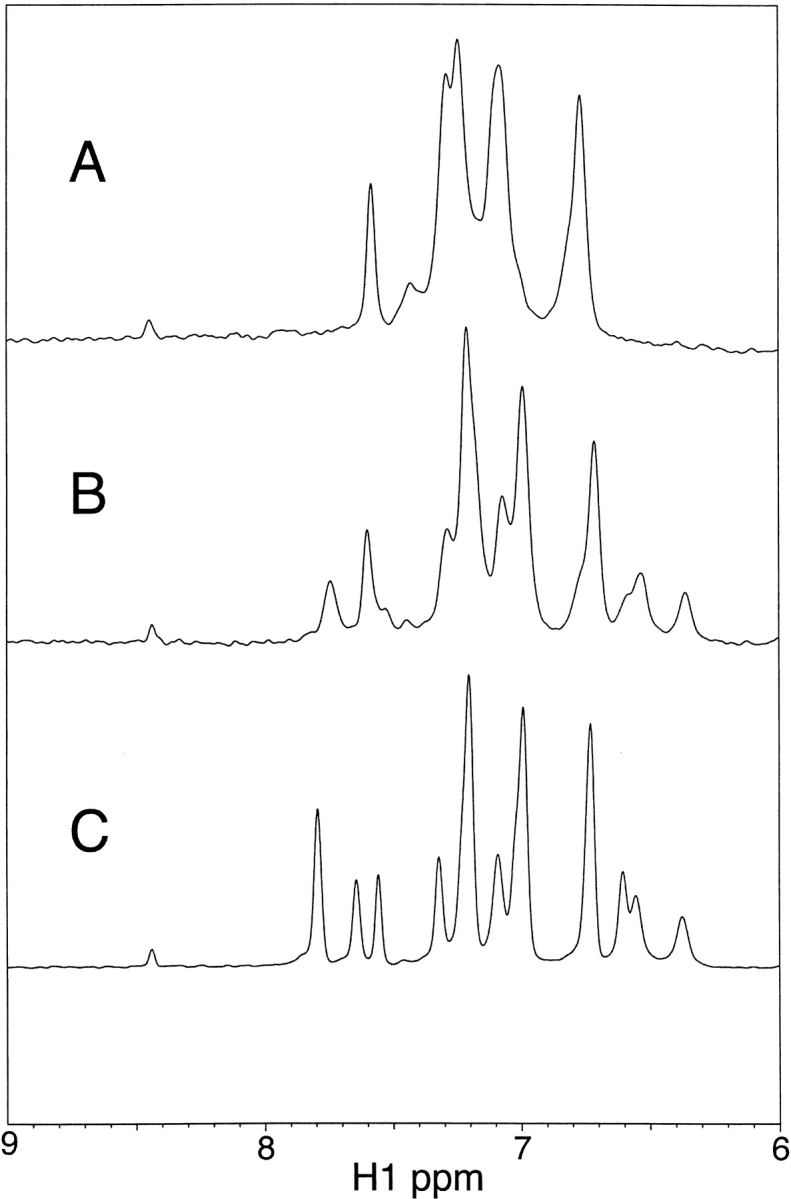
Proton 1D NMR spectra of 100 μM I6T53 in 50 mM sodium acetate, pD 5.2, D2O. (A) 79°C;. (B) 70°C;. (C) 25°C.
Figure 4 ▶ shows a comparison of I6T53 fluorescence samples (100 μM) before and after fibril formation at 72°C. The proton 1D NMR measurements in Figure 4 ▶ were both made at 25°C and reveal that following the fluorescence and light scattering measurements carried out over a period of 22 h at 72°C, a second soluble form of I6T53 appears. HSQC and NOESYHSQC spectra (data not shown) of this second conformation, which we call ‘heat-damaged’ are very similar to that of the native state. Although we have not yet identified the nature of the modification to this species, electrospray mass spectroscopy analysis of the soluble material shows two major species—one with the predicted molecular mass for this protein and one with a molecular mass less by 18 mass units. A number of lesser mass peaks are also visible.
Figure 4.
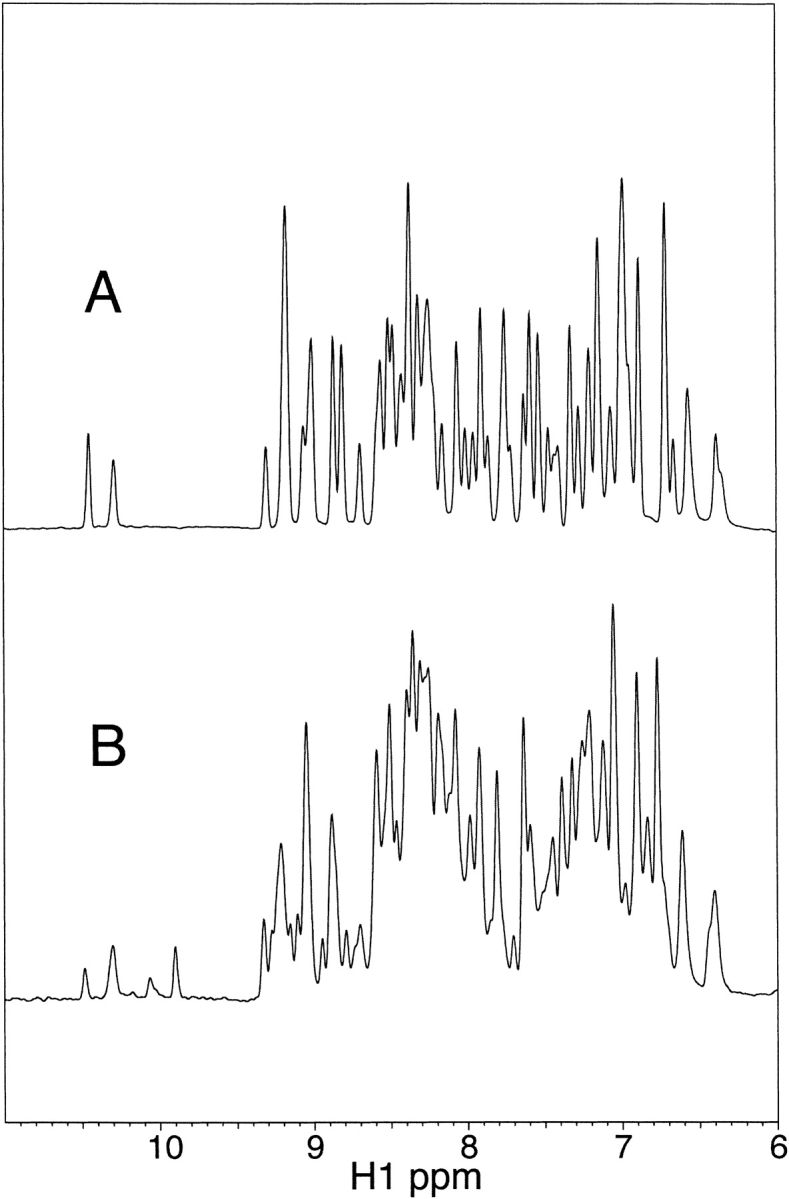
Proton 1D NMR spectra of 100 μM I6T53 in 50 mM sodium acetate, pH 5.2. (A) 25°C. (B) 25°C after heating at the Tm (72°C) 22 h (same sample as used for fluorescence measurements).
Fibril formation was also followed directly using NMR spectroscopy. We found that during the lag phase, 1D spectra collected at the midpoint of thermal unfolding showed only a decrease in signal intensity and a slight broadening at the onset of fibril formation (data not shown). These changes occurred after about 5 h of heating, and we attribute the 3-h increase in lag phase to a difference in the measurement sensitivities and a lack of stirring in the NMR sample tube. We were able to detect the presence of the second heat-damaged form of I6T53 shortly after fibrils appeared, at about 6 h of heating. We do not know at this time whether or not the heat-damaged form is involved in stimulating amyloid fibril formation or even if it is incorporated into fibrils.
NMR diffusion measurements can be used to determine the association state of a protein, and in the case of native I6T53 at 25°C we find that the measured diffusion coefficient, 1.76 cm2/s, correlates well with that calculated for the monomer (Fig. 5 ▶). Unfortunately, because of significant solution convection in the NMR tube at the high temperatures (Esturau et al. 2001), we were unable to make diffusion measurements at the temperatures where fibrils are formed. However, after heating, both the native and heat-damaged protein that remain detectable by NMR diffuse at 25°C with the same mobility as the protein before heating. It is unlikely that protein not bound up in fibrils would exist in an associated state at 72°C and then disassociate to monomers upon cooling to 25°C. This indicates that the soluble protein we can detect by NMR is monomeric; the remainder of I6T53 is bound up in visible fibrils, estimated to be about half the protein based on the decrease in the NMR signal intensity, consistent with previous measurements (Ramírez-Alvarado et al. 2000).
Figure 5.

NMR diffusion measurements. The line represents a linear fit of the diffusion data measured at 25°C for native I6T53 before heating. Diffusion data after heating for the native IT Trp NɛH(▵) and heat-damaged IT Trp‘ NɛH(○) are shown. The diffusion coefficient is the slope; γ, gyromagnetic ratio (hydrogen); δ, duration of gradient pulse; Gz, gradient amplitude; Δ, delay during which molecule diffuses; A, peak volume; Ao, initial peak volume.
Fluorescence and scattering of I6T53 at pH 9.0, 54°C
When the experiment was performed at 100 μM I6T53, 10 mM acetate/borate/citrate pH 9.0, 54°C (melting temperature of I6T53 under these conditions), no increase of scattering or change in fluorescence was observed over the course of 4.7 h. The sample had no visible signs of aggregation, and no amyloid fibrils were detected by electron microscopy. Likewise, no binding of the dye Thioflavine T was evident.
It is possible that these conditions inhibit fibril formation by preventing the creation of a nucleus; we tested this hypothesis with a postseeding experiment, by adding I6T53 amyloid seeds 1/100 (v/v) to the sample which had been incubated for 4.7 h. Addition of seeds induced no changes in scattering or fluorescence, and no binding of Thioflavine T was observed even 5 h postseeding. The postseeding experiment disproves the idea that it is the inhibition of nucleus formation which prevents efficient fibril growth, because I6T53 was not recruited to elongate the added amyloid seeds. The lack of fibril formation under these conditions could be a consequence of destabilizing the amyloidogenic intermediate or of stabilizing the native state, either of which would shift the equilibrium away from fibril formation.
Fluorescence and scattering of I6T53 at pH 9.0, 1 M sodium sulfate, and 74°C
Based on the different conditions that were tested previously (Ramírez-Alvarado et al. 2000), we examined I6T53 100μM, pH 9.0, 1 M sodium sulfate, 74°C (melting temperature of I6T53 under these conditions; Fig. 6A,B ▶). The kinetics of fibril formation followed by 310/350 and scattering are very similar to the kinetics observed at pH 5.2, 72°C. The half-times for the kinetic reactions determined by 310/350 and scattering were 3.9 h.
Figure 6.


Kinetics of fibril formation of I6T53 at pH 9.0, 1 M sodium sulfate. (A) Normalized intrinsic Trp fluorescence intensity as a function of time (excitation 294 nm, ratio of emission intensity at 310 nm and 350 nm). (B) Normalized light scattering of amyloid formation as a function of time (excitation 340 nm, emission at 340 nm). Inset figures show the total length of the amyloid formation experiment under these conditions (15 h). Experimental conditions: 100 μM I6T53, 1 M sodium sulfate in 10 mM sodium acetate, sodium borate, and sodium citrate buffer, pH 9.0, 74°C
Recovery of the biphasic kinetic behavior observed at pH 5.2 suggests that, under these conditions, the intermediate conformation is significantly populated. These results indicate that the addition of 1 M sodium sulfate at pH 9.0 has a stabilizing effect on the amyloidogenic intermediate and thereby shifts the equilibrium towards fibril formation.
Nucleation-dependent amyloid formation: Seeding
In order to understand the role of fibril seeds as precursors and their possible relationship to the intermediate conformations that are observed during the kinetics of amyloid formation of I6T53, we followed the kinetics of autoseeded fibril formation by I6T53, monitoring fluorescence and scattering. The reaction has a very short lag phase of 0.2 h (Fig. 7A,B ▶), suggesting that the seeds bypass the rate-determining steps of fibril formation (initiation) at pH 5.2, 72°C. Intermediate formation is not rate-limiting in these conditions. The presence of amyloid fibrils was confirmed at the endpoint by electron microscopy (Fig. 7C ▶).
Figure 7.


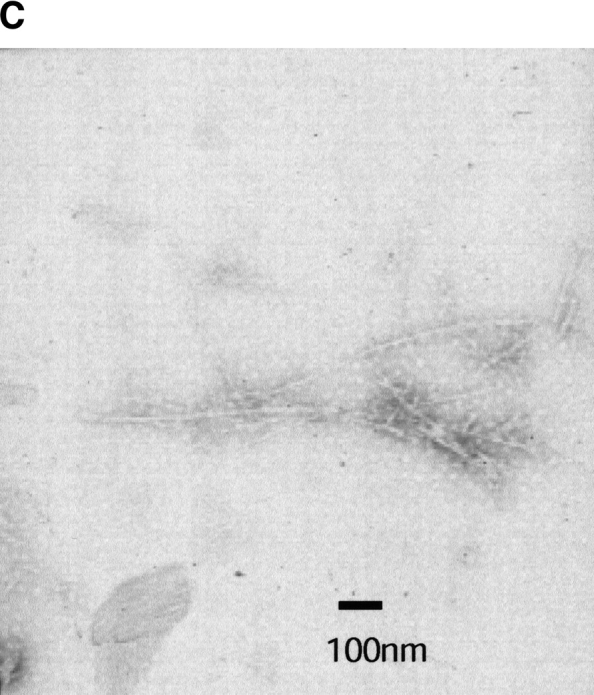
Seeding experiment with I6T53. (A) Normalized intrinsic Trp fluorescence intensity as a function of time (excitation 294 nm, ratio of emission intensity at 310 nm and 350 nm) of self-seed experiment of soluble I6T53 100 μM, with 1/100 (v/v) of I6T53 sonicated fibrils. (B) Normalized light scattering of amyloid formation as a function of time (excitation 340 nm, emission at 340 nm). (C) Electron micrographs of the amyloid fibrils produced by the autoseed reaction.
Discussion
Because the precursor conformations of all amyloidogenic proteins possibly share common features, it is critical to the understanding of amyloid formation that the nature of these intermediates be better characterized. Indeed, there has been speculation that amyloidogenic intermediates rather than mature fibrils are the causative agents of toxicity in cells (Carrell and Lomas 1997; Lansbury 1999; Harper et al. 1999; Bucciantini et al. 2002). β-sheet-containing oligomeric species might bind to any number of cellular targets, thereby triggering a cytotoxic cascade. The short lifetime of partially folded intermediates combined with their low concentration at a given time in the process of amyloid formation makes it difficult to characterize their conformations.
However, there has been some progress recently in the characterization of amyloidogenic intermediates, using experimental conditions in which the intermediate population is maximized. The β1 system described here provides an excellent model because the timescale of I6T53 fibril formation makes the population of conformations through-out the reaction amenable to a variety of spectroscopic methods.
Our results show that within the limits of detection, the protein appears to be monomeric during the lag phase where significant fibril formation has not occurred. The population during the lag phase could be partially folded. Fluorescence spectroscopy shows that the exposure of the tryptophan residue in the lag phase is different from that in the fibrils. The NMR spectra during the lag phase also show that under conditions where fibrils form, both the unfolded and folded states exist. It is often the case that the equilibrium between folded and unfolded protein is slow on the NMR timescale. In fact, two distinct states, native and denatured, have been observed by NMR at the acid-induced denaturation transition acid unfolded midpoint of the F30H mutant of the protein G B1 domain (Sari et al. 2000).
Although there are some studies in which amyloid formation was induced by neutralization of aspartate (Orpiszewski and Benson 1999) or by oxidation damage (Hoyer 1993), a direct correlation of heat damage and amyloid formation has not been described. It is possible that heat damage could trigger the formation of amyloid fibrils in systems in which incubation at high temperatures is required.
The lag phase varied in certain conditions tested in the present study. We found that the kinetics of fibril formation at pH 9.0 are slower than at pH 5.2. No amyloid fibrils were visible by electron microscopy, and no enhancement of the Thioflavine T fluorescence was observed at pH 9.0. It is possible that at pH 9.0 the intermediate conformations are formed very slowly or are very unstable. In contrast, the addition of 1 M sodium sulfate to the reaction at pH 9.0 recovers the two-phase kinetic profile that is very similar to the kinetics at pH 5.2. There are several reports of the effect of salt (in particular sodium sulfate) on the stability and folding kinetics of different proteins. For example, sodium sulfate was shown to stabilize folding intermediates of both the predominantly β-sheet wild-type β1 protein (Park et al. 1997), and the predominantly α-helical protein Im7 (Ferguson et al. 1999). Fink and coworkers suggested that the salt-induced effects on the structure, stability, and folding kinetics of staphylococcal nuclease may be attributed to the binding of counter ions, namely anions, resulting in minimization of intramolecular electrostatic repulsion (Nishimura et al. 2001). Sodium sulfate may stabilize the intermediate population of I6T53 at pH 9.0 in this fashion, thereby facilitating fibrillogenesis. Compared to protein folding, a critical difference in the effect of salt on amyloid formation is that intermolecular interactions are very important in making fibrils, and these interactions could be enhanced by increasing the ionic strength of the solution.
It is possible that the intermediate population that results in amyloid formation is an on-pathway unfolding intermediate (Park et al. 1997). We show that the B1 domain of protein G forms amyloid fibrils at the midpoint of the unfolding transition, where the protein is a mixture of folded and unfolded states in slow exchange, as defined by our NMR measurements.
Interestingly, we did not observe the properties of an intermediate when we incubated I6T53 at pH 9.0, 54°C. Addition of 1 M sodium sulfate regenerates both the 310/350 nm and the scattering properties of the amyloidogenic intermediate present at pH 5.2, 72°C. It is possible that the same stabilizing effect that sodium sulfate has for a wild-type β1 folding intermediate (Park et al. 1997) allows us to observe I6T53 amyloidogenic intermediate. It is also possible that dif ferent conformers mediate fibril nucleation and elongation.
We suggest that the biphasic behavior of I6T53 amyloid formation could be explained by an increased stability of its intermediate conformations relative to those of wild type and the other mutants. We think that the stability of the native state is possibly not the determining factor because the wild type, which is more stable than I6T53, does not show such a long lag phase.
A number of studies have shown that mutations can promote amyloid fibril formation; here we show that protein engineering can also delay the onset of fibril formation. Several questions are raised as a result of these studies: Which are the residues involved in the conformational change? What makes the I6T53 variant different from the other protein variants that lack detectable intermediate conformations? What structural features promote amyloid formation? Further investigation of this phenomenon will provide insights into the nature of amyloidogenic intermediates and should stimulate novel strategies for the prevention of fibril formation.
Materials and methods
Protein preparation
Protein expression and purification was performed as described (Smith et al. 1994). Briefly, ion exchange chromatography (Q-sepharose) with a gradient of NaCl in 20 mM Tris-HCl buffer, pH 8.0 was followed by gel filtration chromatography (Hi-Load Superdex 75, Pharmacia) in 50 mM sodium acetate, pH 5.2. Protein purity was estimated by polyacrylamide gel electrophoresis, and the protein concentration was determined from amino acid analysis performed at the HHMI Biopolymer Laboratory and W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University. The B1 protein framework for this study was described previously (Smith et al. 1994; Merkel et al. 1998). The protein is the B1 domain of IgG-binding protein G (Gronenborn et al. 1991) with a T2Q mutation introduced to ensure homogeneous N-terminal processing. Changes are incorporated on the solvent-exposed surface of the β sheet to provide a homogeneous ‘host’ environment for the ‘guest’ positions. The ‘host’ environment is comprised of A15 and A44 (cross-strand), T4 and T8 (same strand), and T51 and T55 (same strand). In different studies, various combinations were substituted at the ‘guest’ positions (e.g., positions 6 and 53). This study focused on the protein I6T53, in which positions 6 and 53 are coincidentally the residues present in wild-type B1.
Seed preparation
Typically, 100 μM solution of I6T53 was converted to fibrils by agitation at 72°C at pH 5.2 for about 24 h. Seeds were prepared by sonicating the fibril solution for 10 min.
Electron microscopy
Electron microscopy was performed by using a Zeiss JEM-A transmission electron microscope at 80 kV excitation voltage. Fibril solution (6 μL) was air-dried for 5 min on a Formvar/carbon-coated copper grid. The sample was then negatively stained with 6 μL of 1% phosphotungstic acid solution, pH 7.0.
Thioflavine T staining
The Thioflavine T fluorescence analysis was performed as described (Le Vine 1993). Aliquots of soluble protein or fibril suspension (5 μL) were added to 3 μM Thioflavine T in 50 mM sodium acetate, pH 5.2 to a final volume of 1 mL. Excitation spectra were recorded on a Photon Technology International C-61 fluorometer with an excitation wavelength of 450 nm and emission wavelength of 484 nm.
Fluorescence
Fibril formation was recorded on a Photon Technology International C-61 fluorometer fitted with a temperature-controlled cell holder. Glan-Thompson-style plane polarizers were used in anisotropy experiments. The slit widths were 1 nm and 2 nm depending on the fluorescence intensity. The data collection was performed by acquisition of 0.5 points/sec during 2-sec and 25-sec pause with 1000 repeats. The shutters were only open during acquisition in the automatic mode. All fluorescence spectra were measured in a 1-cm pathlength cuvette. The β1 I6T53 variant has a single Trp residue in position 43. The kinetics of fibril formation were followed by changes in the ratio of intrinsic tryptophan emission intensity at 310 and 350 nm, excitation 294 nm.
Light scattering
Light scattering measurements were made using the spectrofluorometer described above, fitted with Glan-Thompson polarizers in parallel orientation. Excitation and emission wavelengths were set to 340 nm.
NMR
NMR data were collected on a Varian Inova 800 MHz NMR spectrometer. Water elimination in proton 1D experiments was achieved using a modified WATERGATE (Piotto et al. 1992; Altieri and Byrd 1995). A spectral width of 11000 Hz was defined by 8192 data points; 128–512 scans were necessary at the low concentrations studied here. Data were processed with NMRpipe (Delaglio et al. 1995), using a gaussian window of 10 Hz broadening and zero filling to double the size. NMR diffusion measurements were made at 25°C, and peaks were integrated using the Varian software VNMR6.1C. The pulse sequence used for these experiments was essentially that of Lapham et al. (1997), based on the study of Tanner (1970).
Mass spectrometry
Mass spectra were collected using a Micromass LCT electrospray time of flight spectrometer in nano-ESI mode.
Acknowledgments
We thank Andrew Miranker, Thomas Magliery, Shae Padrick, Steve Marino, Gary Sarkis, Sheila Jaswal, Enrique de la Cruz, and the Regan lab for thorough manuscript revision, comments, and helpful discussion. Shae Padrick is gratefully acknowledged for performing the mass spectroscopy analysis. M.R.-A. was a Human Frontier Science Program long-term fellow. This work was supported by NIH grant GM-57265.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0227403.
References
- Alexander, P., Fahnestock, S., Lee, T., Orban, J., and Bryan, P. 1992a. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: Why small proteins tend to have high denaturation temperatures. Biochemistry 31 3597–3603. [DOI] [PubMed] [Google Scholar]
- Alexander, P., Orban, J., and Bryan, P. 1992b. Kinetic analysis of folding and unfolding the 56 amino acid IgG-binding domain of streptococcal protein G. Biochemistry 31 7243–7248. [DOI] [PubMed] [Google Scholar]
- Altieri, A.S. and Byrd, R.A. 1995. Randomization approach to water suppression in multidimensional NMR using pulsed field gradients. J. Magn. Reson. B. 107 260–266. [DOI] [PubMed] [Google Scholar]
- Blake, C. and Serpell, L. 1996. Synchrotron X-ray studies suggests that the core of the transthyretin amyloid fibril is a continuous β-sheet helix. Structure 4 989–998. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M., Radford, S.E., Blake, C.C., et al. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C.M., and Stefani, M. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416 507–511. [DOI] [PubMed] [Google Scholar]
- Canet, D., Sunde, M., Last, A.M., Miranker, A., Spencer, A., Robinson, C.V., and Dobson, C.M. 1999. Mechanistic studies of the folding of human lysozyme and the origin of amyloidogenic behavior in its disease-related variants. Biochemistry 38 6419–6427. [DOI] [PubMed] [Google Scholar]
- Canet, D., Last, A.M., Tito, P., Sunde, M., Spencer, A., Archer, D.B., Redfield, C., Robinson, C.V., and Dobson, C.M. 2002. Local cooperativity in the unfolding of an amyloidogenic variant of human lysozyme. Nat. Struct. Biol. 9 308–315. [DOI] [PubMed] [Google Scholar]
- Carrell, R.W. and Lomas, D.A. 1997. Conformational disease. Lancet 350 134–138. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Webster, P., Taddei, N., Clark, A., Stefani, M., Ramponi, G., and Dobson, C.M. 1999. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. 96 3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Mangione, P., Andreola, A., Giorgetti, S., Stefani, M., Dobson, C.M., Bellotti, V., and Taddei, N. 2001. Detection of two partially structured species in the folding process of the amyloidogenic protein β2-microglobulin. J. Mol. Biol. 307 379–391. [DOI] [PubMed] [Google Scholar]
- Damas, A.M., Ribeiro, S., Lamzin, V.S., Palha, J.A., and Saraiva, M.J. 1996. Structure of Val122Ile variant transthyretin—A cardiomyopathic mutant. Acta Crystallogr. D Biol. Crystallogr. 52 966–972. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Esturau, N., Sanchez-Ferrando, F., Gavin, J.A., Roumestand, C., Delsuc, M.A., and Parella, T. 2001. The use of sample rotation for minimizing convection effects in self-diffusion NMR measurements. J. Magn. Reson. 153 48–55. [DOI] [PubMed] [Google Scholar]
- Fandrich, M., Fletcher, M.A., and Dobson, C.M. 2001. Amyloid fibrils from muscle myoglobin. Nature 410 165–166. [DOI] [PubMed] [Google Scholar]
- Ferguson, N., Capaldi, A.P., James, R., Kleanthous, C., and Radford, S.E. 1999. Rapid folding with and without populated intermediates in the homologous four-helix proteins Im7 and Im9. J. Mol. Biol. 286 1597–608. [DOI] [PubMed] [Google Scholar]
- Gallagher, T., Alexander, P., Bryan, P., and Gilliland, G.L. 1994. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry 33 4721–4729. [PubMed] [Google Scholar]
- Gronenborn, A.M., Filpula, D.R., Essig, N.Z., Achari, A., Whitlow, M., Wingfield, P.T., and Clore, G.M. 1991. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253 657–661. [DOI] [PubMed] [Google Scholar]
- Gross, M., Wilkins, D.K., Pitkeathly, M.C., Chung, E.W., Higham, C., Clark, A., and Dobson, C.M. 1999. Formation of amyloid fibrils by peptides derived from the bacterial cold shock protein CspB. Protein Sci. 8 1350–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijarro, J.I., Sunde, M., Jones, J.A., Campbell, I.D., and Dobson, C.M. 1998. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. 95 4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, J.A., Steinrauf, L.K., Braden, B.C., Liepnieks, J., Benson, M.D., Holmgren, G., Sandgren, O., and Steen, L. 1993. The x-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30 → Met variant to 1.7Å resolution. J. Biol. Chem. 268 2416–2424. [PubMed] [Google Scholar]
- Harper, J.D., Wong, S.S., Lieber, C.M., and Lansbury, Jr., P.T. 1999. Assembly of Ab amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 38 8972–8980. [DOI] [PubMed] [Google Scholar]
- Hoyer, S. 1993. Brain oxidative energy and related metabolism, neuronal stress and Alzheimer’s disease: A speculative synthesis. J. Geriatr. Psychiatry Neurol. 6 3–13. [DOI] [PubMed] [Google Scholar]
- Hurle, M.R., Helms, L.R., Li, L., Chan, W., and Wetzel, R. 1994. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci 91 5446–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez, J.L., Guijarro, J.I., Orlova, E., Zurdo, J., Dobson, C.M., Sunde, M., and Saibil, H.R. 1999. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 18 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J.W. 1996. Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 6 11–17. [DOI] [PubMed] [Google Scholar]
- ———. 1998. The alternative conformations of amyloidogenic proteins and their multistep assembly pathways. Curr. Opin. Struct. Biol. 8 101–106. [DOI] [PubMed] [Google Scholar]
- Kelly, J.W. and Lansbury, P.T. 1994. A chemical approach to elucidate the mechanism of transthyretin and β-protein amyloid fibril formation. Amyloid 1 186–205. [Google Scholar]
- Konno, T., Murata, K., and Nagayama, K. 1999. Amyloid-like aggregates of a plant protein: A case of a sweet-tasting protein, monellin. FEBS Lett. 454 122–126. [DOI] [PubMed] [Google Scholar]
- Krebs, M.R., Wilkins, D.K., Chung, E.W., Pitkeathly, M.C., Chamberlain, A.K., Zurdo, J., Robinson, C.V., and Dobson, C.M. 2000. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 300 541–549. [DOI] [PubMed] [Google Scholar]
- Lai, Z., Colon, W., and Kelly, J.W. 1996. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35 6470–6482. [DOI] [PubMed] [Google Scholar]
- Lansbury, Jr., P.T. 1999. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. 96 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapham, J., Rife, J.P., Moore, P.B., and Crothers, D.M. 1997. Measurement of diffusion constants for nucleic acids by NMR. J. Biomol. NMR. 10 255–262. [DOI] [PubMed] [Google Scholar]
- Le Vine, H. 1993. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 2 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinovich, S.V., Brew, S.A., Aota, S., Akiyama, S.K., Haudenschild, C., and Ingham, K.C. 1998. Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol. 280 245–258. [DOI] [PubMed] [Google Scholar]
- Liu, K., Cho, H.S., Lashuel, H.A., Kelly, J.W., and Wemmer, D.E. 2000. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Biol. 7 754–757. [DOI] [PubMed] [Google Scholar]
- McParland, V.J., Kad, N.M., Kalverda, A.P., Brown, A., Kirwin-Jones, P., Hunter, M.G., Sunde, M., and Radford, S.E. 2000. Partially unfolded states of β2-microglobulin and amyloid formation in vitro. Biochemistry 39 8735–8746. [DOI] [PubMed] [Google Scholar]
- Merkel, J.S. and Regan, L. 1998. Aromatic rescue of glycine in β-sheets. Fold Des. 3 449–455. [DOI] [PubMed] [Google Scholar]
- Merkel, J.S., Sturtevant, J.M., and Regan, L. 1999. Side-chain interactions in parallel β-sheets: The energetics of cross-strand pairings. Structure Fold Des. 7 1333–1343. [DOI] [PubMed] [Google Scholar]
- Morgan, C.J., Gelfand, M., Atreya, C., and Miranker, A.D. 2001. Kidney dialysis-associated amyloidosis: A molecular role for copper in fiber formation. J. Mol. Biol. 309 339–345. [DOI] [PubMed] [Google Scholar]
- Nishimura, C., Uversky, V.N., and Fink, A.L. 2001. Effects of salts on the stability and folding of staphylococcal nuclease. Biochemistry 40 2113–2128. [DOI] [PubMed] [Google Scholar]
- Ohnishi, S., Koide, A., and Koide, S. 2000. Solution conformation and amyloid-like fibril formation of a polar peptide derived from a β-hairpin in the OspA single-layer β-sheet. J. Mol. Biol. 301 477–489. [DOI] [PubMed] [Google Scholar]
- Orpiszewski, J., and Benson, M.D. 1999. Induction of β-sheet structure in amyloidogenic peptides by neutralization of aspartate: A model for amyloid nucleation. J. Mol. Biol. 289 413–428. [DOI] [PubMed] [Google Scholar]
- Park, S.H., O’Neil, K.T., and Roder, H. 1997. An early intermediate in the folding reaction of the B1 domain of protein G contains a native-like core. Biochemistry 36 14277–14283. [DOI] [PubMed] [Google Scholar]
- Park, S.H., Shastry, M.C., and Roder, H. 1999. Folding dynamics of the B1 domain of protein G explored by ultrarapid mixing. Nat. Struct. Biol. 6 943–947. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 2 661–665. [DOI] [PubMed] [Google Scholar]
- Ramírez-Alvarado, M., Merkel, J.S., and Regan, L. 2000. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. 97 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari, N., Alexander, P., Bryan, P.N., and Orban, J. 2000. Structure and dynamics of an acid-denatured protein G mutant. Biochemistry 39 965–977. [DOI] [PubMed] [Google Scholar]
- Sebastiao, M.P., Saraiva, M.J., and Damas, A.M. 1998. The crystal structure of amyloidogenic Leu55 → Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 273 24715–24722. [DOI] [PubMed] [Google Scholar]
- Serpell, L.C., Sunde, M., and Blake, C.C. 1997. The molecular basis of amyloidosis. Cell. Mol. Life Sci. 53 871–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C.K., and Regan, L. 1995. Guidelines for protein design: The energetics of β-sheet side chain interactions. Science 270 980–982. [DOI] [PubMed] [Google Scholar]
- Smith, C.K., Withka, J.M., and Regan, L. 1994. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry 33 5510–5517. [DOI] [PubMed] [Google Scholar]
- Sunde, M., Serpell, L.C., Bartlam, M., Fraser, P.E., Pepys, M.B., and Blake, C.C. 1997. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273 729–739. [DOI] [PubMed] [Google Scholar]
- Takano, K., Funahashi, J., and Yutani, K. 2001. The stability and folding process of amyloidogenic mutant human lysozymes. Eur. J. Biochem. 268 155–159. [DOI] [PubMed] [Google Scholar]
- Tanner, J.E. 1970. Use of the stimulated echo in NMR diffusion studies. J. Chem. Phys. 52 2523–2526. [Google Scholar]
- Terry, C.J., Damas, A.M., Oliveira, P., Saraiva, M.J., Alves, I.L., Costa, P.P., Matias, P.M., Sakaki, Y., and Blake, C.C. 1993. Structure of Met30 variant of transthyretin and its amyloidogenic implications. EMBO J. 12 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas, V., Zurdo, J., Filimonov, V.V., Aviles, F.X., Dobson, C.M., and Serrano, L. 2000. Protein engineering as a strategy to avoid formation of amyloid fibrils. Protein Sci. 9 1700–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J., Schell, M., Murphy, C., Hrncic, R., Stevens, F.J., and Solomon, A. 1999. Thermodynamic instability of human l6 light chains: Correlation with fibrillogenicity. Biochemistry 38 14101–14108. [DOI] [PubMed] [Google Scholar]
- West, M.W., Wang, W., Patterson, J., Mancias, J.D., Beasley, J.R., and Hecht, M.H. 1999. De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. 96 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutani, K., Takayama, G., Goda, S., Yamagata, Y., Maki, S., Namba, K., Tsunasawa, S., and Ogasahara, K. 2000. The process of amyloid-like fibril formation by methionine aminopeptidase from a hyperthermophile, Pyrococcus furiosus. Biochemistry 39 2769–2777. [DOI] [PubMed] [Google Scholar]