

Abstract
A solid-state NMR approach for simultaneous resonance assignment and three-dimensional structure determination of a membrane protein in lipid bilayers is described. The approach is based on the scattering, hence the descriptor “shotgun,” of 15N-labeled amino acids throughout the protein sequence (and the resulting NMR spectra). The samples are obtained by protein expression in bacteria grown on media in which one type of amino acid is labeled and the others are not. Shotgun NMR short-circuits the laborious and time-consuming process of obtaining complete sequential assignments prior to the calculation of a protein structure from the NMR data by taking advantage of the orientational information inherent to the spectra of aligned proteins. As a result, it is possible to simultaneously assign resonances and measure orientational restraints for structure determination. A total of five two-dimensional 1H/15N PISEMA (polarization inversion spin exchange at the magic angle) spectra, from one uniformly and four selectively 15N-labeled samples, were sufficient to determine the structure of the membrane-bound form of the 50-residue major pVIII coat protein of fd filamentous bacteriophage. Pisa (polarity index slat angle) wheels are an essential element in the process, which starts with the simultaneous assignment of resonances and the assembly of isolated polypeptide segments, and culminates in the complete three-dimensional structure of the protein with atomic resolution. The principles are also applicable to weakly aligned proteins studied by solution NMR spectroscopy.
[The structure we determined for the membrane-bound form of the Fd bacteriophage pVIII coat protein has been deposited in the Protein Data Bank as PDB file 1MZT.]
Keywords: Solid-state NMR, membrane protein, fd coat protein, protein structure determination, Pisa wheels
Solid-state NMR spectroscopy of aligned samples is a general method for determining the structures of proteins in biological supramolecular structures (Opella et al. 1987). In this article, we demonstrate that this method can be applied to membrane proteins in aligned lipid bilayers in a manner conducive to the high throughput required for structural genomics. Membrane proteins can be expressed and isotopically labeled in bacteria, isolated, purified, and reconstituted into fully hydrated lipid bilayers, which can then be aligned to a degree comparable to that observed in single crystals of small molecules (Marassi et al. 1997). “Shotgun” NMR, named for the scattered distribution of the isotopically labeled sites in the protein and the resonances in the spectra, uses the orientational information in the solid-state NMR spectra of aligned samples to simultaneously assign resonances and obtain orientational restraints for structure determination.
The alignment of samples along the direction of the applied magnetic field results in solid-state NMR spectra with resonances characterized by orientationally dependent frequencies. Because nearly all residues in helical membrane proteins are immobile on the millisecond timescale of the chemical shift and heteronuclear dipole–dipole interactions, the orientational information inherent to these anisotropic nuclear spin interactions is fully manifested in high-resolution solid-state NMR spectra, and can be used for structure determination (Cross and Opella 1985; Ketchem et al. 1993; Opella et al. 1999; Wang et al. 2001). The alignment of membrane proteins in lipid bilayers is fixed by the preparation and placement of the sample in the magnetic field; therefore, each measured frequency reflects the orientation of a specific site in the protein with respect to the bilayer. Peptide plane orientations are determined directly from the NMR frequencies, and the dihedral angles φ and ξ, which describe the three-dimensional structure of the protein backbone, are calculated from the orientations of the peptide planes joined at the α-carbons.
The PISEMA (polarization inversion spin exchange at the magic angle) experiment (Wu et al. 1994) yields high-resolution 1H–15N dipolar coupling/15N chemical shift separated local field spectra (Waugh 1976) of 15N labeled proteins in aligned samples (Marassi et al. 1997). The orientational dependencies of these two anisotropic spin interactions serve as both the mechanisms for resolution among amide resonances, and the sources of orientational restraints for backbone structure determination. The PISEMA spectra of aligned proteins display distinctive resonance patterns that are a consequence of the direct mapping of protein structure onto NMR frequencies. These wheel-like patterns, called Pisa (polarity index slant angle) wheels, are sensitive indices of secondary structure and topology, and mirror helical wheel projections of residues in both α-helices (Marassi and Opella 2000; Wang et al. 2000) and β-strands (Marassi 2001). Similar patterns are also observed in the solution NMR spectra of weakly aligned proteins (unpublished results). The tilt (slant angle) of a helix can be determined by inspection of a Pisa wheel without resonance assignments, and only a limited amount of assignment information is needed to determine the rotation angle (polarity index) of the helix, for example, one sequential assignment or the pattern from one selectively labeled sample. This information is sufficient to characterize the architecture of a membrane protein, and also provides the starting point for the determination of the three-dimensional structure.
Previous determinations of protein structures by solid-state NMR utilized on orientational restraints derived from the frequencies of independently assigned resonances (Cross and Opella 1985; Ketchem et al. 1993; Opella et al. 1999; Wang et al. 2001). In contrast, the Shotgun NMR approach relies solely on the spectra from one uniformly, and several selectively, 15N-labeled samples, and on the fundamental symmetry properties of Pisa wheels to enable the simultaneous sequential assignment of resonances and the measurement of the orientationally dependent frequencies. Crucial to Shotgun NMR is the fact that each resonance frequency in these spectra reflects molecular orientation relative to a fixed and external reference axis, defined by the direction of the applied magnetic field. This enables the structures of isolated polypeptide segments of the protein to be determined independently from each other, without propagating any associated experimental errors or uncertainties in the magnitudes and molecular orientations of the principal elements of the spin-interaction tensors. The method is demonstrated here for a highly aligned sample, using solid-state NMR experiments, but it is also applicable to weakly aligned samples using solution NMR experiments (unpublished results).
The membrane-bound form of the major pVIII coat protein of the filamentous fd bacteriophage resides within the inner membrane of infected Escherichia coli before incorporation into virus particles that are extruded through the cell membrane. The structure of the membrane-bound form of the protein has been extensively studied in micelle samples by solution NMR spectroscopy (Cross and Opella 1980; Henry and Sykes 1992; McDonnell et al. 1993; Van de Ven et al. 1993; Williams et al. 1996; Almeida and Opella 1997; Papavoine et al. 1998), as well as with solid-state NMR experiments on bilayer samples (Leo et al. 1987; Bogusky et al. 1988; Marassi et al. 1997). The results are in agreement in showing that the protein has two distinct α-helical segments, a short amphipathic in-plane (IP) helix that rests on the membrane surface, a longer hydrophobic transmembrane (TM) helix, and few mobile residues near the N and C termini. The same protein in intact virus particles has also been the subject of many structural studies, including solid-state NMR spectroscopy (Cross and Opella 1983, 1985; Opella et al. 1987) and X-ray fiber diffraction (Glucksman et al. 1992; Marvin et al. 1994). In this article, we describe all of the structured residues of the membrane-bound form of fd coat protein in lipid bilayers with atomic resolution.
Results
NMR spectroscopy
Structure determination begins with the acquisition of two-dimensional PISEMA spectra from one uniformly 15N-labeled (Fig. 1A ▶) and four selectively 15N-labeled (Fig. 2 ▶) samples of fd coat protein in aligned lipid bilayers; the labeling patterns are illustrated at the top of Figure 1 ▶. The NMR spectra of selectively labeled samples are routinely used to assign resonances to types of amino acids in both solution NMR and solid-state NMR studies of proteins. However, in Shotgun NMR they provide much additional information, starting with the clear segregation of PISEMA spectra of helical membrane proteins into regions for the resonances from IP (lower right quadrant) and TM (upper left quadrant) residues. For example, of the 10 resonances observed in the spectrum of 15N-Ala-labeled coat protein, five are in the IP region of the spectrum, including that from Ala 18, which establishes that the IP helix extends at least to residue 18 (Fig. 2A ▶); three resonances are in the TM region; one is isotropic because of motional averaging, which demonstrates that Ala 49 next to the C terminus is mobile on the millisecond timescale; and the N-terminal Ala 1 gives a peak at 35 ppm (not shown in these spectra). Of the four Gly resonances (Fig. 2B ▶), three are in the TM region, and one, assigned to Gly 3 (Bogusky et al. 1987), is from a residue near the mobile N terminus. The chemical shift frequencies of the Gly resonances are upfield of the others, as expected based on the differences in the magnitudes of the principal values of the amide 15N chemical shift tensors for this amino acid (Oas et al. 1987). The two Leu resonances in the spectrum in Figure 2C ▶ are straightforwardly assigned to Leu 14 in the IP region and Leu 41 in the TM region (Marassi et al. 1997). As expected, the resonances from the four valine residues in the protein are in the TM region of the spectrum (Fig. 2D ▶).
Figure 1.
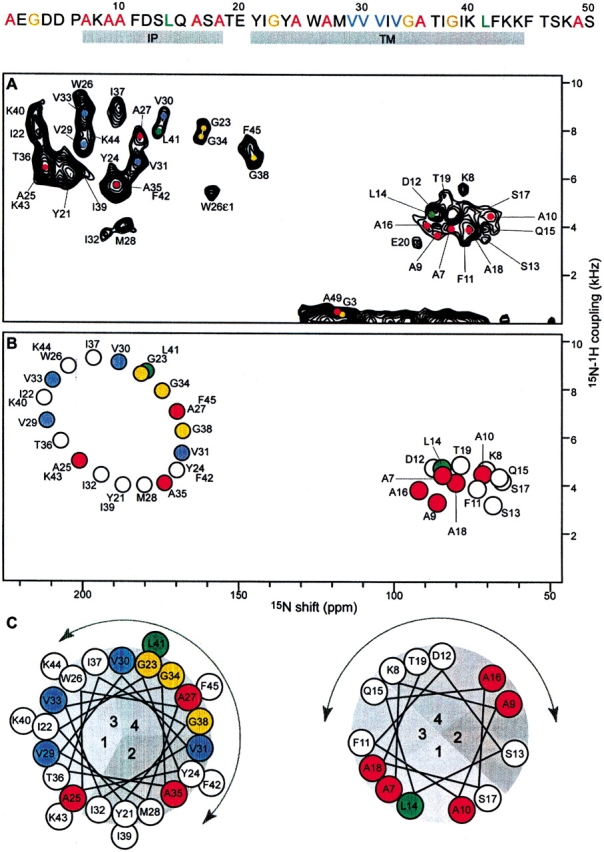
Two-dimensional 1H/15N PISEMA spectra of fd coat protein in aligned lipid bilayers. The amino acid sequence of the protein is shown at the top. (A) Experimental PISEMA spectrum obtained from uniformly 15N-labeled fd coat protein in lipid bilayers. The resonance assignments are marked. (B) Pisa wheel PISEMA spectrum calculated from a protein model with the IP helix parallel to within 3° of the membrane surface (τ = 87°), and the TM helix crossing the membrane at an angle τ = 30°. The helical and Pisa wheel rotations are set to match the resonances in the experimental PISEMA spectra from the Ala (red), Gly (gold), Leu (green), and Val (blue) residues. (C) Helical wheel representations of the protein IP and TM helices. The curved arrows span the N-terminal periplasmic side of the membrane.
Figure 2.

Experimental and calculated 1H/15N PISEMA spectra for the TM region of the fd coat protein, in aligned lipid bilayers selectively 15N-labeled with (A) Ala, (B) Gly, (C) Leu, and (D) Val. The resonances were assigned to specific amino acids in the sequence by comparison of the experimental spectrum (top) with the Pisa wheel spectrum (bottom). In each panel, the Pisa wheel PISEMA spectrum was calculated from a protein model with the IP helix tilted by τ = 87°, and the TM tilted by τ = 30°.
The observation of Pisa wheels in both the IP and TM regions of the spectrum of the uniformly 15N-labled sample (Fig. 1A ▶), by itself, leads to an initial trial model of the protein with two α-helices, one crossing the membrane with a tilt angle (τ) of 30°, the other resting within 3° of parallel to the membrane surface (τ = 87°). This model is sufficient for the calculation of the idealized PISEMA spectrum shown in Figure 1B ▶. Each helix in the trial model is separately rotated around its axis until the resonance pattern in its calculated Pisa wheel spectrum qualitatively matches the resonances in the experimental spectra of selectively labeled samples. This is illustrated with the helical wheel projections of the two helices in Figure 1C ▶, and with the four Pisa wheel spectra in Figure 2 ▶. This procedure yields both the rotation of the two helices about their long axes (ρ), as well as the sequential assignments of the resonances observed in the experimental spectra. Up to this stage, starting with no independently assigned resonances, the procedure has determined the tilt and rotation of both helices with high precision, the lengths of both helices within a few residues, the backbone dynamics qualitatively, and the sequential assignments of resonances from 19 out of the 48 amide backbone sites in the protein.
In the second step, the 15N chemical shift and 1H–15N dipolar coupling frequencies are measured for each resonance in the experimental PISEMA spectrum of the uniformly 15N-labeled sample, and used to calculate the orientations of their corresponding peptide planes. These frequencies provide the restraints for three-dimensional structure determination, because they depend on the orientation of the corresponding molecular site with respect to the direction of the applied magnetic field, and on the magnitudes and orientations of the principal elements of the spin-interaction tensors at that site. The 15N chemical shift frequency is given by:
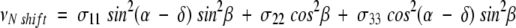 |
(1) |
where σ11, σ22, and σ33 are the principal elements of the 15N chemical shift tensor, and δ is the angle between σ33 and the NH bond. The 1H–15N dipolar coupling frequency is given by:
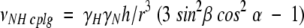 |
(2) |
where γH and γN are the gyromagnetic ratios of the nuclei, h is Planck’s constant, and r is the NH bond length. In both equations, α and β are the polar angles that describe the peptide plane orientation in the magnetic field: α is the angle between the NH bond and the projection of the magnetic field direction on the peptide plane, and β is the angle between the normal to the peptide plane and the direction of the magnetic field (Tycko et al. 1986; Opella et al. 1987). The amide 15N chemical shift tensor and the NH bond length are well characterized in model peptides (Oas et al. 1987; Wu et al. 1995), and can be used to extract α/β orientational restraints from the resonance frequencies using Equations 1 and 2.
The symmetry properties of the spin-interaction tensors give rise to degeneracies in the angular restraints. Each equation yields combinations of the angles α and β that are consistent with a frequency measurement, and that trace out a line on the α/β surface. The actual orientation of a peptide plane must satisfy both Equations 1 and 2 and corresponds to a single α/β point. The orientation is determined for 0 < α < 180 and 0 < β < 90, so that any one of the four orientations (α/β), (α/180 − β), (180 + α/β), and (180 + α/180 − β) are consistent with the experimental data. In addition, because the signs of the 1H–15N dipolar couplings are not determined in these experiments, resonances with couplings smaller than the half-maximal value (∼5 kHz as displayed) can have two possible α/β solutions, resulting in up to eight symmetry-related peptide plane orientations; however, because the dipolar couplings for TM helices with tilt angles <40° are positive, and those for IP helices are negative, the set of restraints is reduced to four symmetry-related orientations for each plane in the initial structural model.
The next step involves the calculation of the φ and ξ dihedral angles from the α/β orientational restraints of pairs of connected peptide planes (Opella et al. 1987; Quine and Cross 2000; Opella et al. 2001). Because at this stage of the analysis only the selectively labeled resonances are definitively assigned, we first perform this calculation for all combinations of α/β restraint sets, where for n resonances, there are a total of (n2 − n) sets of φ/ξ. Each dipeptide combination has two pairs of dihedral angles, each of which satisfies the requirement for tetrahedral geometry and L-amino acid chirality at the α-carbon, and because each peptide plane has four possible orientations, this results in 32 possible φ/ξ sets. However, only 16 of these are unique, because we do not differentiate between positive and negative magnetic field directions. Typically, only one of these is energetically favorable and can be identified using a Ramachandran map.
Orientational ambiguities can also be resolved using additional properties of Pisa wheels (Marassi and Opella 2002). In an α-helix, the orientation of each peptide plane can be predicted from the angles τ and ρ, so that it is possible to select a single orientation, out of the symmetry-related set of four, from its resonance position in the NMR Pisa wheel (Marassi and Opella 2002). This is illustrated in Figure 1C ▶, in which the pie-shaped sections labeled 1, 2, 3, and 4 in the helical wheels (Pisa pies), correspond, respectively, to the four orientations 1 (α/β), 2 (α/180 − β), 3 (180 + α/β), and 4 (180 + α/180 − β). For example, the Val 29 peptide plane has an orientation defined by 1 (α/β). It is connected to the Val 30 peptide plane with orientation 4 (180 + α/180 − β), which, in turn, connects to Val 31 with orientation 2 (α/180 − β). It is the ability to resolve the orientational ambiguities that enables the sequential assignment of the remaining resonances in the spectrum of the uniformly 15N-labeled protein, including those that are not represented in spectra of selectively 15N-labeled samples.
The chemical shift and dipolar coupling frequencies of the PISEMA resonances provide the input for protein structure determination. We begin by constructing isolated segments of structure for the assigned resonances in the sequences: Ala 9–Ala 10, Val 29–Val 30–Val 31, and Val 33–Gly 34. These segments constitute the starting point in a search for the peptide planes and associated resonances that link them. For example, a single residue, Ile 32, whose resonance is unassigned at this stage, separates the peptide segments Val 29–Val 30–Val 31 and Val 33–Gly 34. Because the resonance of Ile 32 falls in section 2 of the helical wheel (Fig. 1C ▶), its peptide plane must have an orientation defined by 2 (α/180 − β), and it must have helical dihedral angles to its neighboring peptide planes. The Ile 32 resonance is assigned by searching the entire set of calculated dihedral angles for a peptide plane combination that satisfies these conditions. The resonances for Met 28 linking the assigned Ala 27 to the Val 29 segment and for Trp 26 linking the assigned Ala 25 and Ala 27 are obtained in a similar fashion. The search is performed for all unlinked planes and segments of planes, until all resonances have been assigned, including those of the turn connecting the two helices, and the indole nitrogen of Trp 26 (Ramamoorthy et al. 1997). Because resonance assignment is accompanied by determination of the calculated restraints and dihedral angles, the structure of the protein is determined.
Structure of a membrane protein in lipid bilayers
The three-dimensional structure of the membrane-bound form of fd coat protein in lipid bilayers is shown in Figure 3 ▶. The 16-Å-long IP helix (residues 8–18) is amphipathic and rests on the membrane surface, with the boundary separating the polar and apolar residues parallel to the lipid bilayer surface, and the apolar residues facing the hydrocarbon core of the lipid bilayer (Fig. 3C ▶). The aromatic residues, Phe 11 in the IP helix, and Tyr 21 in the TM helix, are near this boundary region, and approximately equidistant from the hydrophobic central core of the lipid bilayers (Fig. 3A ▶).
Figure 3.

Structure of the membrane-bound form of fd coat protein in lipid bilayers, with the IP helix in magenta, the TM helix in blue, and the short connecting turn in yellow. The flexible N and C termini are not shown. The dashed gray lines mark the lipid–water boundary. (A) Side view showing the 26° tilt of the TM helix. The Trp 26 side chain is shown in its experimentally determined orientation. The direction of the applied magnetic field is parallel to the arrow. The Lys 40, Lys 43, and Lys 44 side chains were modeled in MolMol and face the cytoplasmic side of the membrane. (B) Front view. (C) View of the IP helix down from the C terminus. The α-carbons are shown as spheres. The dashed gray line representing the membrane–water interface also marks the boundary between hydrophilic (colored) and hydrophobic (gray) residues. The coordinates have been deposited to the Protein Data Bank (PDB file 1MZT).
The 35-Å-long TM helix (residues 21–45) crosses the membrane at an angle of 26° up to residue Lys 40, where the helix tilt changes to 16°. A similar change in helix direction at this site was also observed in the structure of the protein in the phage particles determined by solid-state NMR spectroscopy (Cross and Opella 1985). The helix tilt accommodates the thickness of the phospholipid bilayer, which is ∼31 Å for the lipids, palmitoyl-oleoyl-phosphatidylcholine and -phosphatidylglycerol, used in this study, and typical of E. coli membrane components. Tyr 21 and Phe 45 at the lipid–water interfaces delimit the TM helix. The Trp 26 side chain intercalates in the hydrophobic bilayer interior (Fig. 3A ▶) with an orientation relative to the hydrophobic helix similar to that found for the coat protein in bacteriophage particles (Cross and Opella 1983). The indole NH bond points toward the N terminus and into the lipid–water interface, where it can hydrogen-bond with either water or the phospholipid head groups. Interfacial indole side chains oriented in this way are likely to play a role in determining membrane protein orientation (Schiffer et al. 1992). The TM helix is rotated so that the charged residues, Lys 40, Lys 43, and Lys 44, and the polar residue Thr 36, face the C-terminal, cytoplasmic side of the protein (Fig. 3A ▶). This topology may have functional significance for the bacteriophage assembly process, during which the cytoplasmic, single-stranded, phage DNA is extruded through the inner bacterial membrane after being enclosed within a tube of coat protein subunits. The TM helix orientation is such that it enables the charged amino groups of the lysine side chains to emerge from the membrane interior, and become available at the membrane surface for DNA binding during bacteriophage extrusion.
The TM and IP helices are connected by a short turn (Thr 19–Glu 20) that differs from the longer loop (residues 17–26) determined for the same protein in lipid micelles (Almeida and Opella 1997). This may be due to differences between the micelle and bilayer environments, or to the limitations with working with the few long-range NOE restraints observed in solution NMR studies of helical membrane proteins. Both reasons strongly support an argument in favor of the use of lipid bilayer samples for structure determination of membrane proteins. For the structure determination of the turn between the two helices, the 16 sets of calculated φ/ξ dihedral angles were reduced to 8 for both Thr 19 and Glu 20, because of the restraints imposed by the peptide plane orientations of the flanking amino acid residues, Ala 18 and Tyr 21, and linking the Thr 19 and Glu 20 peptide planes resulted in 16 possible conformations for the turn. Although the tilt and rotation of the IP and TM helices are determined independently of the connecting turn, their relative orientations are defined by the turn geometry, and are different for each turn conformation. The resulting structures can be grouped into four families (Fig. 4A ▶–D), each with a similar (within 10°) relative orientation of the IP and TM helices. Ten conformations including one conformation in family A, one in family B, and all conformations in family C and family D, could be discarded because they have steric clashes between the IP and TM helices. The six remaining structures belong to family A or B (Fig. 4A,B), and only one structure in family A has the most favorable geometry at the connecting turn, and it has been deposited in the Protein Data Bank (PDB file 1MZT), whereas the other structures have less realistic values for the backbone dihedral angles φ/ξ, that map in forbidden regions of the Ramachandran map. The resulting structure, shown in Figure 3 ▶, is particularly appealing because the dihedral angles of the connecting turn do not disrupt the sense of helix winding as the protein structure makes the transition from the N-terminal IP helix to the C-terminal TM helix. This is suggestive of a helix-wind-up conformational change during the phage assembly process, whereby tightening the winding around the connecting turn forces the IP helix to tilt up and to form the single, continuous, long α-helix observed in the phage-bound protein structure (Opella et al. 1987; Glucksman et al. 1992; Marvin et al. 1994). This is also consistent with the model for bacteriophage assembly proposed by Papavoine et al. (1998).
Figure 4.
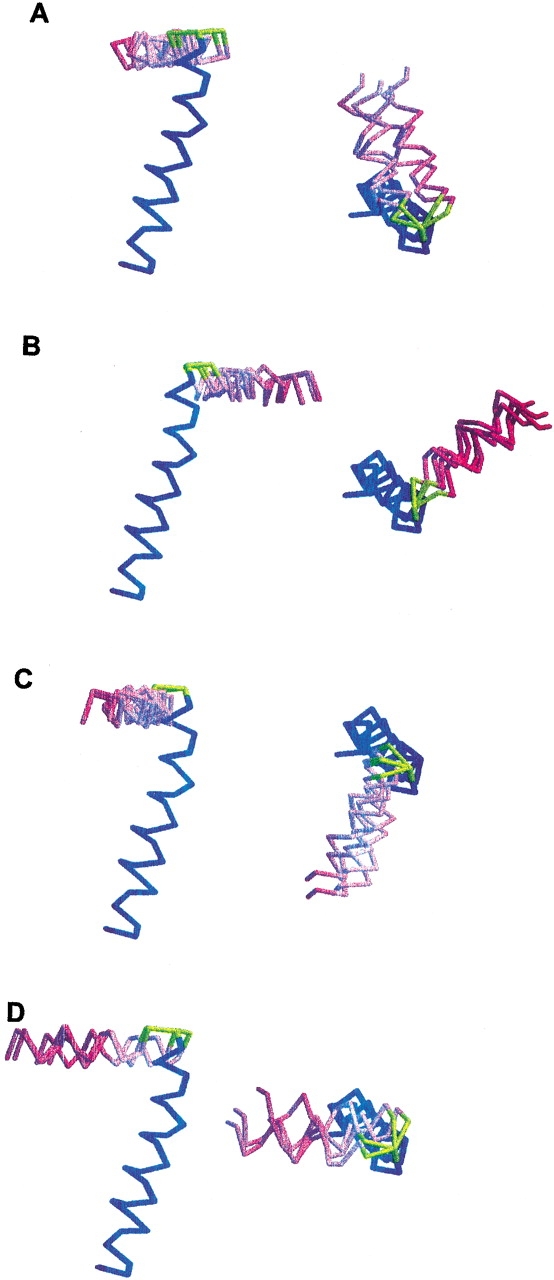
Side and top views of the four structural families obtained for the 16 different geometries of the connecting turn (yellow). Each family (A–D) contains four structures in which the relative orientations of the IP (magenta) and TM (blue) helices are similar and defined by the turn geometry. Family A (A) contains the structure with the most favorable connecting turn geometry (Thr 19 φ/ξ = −93/−82; Glu 20 φ/ξ = −37/132; PDB file 1MZT). Family B (B), family C (C), and family D (D) contain structures with unfavorable connecting turn geometries.
Discussion
The ability to calculate a backbone structure solely from the orientationally dependent 15N chemical shift and 1H–15N dipolar coupling frequencies measured in PISEMA spectra of uniformly and selectively 15N-labeled proteins paves the way for rapid automated methods of protein structure determination. In the Shotgun NMR approach to structure determination, a structural model of the protein is first generated by pattern recognition of the resonances in the PISEMA spectrum of a uniformly 15N-labeled sample, which is then used to calculate an idealized spectrum and model of the protein. The data from a few selectively 15N-labeled samples enable the tilt and rotation of the helices to be determined, and in a more detailed analysis to simultaneously assign all of the resonances and calculate the three-dimensional structure of the protein. Thus, for the fd coat protein, whereas the initial structural model is determined solely from Pisa wheels and consists of two ideal α-helices, the final structure resolves a distinct kink near residue 39, which changes the direction of the TM helix. The quality of the structure cannot be described in terms of a conventional RMSD, because a unique structure is determined from the experimental data. For comparison, the ability to identify subtle structural features, such as a kink in an otherwise uniform helix, can be accomplished in structures determined by X-ray crystallography only when the resolution is 2Å or better. Thus, the structures determined by solid-state NMR of aligned samples have an effective resolution that rivals that of the best experimental determinations of proteins by other methods.
There are two classes of membrane proteins with structures dominated by α-helices or β-strands. Although the class of protein is generally known, before structure determination is initiated, from sequence analysis or other spectroscopic methods, solid-state NMR of aligned samples is also able to distinguish between these two cases. The Shotgun NMR approach, as described here, relies on being able to detect Pisa wheel patterns of resonances that reflect the protein structure in the spectra of oriented proteins. In the case of fd coat protein, the spectra reflect helical structure; however, regular Pisa wheel patterns are also predicted for β-strands (Marassi 2001), so there is every reason to believe that the Shotgun NMR approach will be equally applicable to this class of proteins. The structure determinations of the turns and loops are, in principle, more complex, but enormous simplifications result from the structural constraints imposed by the flanking secondary-structure elements. For example, in the case of fd coat protein, the tilts and rotations of the two helices place strong steric restraints on the allowed conformations of residues in the turn connecting the two helices, thus limiting the result to a single structure of the backbone. The structures of nitrogen-containing side chains are also readily determined, and extensions of the approach to 13C will enable the structures of all side chains to be included.
The Shotgun NMR approach for simultaneously assigning spectra and determining structures by solid-state NMR of aligned samples is generally applicable. A total of five two-dimensional PISEMA spectra were sufficient for structure determination of this 50-residue protein. Simulations and unpublished experimental results indicate that this approach can be applied directly to proteins two to three times larger and, with the use of higher dimensional experiments and stronger magnetic fields, to substantially larger proteins. Aligned bilayer samples can be prepared from proteins that are expressed and labeled in bacteria or other systems, only a few experimental spectra are required, and data analysis methods can be automated. Therefore, this method has the potential for high-throughput structure determination of membrane proteins.
Materials and methods
The preparation of samples of 15N-labeled fd coat protein in aligned lipid bilayers and the details of NMR experiments have been described (Marassi et al. 1997). NMR spectra were obtained on a Chemagnetics-Otsuka Electronics spectrometer with a wide bore Oxford 400/89 magnet, using a home-built flat-coil probe double-tuned to the resonance frequencies of 1H at 400.4 MHz, and 15N at 40.6 MHz. The structure calculations were performed using a suite of Fortran programs developed in our laboratories. Peptide plane orientations were calculated from the input NMR frequencies and the values of the spin-interaction tensors, in terms of α/β polar angles, using Equations 1 and 2 in the program RESTRICT. Dihedral angles were calculated by constraining tetrahedral geometry around the common α-carbon of adjoining peptide planes, of known orientation (Opella et al. 1987, 1999; Quine and Cross 2000) using the program ABtoTOR. The program uses standard peptide plane geometry and the dihedral angle ω = 180°. The principal values and molecular orientation of the amide 15N chemical shift tensor were σ11 = 64 ppm; σ22 = 77 ppm; σ33 = 217 ppm; δ = 17°, and the NH bond distance was 1.07 Å (Wu et al. 1995). The values and orientation of the amide 15N chemical shift tensor for Gly residues were σ11 = 41 ppm; σ22 = 64 ppm; σ33 = 210 ppm; and δ = 18° (Oas et al. 1987). PDB coordinates were calculated, from the resulting protein structure and orientation in the lipid bilayers, using the programs TORtoPDB and ROTPDB. Calculation of spectra from structural coordinates was performed using the program PDBtoNMR. The structure was rendered using MolMol (Koradi et al. 1996) and RasMol (Sayle and Milner-White 1995). The coordinates have been deposited in the Protein Data Bank (PDB file 1MZT).
The orientation of the Trp 26 indole side chain was obtained using Equations 1 and 2, in which α was redefined as the angle between the indole NH bond and the projection of the magnetic field direction on the aromatic plane, and β as the angle between the normal to the aromatic indole plane and the direction of the magnetic field. The values and orientation of the indole 15Nɛ1 chemical shift tensor were σ11 = 61 ppm; σ22 = 130 ppm; σ33 = 181 ppm; δ = 0°, and the NH bond distance was 1.07 Å (Ramamoorthy et al. 1997). Of the four possible symmetry-related orientations, two lead to atomic overlap. The remaining two orientations each have the indole NH bond pointing toward the protein N terminus or away from it. We chose the first because it places the NH bond at the lipid–water interface, and enables hydrogen-bonding with water.
Acknowledgments
This research was supported by grants R37GM24266, RO1CA82864, RO1GM29754, and PO1GM56538 from the National Institutes of Health, and grants DAMD17-00-1-0506 and DAMD17-02-1-0313 from the Department of the Army. It used the Resource for Solid-State NMR of Proteins, supported by grant P41RR09731, from the Biomedical Research Technology Program, National Center for Research Resources, National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0211503.
References
- Almeida, F.C. and Opella, S.J. 1997. fd coat protein structure in membrane environments: Structural dynamics of the loop between the hydrophobic trans-membrane helix and the amphipathic in-plane helix. J. Mol. Biol. 270 481–495. [DOI] [PubMed] [Google Scholar]
- Bogusky, M.J., Schiksnis, R.A., Leo, G.C., and Opella, S.J. 1987. Protein backbone dynamics by solid state and solution 15N NMR spectroscopy. J. Magn. Reson. 72 186–190. [Google Scholar]
- Bogusky, M.J., Leo, G.C., and Opella, S.J. 1988. Comparison of the dynamics of the membrane bound form of fd coat protein in micelles and in bilayers. Proteins: Struct. Funct. Genetics 4 123–130. [DOI] [PubMed] [Google Scholar]
- Cross, T.A. and Opella, S.J. 1980. Structural properties of fd coat protein in sodium dodecyl sulfate micelles. Biochem. Biophys. Res. Commun. 92 478–484. [DOI] [PubMed] [Google Scholar]
- ———. 1983. Protein structure by solid-state NMR. J. Am. Chem. Soc. 105 306–308. [Google Scholar]
- ———. 1985. Protein structure by solid-state NMR: Residues 40–45 of bacteriophage fd coat protein. J. Mol. Biol. 182 367–381. [DOI] [PubMed] [Google Scholar]
- Glucksman, M.J., Bhattacharjee, S., and Makowski, L. 1992. Three-dimensional structure of a cloning vector: X-ray diffraction studies of filamentous bacteriophage M13 at 7 Å resolution. J. Mol. Biol. 226 455–470. [DOI] [PubMed] [Google Scholar]
- Henry, G.D. and Sykes, B.D. 1992. Assignment of amide 1H and 15N NMR resonances in detergent-solubilized M13 coat protein: A model for the coat protein dimer. Biochemistry 31 5284. [DOI] [PubMed] [Google Scholar]
- Ketchem, R.R., Hu, W., and Cross, T.A. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 261 1457–1460. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Leo, G.C., Colnago, L.A., Valentine, K.G., and Opella, S.J. 1987. Dynamics of fd coat protein in lipid bilayers. Biochemistry 26 854–862. [DOI] [PubMed] [Google Scholar]
- Marassi, F.M. 2001. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys. J. 80 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi, F.M. and Opella, S.J. 2000. A solid-state NMR index of membrane protein helical structure and topology. J. Magn. Reson. 144 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2002. Using Pisa pies to resolve ambiguities in angular constraints from PISEMA spectra of aligned proteins. J. Biomol. NMR 23 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi, F.M., Ramamoorthy, A., and Opella, S.J. 1997. Complete resolution of the solid-state NMR spectrum of a uniformly 15N-labeled membrane protein in phospholipid bilayers. Proc. Natl. Acad. Sci. 94 8551–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin, D.A., Hale, R.D., Nave, C., and Citterich, M.A. 1994. Molecular models and structural comparisons of native and mutant class I filamentous bacteriophages Ff (fd, f1, MI3), Ifl and Ike. J. Mol. Biol. 235 260–286. [DOI] [PubMed] [Google Scholar]
- McDonnell, P.A., Shon, K., Kim, Y., and Opella, S.J. 1993. fd coat protein structure in membrane environments. J. Mol. Biol. 233 447–463. [DOI] [PubMed] [Google Scholar]
- Mesleh, M.F., Veglia, G., DeSilva, T.M., Marassi, F.M., and Opella, S.J., 2002. Dipolar waves as NMR maps of protein structure. J. Am. Chem. Soc. 124 4206–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oas, T.G., Hartzell, C.J., Dahlquist, W., and Drobny, G.P. 1987. The amide 15N chemical shift tensors of four peptides determined from 13C dipole-coupled chemical shift powder patterns. J. Am. Chem. Soc. 109 5962–5966. [Google Scholar]
- Opella, S.J., Stewart, P.L., and Valentine, K.G. 1987. Protein structure by solid-state NMR Spectroscopy. Q. Rev. Biophys. 19 7–49. [DOI] [PubMed] [Google Scholar]
- Opella, S.J., Marassi, F.M., Gesell, J.J., Valente, A.P., Kim, Y., Oblatt-Montal, M., and Montal, M. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat. Struct. Biol. 6 374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella, S.J., Ma, C., and Marassi, F.M. 2001. NMR of membrane associated peptides and proteins. Methods Enzymol. 339 285–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavoine, C.H.M., Christiaans, B.E.C., Folmer, R.H.A., Konings, R.N.H., and Hilbers, C.W. 1998. Solution structure of the M13 major coat protein in detergent micelles: A basis for a model of phage assembly involving specific residues. J. Mol. Biol. 282 401–419. [DOI] [PubMed] [Google Scholar]
- Quine, J.R. and Cross, T.A. 2000. Transmembrane protein structure from NMR. Concepts Magn. Reson. 12 71. [Google Scholar]
- Ramamoorthy, A., Wu, C.H., and Opella, S.J. 1997. Magnitudes and orientations of the principal elements of the 1H chemical shift, 1H–15N dipolar coupling, and 15N chemical shift interaction tensors in 15N-tryptophan and 15N-histidine side chains determined by three-dimensional solid-state NMR spectroscopy. J. Am. Chem. Soc. 119 10479–10486. [Google Scholar]
- Sayle, R. and Milner-White, E.J. 1995. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 20 374–376. [DOI] [PubMed] [Google Scholar]
- Schiffer, M., Chang, C.H., and Stevens, F.J. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5 213–214. [DOI] [PubMed] [Google Scholar]
- Tycko, R., Stewart, P.L., and Opella, S.J. 1986. Peptide plane orientations determination by fundamental and overtone nitrogen 14 NMR. J. Am. Chem. Soc. 108 5419–5425. [Google Scholar]
- Van de Ven, F.J.M., Os, J.W.M., Aelen, J.M.A., Wymenja, S.S., Remerowski, M.L., Konings, C.W., and Hilbers, R.N.H. 1993. Assignment of 1H and backbone 13C resonances in detergent-solubilized M13 coat protein via multinuclear multidimensional NMR: A model for the coat protein monomer. Biochemistry 32 8322. [DOI] [PubMed] [Google Scholar]
- Wang, J., Denny, J., Tian, C., Kim, S., Mo, Y., Kovacs, F., Song, Z., Nishimura, K., Gan, Z., Fu, R., et al. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144 162–167. [DOI] [PubMed] [Google Scholar]
- Wang, J., Kim, S., Kovacs, F., and Cross, T.A. 2001. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 10 2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh, J.S. 1976. Uncoupling of local field spectra in nuclear magnetic resonance: Determination of atomic positions in solids. Proc. Natl. Acad. Sci. 73 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K.A., Farrow, N.A., Deber, C.M., and Kay, L.E. 1996. Structure and dynamics of bacteriophage IKe major coat protein in MPG micelles by solution NMR. Biochemistry 35 5145–5157. [DOI] [PubMed] [Google Scholar]
- Wu, C., Ramamoorthy, A., and Opella, S.J. 1994. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. A 109 270–272. [Google Scholar]
- Wu, C., Ramamoorthy, A., Gierasch, L.M., and Opella, S.J. 1995. Simultaneous characterization of the amide 1H chemical shift, 1H–15N dipolar, and 15N chemical shift interaction tensors in a peptide bond by three-dimensional solid-state NMR spectroscopy. J. Am. Chem. Soc. 117 6148–6149. [Google Scholar]