

Abstract
Glu-60 of the zinc-dependent Thermoanaerobacter brockii alcohol dehydrogenase (TbADH) is a strictly conserved residue in all members of the alcohol dehydrogenase (ADH) family. Unlike most other ADHs, the crystal structures of TbADH and its analogs, ADH from Clostridium beijerinckii (CbADH), exhibit a unique zinc coordination environment in which this conserved residue is directly coordinated to the catalytic zinc ion in the native form of the enzymes. To explore the role of Glu-60 in TbADH catalysis, we have replaced it by alanine (E60A-TbADH) and aspartate (E60D-TbADH). Steady-state kinetic measurements show that the catalytic efficiency of these mutants is only four- and eightfold, respectively, lower than that of wild-type TbADH. We applied X-ray absorption fine-structure (EXAFS) and near-UV circular dichroism to characterize the local environment around the catalytic zinc ion in the variant enzymes in their native, cofactor-bound, and inhibited forms. We show that the catalytic zinc site in the studied complexes of the variant enzymes exhibits minor changes relative to the analogous complexes of wild-type TbADH. These moderate changes in the kinetic parameters and in the zinc ion environment imply that the Glu-60 in TbADH does not remain bound to the catalytic zinc ion during catalysis. Furthermore, our results suggest that a water molecule replaces this residue during substrate turnover.
Keywords: Alcohol dehydrogenase, metalloenzyme, glutamate, active site, X-ray absorption
Interconversions of alcohols, aldehydes, and ketones are essential processes in both prokaryotes and eukaryotes. Several catalytic mechanisms for alcohol dehydrogenases (ADHs) have been proposed based on accumulating structural and spectroscopic evidence gathered from the most-studied enzyme, horse liver ADH (HLADH; Bertini 1986; Wilkinson 1987). The oxidation of alcohols requires a net removal of two hydrogen atoms from the substrate. This dehydrogenation process is known to proceed by coupled processes of proton abstraction and hydride ion transfer. The two main classes of structural mechanisms based on a proton relay system that have been proposed for HLADH differ specifically in the hypothesized coordination of the zinc ion during catalysis. One mechanism involves the displacement of the zinc-bound water by the alcohol substrate, and so, the zinc ion remains tetrahedrally coordinated during catalysis (Dunn et al. 1975; Eklund et al. 1982). Alternatively, the substrate molecule is added to the tetrahedral zinc ion to form penta-coordinated zinc (Dworschack and Plapp 1977; Makinen et al. 1983).
Thermoanaerobacter brockii ADH (TbADH) is a medium-chain, NADP+-linked (Lamed and Zeikus 1981) class-A enzyme (Peretz et al. 1993) that reversibly catalyzes the oxidation of secondary alcohols to the corresponding ketones. TbADH is a tetramer comprising four identical subunits, each containing 352-amino-acid residues (molecular weight of ∼38,000 D per subunit). It is an important and interesting ADH, because it is stable at high temperatures (up to 85°C), tolerates organic solvents well, shows broad specificity and high reactivity toward secondary alcohols, and has low reactivity toward primary alcohols. The overall three-dimensional structures of TbADH and its analog, ADH from Clostridium beijerinckii (CbADH), were studied by X-ray crystallography (Korkhin et al. 1998). CbADH and TbADH have 75% sequence identity and similar three-dimensional structures.
The crystal structure of apo-CbADH (cofactor free) shows that the single zinc ion in the catalytic site is bound to Cys-37, His-59, Asp-150, and Glu-60. Unlike most reported structures of apo ADHs (e.g., HLADH; Colonna-Cesari et al. 1986), human glutathione-dependent formaldehyde dehydrogenase (FDH; Sanghani et al. 2002), the conserved Glu-60 residue is bound directly to the zinc ion instead of a water molecule (Korkhin et al. 1998). This residue is strictly conserved in all ADH family, in which it is designated as Glu-68 (Sun and Plapp 1992). Interestingly, the crystal structures of CbADH and TbADH, complexed with NADP+ (holo-CbADH and holo-TbADH, respectively), show that the Glu-60 ligand is overtaken by a slight conformational change (Fig. 1 ▶). This results in an increase in the interatomic distance (3.36 to 5.36 Å) between the catalytic zinc ion and the nearest oxygen of Glu-60 (Korkhin et al. 1998); the zinc ion remained coordinated to the rest of its first-shell ligand residues. However, extended X-ray absorption fine-structure (EXAFS) studies on holo-TbADH (in solution) showed that the metal ion maintained its tetrahedral coordination with one Zn-S and three Zn-O/N contributions from its first-shell ligand residues (Kleifeld et al. 2000a). Interestingly, a direct coordination of the conserved Glu-60 to the catalytic zinc ion in other ADH family members was recently observed in the crystal structures of the sorbitol dehydrogenase (Johansson et al. 2001), and ADH from the hyperthermophilic archaeon Sulfolobus solfataricus (Esposito et al. 2002). This raised the notion that the conserved Glu residue in these enzymes is essential for catalysis.
Figure 1.

Schematic view of the active site in TbADH. The active site representation of TbADH in the ternary complex with NADP+ (PDB: 1YKF). The catalytic zinc ion is bound to four protein residues: Cys-37, His-59, Asp-150, and Glu-60. The binding of Glu-60 to the zinc ion is not conclusive in the holo-TbADH crystal structure (Korkhin et al. 1998).
The role of the analogous conserved Glu-68 residue in the catalysis of HLADH and yeast ADH (YADH) was studied by both experimental (Ganzhorn and Plapp 1988) and theoretical approaches (Ryde 1995a,b). Substitution of this residue in YADH with glutamine resulted in a severe loss in enzyme activity (100-fold lower than wild-type; Ganzhorn and Plapp 1988), indicating that the conserved glutamate residue is important in the catalysis of ADHs, although not directly bound to the catalytic zinc ion. The proposed catalytic role of this residue was derived from theoretical calculations, which indicated that the conserved Glu-68 residue in HLADH is intermittently coordinated to the catalytic zinc ion during catalysis to facilitate the binding of the alcohol substrate and product release (Ryde 1995a,b). Similar ligand exchange mechanism was proposed based on the crystal structures of the apo, binary, and inhibitory ternary complexes of FDH (Sanghani et al. 2002). However, in contrast to the other well-studied members of the ADH family, FDH obeys a random bi-bi kinetic mechanism rather than an ordered bi-bi mechanism (Sanghani et al. 2000).
It was suggested, albeit inconclusively, that the catalytic mechanism of TbADH may be different than the one assigned for the HLADH (Korkhin et al. 1998). The results on the role of Glu-68 in HLADH and YADH, together with the crystallographic evidence of the binding of Glu-60 to the catalytic zinc ion in TbADH (Korkhin et al. 1998), inspired us to look into the catalytic role of this residue in TbADH. Specifically, we replaced the Glu-60 residue in TbADH with alanine (E60A-TbADH) and with aspartate (E60D-TbADH). In the present study, we report comparative studies of these variant and wild-type enzymes by using X-ray absorption spectroscopy near-UV circular dichroism (CD), kinetics, and modeling experiments.
Results
Protein purification and initial analysis
Overexpression of E60A-TbADH and E60D-TbADH in Escherichia coli yielded intact enzymes. The mutated proteins were obtained in similar yields as the wild-type (∼200 mg/L). All enzymes used in this study were purified to homogeneity as judged by SDS-PAGE (Laemmli 1970). The purified protein migrated as a single homogeneous band with an estimated molecular weight of 38 kD (data not shown). The enzymes were extensively dialyzed against metal-free buffer (see Materials and Methods) and analyzed for zinc content by using inductively coupled plasma atomic emission spectroscopy (ICP-AES). Both mutants contain one zinc atom/subunit, which is the same amount of zinc as the wild-type enzyme (Bogin et al. 1997). The activity of the mutated enzymes and their structural-function relationships were characterized in the presence of their cofactor NADP+ and the inhibitor dimethyl sulfoxide (DMSO; Perlman and Wolff 1968; Kleifeld et al. 2000a).
Kinetic characterization of TbADH mutants
Initial velocity studies provided the kinetics constants for 2-propanol oxidation (Table 1). The E60A-TbADH and the E60D-TbADH mutations decreased the turnover number by only twofold and eightfold, respectively. The Michaelis constants of E60A-TbADH for NADP+ (Ka) and 2-propanol (Kb) were about twofold higher than the corresponding constants of wild-type TbADH (Table 1). Interestingly, in the E60D-TbADH mutant, the Ka decreased eightfold, whereas the Kb remained the same as for wild-type TbADH. Thus, in E60A-TbADH and E60D-TbADH, the enzymatic efficiency (kcat/Kb) was reduced only by approximately fourfold and eightfold, respectively. In addition, the specific activity of the variant enzymes in elevated temperatures exhibited the same trend as observed by the kinetics (Fig. 2 ▶).
Table 1.
Kinetic constants for mutant enzymes
| Kinetic constants | Wild-type | E60A | E60D |
| Ka, μM | 83 | 140 | 11 |
| Kb, mM | 0.22 | 0.43 | 0.20 |
| Kia, μM | 9 | 67 | 2.5 |
| kcat μmole min−1 mg−1 | 48 | 23 | 5.7 |
| kcat/Kb, min−1 | 218 | 53.5 | 28.5 |
Kinetic constants were determined at 40°C in 150 mM Tris-HCl buffer (pH 9.0) by varying the substrate and cofactor concentrations in initial velocity studies as described in Materials and Methods. Ka and Kb are the Michaelis constants for NADP+ and 2-propanol, respectively. Kia is the dissociation constant for NADP+; kcat is the turnover number for 2-propanol oxidation. Errors for fitted parameters were in the range of 5% to 10%.
Figure 2.

Kinetic measurement of wild-type TbADH, E60A-TbADH, and E60D-TbADH. Enzymatic activity was measured after the absorption increase at 340 nm (ɛ340 = 6.2 mM−1 cm−1) for the formation of NADPH. One unit of ADH activity is defined as the amount of enzyme that catalyzes the formation of 1 μmole of NADPH per minute under initial velocity conditions. The specific activity for oxidation of 2-butanol by wild-type TbADH (black), E60A-TbADH (empty), and E60D-TbADH (hatched) was monitored at different temperatures from 25°C to 60°C. Reaction conditions were 50 nM enzyme, 0.5 mM NADP+ (above saturation concentration), 150 mM 2-butanol, and 100 mM Tris-HCl (pH 9.0) in total volume of 1 ml.
All together, these results show that substitution of the Glu-60 with the "inert" alanine and the charged aspartate residues resulted in mild changes in binding affinities (Ka, Kb) and turnover numbers. In contrast to similar mutations in the conserved Glu residue in YADH (Ganzhorn and Plapp 1988), it failed to significantly reduce enzyme activity. Importantly, replacement of the other zinc binding ligands in TbADH (Asp-150, Cys-37, and His-59) to alanine resulted in complete abolishment of activity and hindered the reconstitution of the catalytic zinc ion to the enzyme (Bogin et al. 1997).
X-ray absorption spectroscopy studies
The role of Glu-60 in TbADH reaction mechanism was further studied by EXAFS spectroscopy. EXAFS is a valuable technique for structural elucidation of a variety of metal sites in metalloproteins (Scott 1985; Kleifeld et al. 2000b, 2001b). The technique measures the transition of core electronic states of the metal to excited electronic states or continuum states. X-ray absorption spectroscopy (XAS) studies can be conducted in any state of the matter, for example, gas, liquid, or crystalline phase. Spectral analysis near the electronic transition (XANES) provides information on the charge state and geometry of the metal. Spectral analysis above the absorption edge, in the EXAFS region, provides complementary structural information, such as coordination numbers, types, and distances from neighboring atoms to the central (absorbing) atom. In addition, XAS is an excellent structural tool to probe the d10 zinc ion, which is generally spectroscopically silent (Scott 1985).
EXAFS analysis
We studied the local structure around the catalytic zinc ion in various complexes of E60A-TbADH and E60D-TbADH, with the cofactor NADP+ and the inhibitor DMSO (see Materials and Methods). Standard curve-fitting procedures were used to fit the FEFF7 rhetorical models to the real and imaginary parts of the Fourier-transformed χ(k) (Rehr et al. 1991; Stern et al. 1995; Zabinsky et al. 1995). The results of the EXAFS data analysis and the fitting parameters are described in Table 2 and Figure 3 ▶.
Table 2.
EXAFS curve-fitting results
| Zn-N/O | Zn-N/O | |||||||||
| Fit # | Sample | Reduced χ2 | ΔE0 | N | R [Å] | σ2 | ΔE0 | N | R [Å] | σ2 |
| Wild-type TbADHa | 0 [F] | 1 [F] | 1.81 (3) | 1.6E–05 | 0.0 [F] | 2 [F] | 2.00 (2) | 3.5E–0.3 | ||
| Wild-type TbADH-complexa | −2.7 [F] | 2 [F] | 1.83 (3) | 1.6E–05 | −2.7 [F] | 2 [F] | 2.05 (3) | 1.4E–0.3 | ||
| 1 | E60A | 7.5 | 0.8 [F] | 3 [F] | 2.00 (1) | 3.3E–03 | ||||
| 2 | E60A | 12.6 | 0.8 [F] | 4 [F] | 2.02 (1) | 8.6E–03 | ||||
| 3 | E60A | 8.2 | 0.8 [F] | 3.0 (2) | 2.01 [F] | 4.6E–03 | ||||
| 4 | E60A-Complex | 8.7 | 0.2 [F] | 1 [F] | 1.90 (3) | 2.5E–04 | 0.2 [F] | 2 [F] | 1.99 (5) | 1.0E–06 |
| 5 | E60A-Complex | 3.2 | 0.2 [F] | 1 [F] | 1.87 (1) | 3.5E–03 | 0.2 [F] | 3 [F] | 1.97 (1) | 1.4E–03 |
| 6 | E60A-Complex | 3.0 | 0.2 [F] | 1 [F] | 1.86 (1) | 1.2E–02 | 0.2 [F] | 4 [F] | 1.98 (1) | 2.1E–03 |
| 7 | E60A-Complex | 2.3 | 0.2 [F] | 1 [F] | 1.87 (2) | 7.3E–03 | 0.2 [F] | 3.5 (1.0) | 1.98 [F] | 2.1E–03 |
| 8 | E60D | 8.7 | 3.3 [F] | 3 [F] | 1.98 (1) | 2.0E–03 | ||||
| 9 | E60D | 17.3 | 3.3 [F] | 4 [F] | 1.99 (1) | 3.8E–03 | ||||
| 10 | E60D | 39.3 | 3.3 [F] | 5 [F] | 2.00 (1) | 6.9E–03 | ||||
| 11 | E60D | 9.5 | 3.3 [F] | 3.0 (2) | 1.99 [F] | 6.2E–03 | ||||
| 12 | E60D-Complex | 28 | −2.5 [F] | 1 [F] | 1.86 (8) | 2.4E–03 | −2.5 [F] | 2 [F] | 1.99 (5) | 1.0E–06 |
| 13 | E60D-Complex | 6.0 | −2.5 [F] | 1 [F] | 1.80 (2) | 3.0E–06 | −2.5 [F] | 3 [F] | 1.97 (1) | 8.0E–04 |
| 14 | E60D-Complex | 9.5 | −2.5 [F] | 1 [F] | 1.77 (2) | 3.0E–03 | −2.5 [F] | 4 [F] | 1.97 (1) | 4.8E–03 |
| 15 | E60D-Complex | 7.2 | −2.5 [F] | 1 [F] | 1.81 (2) | 1.0E–06 | −2.5 [F] | 3.0 (3) | 1.98 [F] | 1.3E–03 |
Table 2.
Continued
| Zn-S | Zn-C | Zn-C | |||||||||
| ΔE0 | N | R [Å] | σ2 | ΔE0 | N | R [Å] | σ2 | ΔE0 | N | R [Å] | σ2 |
| 0 [F] | 1 [F] | 2.24 (1) | 1.0E–06 | ||||||||
| −2.7 [F] | 1 [F] | 2.25 [F] | 1.0E–04 | ||||||||
| 0.8 [F] | 1 [F] | 2.25 (2) | 4.4E–03 | 7.0 [F] | 2 [F] | 2.77 (1) | 3.3E–03 | 7.0 [F] | 3 [F] | 3.08 [F] | 3.3E–03 |
| 0.8 [F] | 1 [F] | 2.23 (2) | 1.6E–03 | 7.0 [F] | 2 [F] | 2.77 (2) | 2.0E–04 | 7.0 [F] | 3 [F] | 3.08 [F] | 1.8E–03 |
| 0.8 [F] | 1 [F] | 2.24 (2) | 2.5E–06 | 7.0 [F] | 2 [F] | 2.77 (2) | 6.0E–04 | 7.0 [F] | 3 [F] | 3.08 [F] | 2.0E–03 |
| 0.2 [F] | 1 [F] | 2.30 (3) | 3.6E–03 | 0.2 [F] | 1 [F] | 2.71 [F] | 3.0E–03 | 0.2 [F] | 2 [F] | 2.95 [F] | 3.0E–03 |
| 0.2 [F] | 1 [F] | 2.27 (2) | 3.6E–03 | 0.2 [F] | 1 [F] | 2.71 [F] | 3.6E–03 | 0.2 [F] | 2 [F] | 2.95 [F] | 3.6E–03 |
| 0.2 [F] | 1 [F] | 2.27 (1) | 2.2E–03 | 0.2 [F] | 1 [F] | 2.71 [F] | 4.5E–03 | 0.2 [F] | 2 [F] | 2.95 [F] | 4.5E–03 |
| 0.2 [F] | 1 [F] | 2.27 (1) | 2.2E–03 | 0.2 [F] | 1 [F] | 2.71 [F] | 4.1E–03 | 0.2 [F] | 2 [F] | 2.95 [F] | 4.1E–03 |
| 3.3 [F] | 1 [F] | 2.30 (1) | 1.0E–06 | −6.0 [F] | 2 [F] | 2.83 (2) | 3.9E–03 | ||||
| 3.3 [F] | 1 [F] | 2.29 (1) | 3.2E–03 | −6.0 [F] | 2 [F] | 2.88 (4) | 3.6E–03 | ||||
| 3.3 [F] | 1 [F] | 2.26 (1) | 4.8E–03 | −6.0 [F] | 2 [F] | 2.91 (6) | 5.0E–03 | ||||
| 3.3 [F] | 1 [F] | 2.30 (1) | 1.0E–06 | −6.0 [F] | 2 [F] | 2.83 (2) | 2.8E–03 | ||||
| −2.5 [F] | 1 [F] | 2.26 (3) | 1.8E–03 | 7.2 [F] | 2 [F] | 3.03 (9) | 1.2E–02 | ||||
| −2.5 [F] | 1 [F] | 2.28 (1) | 3.8E–03 | 7.2 [F] | 2 [F] | 3.05 (2) | 1.2E–02 | ||||
| −2.5 [F] | 1 [F] | 2.26 (1) | 5.5E–03 | 7.2 [F] | 2 [F] | 3.06 (5) | 1.4E–02 | ||||
| −2.5 [F] | 1 [F] | 2.26 (1) | 5.0E–03 | 7.2 [F] | 2 [F] | 3.04 (5) | 1.4E–02 | ||||
Results of EXAFS curve-fitting analysis of apo-E60A (fits 1–3), apo-E60D (fits 8–11); E60A-TbADH in complex with NADP+ + DMSO, designated as E60A-complex (fits 4–7); and E60D-TbADH in complex with NADP+ + DMSO, designated as E60D-complex (fits 12–15). F stands for fixed and indicates that the respective parameter was fixed in the fit model; R, distance of atoms from the zinc ion in Angstoms; N, the number of atoms; σ2, the Debye-Waller factor; ΔE0, the correction to the energy origin. Fixing of specific parameter was executed only after varying it and choosing its best value. The uncertainties of the R parameter are shown in parentheses.
a Fitting results of wild-type TbADH and wild-type TbADH in complex with NADP+ + DMSO (designated wild-type TbADH-complex) are from Kleifeld et al. (2000a).
Figure 3.

EXAFS curve-fitting results of the zinc site in E60A-TbADH and E60D-TbADH in their apo-forms and in complex with NADP+ + DMSO. EXAFS curve-fitting analysis of apo-E60A-TbADH (A), apo-E60D-TbADH (B), holo-E60A-TbADH + DMSO (C), and holo-E60D + DMSO (D) to simulated theoretical zinc-ligand contributions of TbADH active site. The solid line indicates the experimental data, and the open circle line represents the best fits. Inserts show the best fits in back transformed representation. Experimental data were extracted and normalized by using the UWXAFS analysis package. Theoretical XAFS signal was constructed of apo-CbADH sub-unit B active site data (PDB entry 1PED) and calculated by using FEFF7 (Rehr et al. 1991; Zabinsky et al. 1995). The fitting analysis procedure is described in text. The quality of the fits and parameters are presented in Table 2.
Distance and coordination-number parameters obtained for the various E60A-TbADH and E60D-TbADH complexes were determined by analyzing each data set separately, avoiding the use of heavy constraints. The stability of the bond-distances solutions derived for each structure was examined by varying the initial conditions of the distance and thermal disorder (Debye-Waller) parameters, while examining the contribution to the fit quality parameters: χ2 and the R-factor values. The possibility for a higher coordination number (more than four ligands) at the active zinc site was examined by either varying or fixing and stepping the coordination number, while testing the stability of the fitting results (Table 2). The ΔE0 and the reduction factor (S0) were optimized for each fit either by varying or stepping analysis.
The local structure around the catalytic zinc ion in the apo-E60A-TbADH enzyme, determined by our EXAFS data-analysis procedures (Table 2), was best fitted with three Zn-O/N contributions with averaged bond distance of 2.00 ± 0.01 Å and one Zn-S contribution at 2.25 ± 0.02 Å. Any attempt to resolve the Zn-O/N distances failed, owing to high correlation in the ΔR parameters in the fit. The inability of alanine to coordinate the zinc ion and the relative high activity of this mutant in the presence of substrates (Table 1; Fig. 2 ▶) suggest that the forth-coordinated ligand at the zinc ion in E60A-TbADH is water/OH− molecule, which binds the zinc ion via the oxygen molecule of the water/OH− ligand (Table 2). Fitting of E60A-TbADH with models of higher coordination number were unstable and resulted in higher Debye-Waller and χ2 values (Table 2, fit 2). Similarly, the zinc ion in apo-E60D-TbADH was found to be coordinated to three Zn-O/N at averaged bond distance of 1.98 ± 0.01 Å and one Zn-S contribution at 2.30 ± 0.01 Å (Table 2).
To further study the local structure of the catalytic zinc ion in the variant mutants in their ternary complexes, we analyzed the EXAFS of E60A- and E60D-TbADH mutants with the inhibitor DMSO and NADP+. The activity of the variant enzymes toward 2-propanol was inhibited by DMSO, as observed for wild-type TbADH (Kleifeld et al. 2000a; data not shown).
The first coordination shell of the zinc ion in the E60A-TbADH + NADP+ + DMSO (holo-E60A-TbADH + DMSO) complex was best fitted with penta-coordination structure including one Zn-O/N at 1.87 ± 0.01 Å, three Zn-O/N at 1.97 ± 0.01 Å, and one Zn-S at 2.27 ± 0.02 Å. Stepping the coordination number from four to six in these fits resulted in either unacceptable Debye-Waller factors (>0.01 for first-shell coordination) or in higher χ2 values and unstable fits. Varying the number of the Zn-O/N contributions resulted in total number of 3.5 ± 1 (Table 2). which is in agreement with penta-coordination structure within the error limits.
Consistently, the local structure around the zinc ion in the holo-E60D-TbADH + DMSO complex was best fitted to penta-coordination structure, with one Zn-O/N at 1.80 ± 0.02 Å, three Zn-O/N at 1.97 ± 0.01 Å, and one Zn-S contribution at 2.28 ± 0.01 Å. Stepping the coordination number in these fits resulted in high χ2 values for total of four-coordination structure and unstable fits for six-coordination structure. Varying the number of the equidistance Zn-O/N contributions in the fit resulted in 3.0 ± 0.3, which is in good agreement with total coordination number of five. The observed distortions in coordination number and distances detected at the catalytic zinc ion in the variant enzymes on the binding of DMSO and the inhibition of their catalytic activity indicate direct binding of the DMSO molecule to the zinc ion. In addition, the binding of DMSO changes the Zn-O/N bond distance distribution, resulting in one Zn-O/N contribution in the order of 1.83 Å and three Zn-O/N contributions at 1.97 Å. Overall, these results show that the binding of DMSO changes the coordination number of the catalytic zinc complex from four to five. Importantly, similar penta-coordination number was detected in the complex of TbADH + NADP+ + DMSO (holo-TbADH + DMSO; Kleifeld et al. 2000a). Yet, the Zn-O/N distance distribution is different, indicating the effect of the point mutations on the zinc coordination sphere (Table 2, fits 5,13). These results further support the kinetic analysis of the variant enzymes (Table 1) by showing only mild differences in comparison to the wild-type TbADH structures.
Edge spectral analysis
Although difficult to interpret quantitatively, near-edge spectra are very sensitive to the geometry and nature of the local metal environment. Changes in edge position are also a good indicator for changes in coordination number in this system, as we demonstrated before (Kleifeld et al. 2000a). Specifically, the near-edge spectra of the wild-type TbADH were shown to exhibit a distinct shift in edge position to higher energy on the binding of DMSO. Similarly, Figure 4 ▶ demonstrates that the edge position of the variant enzymes is shifted 0.7 to 1.0 eV to higher energy on the binding of DMSO to the catalytic zinc ion. These results support the EXAFS analysis of the various complexes of the variant enzymes, indicating the direct coordination of DMSO to the catalytic zinc ion to form penta-coordinated complex. To further examine the inhibitory effect of DMSO on the catalytic site ternary structure of the E60A-TbADH and E60D-TbADH mutants, we conducted near-UV CD studies in solution.
Figure 4.

Zinc K edge spectra of E60A-TbADH and E60D-TbADH. Raw normalized XAS data in the Zn K-edge region of E60A-TbADH (A) and E60D-TbADH (B) in the apo-from (solid line) and in complex with NADP+ + DMSO (dashed line). The edge position of both mutants was shifted to higher energy on binding of DMSO.
Near-UV CD studies
CD spectroscopy measures differences in the absorption of left-handed polarized light versus right-handed polarized light that arises owing to structural asymmetry. Practically, CD spectroscopy conducted at the far-UV spectral region (190 to 250 nm) probes the peptide bond. The CD spectrum of a protein in the near-UV spectral region (250 to 350 nm) probes the aromatic amino acids and disulfide bonds. Therefore, near UV-CD experiments can provide certain aspects of tertiary structure. Specifically, signals in the region from 255 to 265 nm are attributable to phenylalanine residues. Signals from 265 to 283 nm are attributable to tyrosine, and those from 285 to 300 nm are attributable to tryptophan (Strickland 1974; Woody 1995).
Although the far UV-CD spectra of wild-type TbADH, holo-TbADH, and holo-TbADH + DMSO complexes did not show any significant differences, the near-UV CD spectra of these complexes exhibit distinct changes on cofactor and DMSO binding (Kleifeld et al. 2000a). These studies provided the basis for further investigation of the near-UV CD spectra of the E60A- and E60D-TbADH mutants.
Figure 5 ▶ shows the near-UV CD spectra of various complexes of E60A-TbADH and E60D-TbADH with both NADP+ and DMSO, in which each spectrum is compared with the wild-type TbADH analogous complex. The apo-E60A-TbADH spectrum is distinct in its features when compared with the apo-TbADH spectrum. However, the spectra of E60A-TbADH, holo-E60A-TbADH, and holo-E60A-TbADH + DMSO show similar spectral trends, among the various transitions, as observed for the same complexes of wild-type TbADH (Fig. 5 ▶). On addition of NADP+ (in both holo-E60A-TbADH and holo-TbADH), the tyrosine-associated peak at 275 nm is decreasing, followed by a more significant change at 260 nm, which is assigned to the contribution from the aromatic rings of the cofactor (Kleifeld et al. 2000a). Specifically, the large change at 275 nm is presumably owing to the 120-degree rotation of tyrosine 218, which is induced on cofactor binding, as indicated by crystallography (Korkhin et al. 1998). The addition of DMSO in both complexes of holo-TbADH and holo-E60A-TbADH partially restores their native spectra by slight increase of the peak at 275 nm. The overall ellipticity signal in the spectra of E60A-TbADH complexes is stronger than that of the wild-type TbADH spectra in the tyrosine region. This may be explained by the net decrease in steric effect on the relevant tyrosines in the active site of TbADH, owing to the absence of the Glu-60 residue in the catalytic pocket and the lack of ionic bond to Lys-346.
Figure 5.
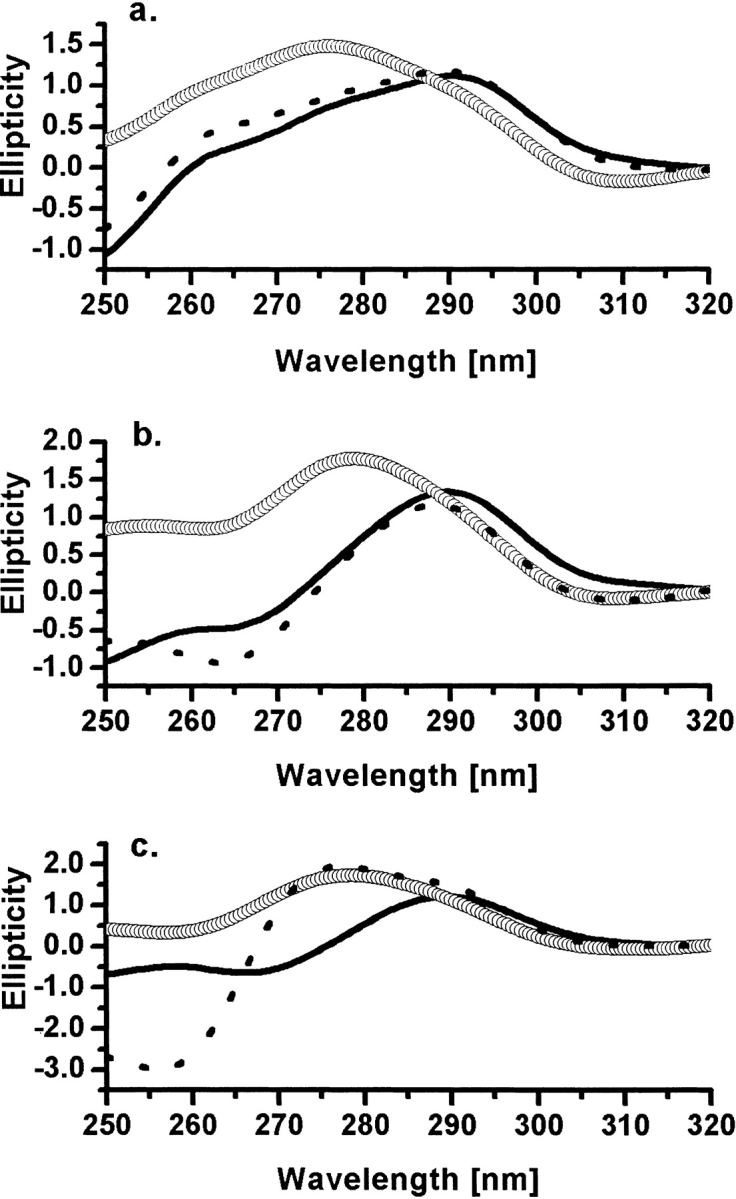
Near-UV CD studies. (A) Apo-forms of wild-type TbADH (solid line), E60A-TbADH (circles), and E60D-TbADH (dashed line). (B) Holo-forms of wild-type TbADH (solid line), E60A-TbADH (circles), and E60D-TbADH (dashed line). (C) Ternary complex with NADP+ + DMSO of wild-type TbADH (solid line), E60A-TbADH (open circles), and E60D-TbADH (dashed line). Spectra were recorded at 25°C. The relevant background spectra were subtracted from the raw spectra of each sample, and the residual spectra were normalizing to the signal at 315 nm.
The near-UV CD spectra of wild-type TbADH and E60D-TbADH are similar in both native and cofactor-bound forms. However, the addition of DMSO to holo-E60D-TbADH resulted in distinct spectral features. These results may support the kinetic analysis of this mutant, indicating stronger binding of the cofactor in the catalytic pocket, possibly owing to enhanced charge stabilization by the aspartate residue.
Modeling studies
To gain better understanding of the conformational changes that are taking place in the active site of E60A-TbADH and E60D-TbADH, we preformed modeling studies by using the structures of apo- and holo-CbADH owing to their better atomic resolution (Korkhin et al. 1998). In the crystal structure of holo-CbADH (Protein Data Bank:1KEV subunit A), there are two water molecules adjacent to the catalytic zinc ion: HOH(21) at 4.86 Å from the zinc ion and HOH(25) at 4.45 Å. Examination of the different possible rotomers of Glu-60 reveals that that small dislocation of this residue creates open space near the catalytic zinc ion that can be filled by one of these water molecules. The elimination of Glu-60 by E60A mutation should enlarge this space and allow the diffusion of HOH(21) in the pocket. This presumably will bring HOH(21) to coordinate with the zinc ion, resulting in minor conformational changes of the other zinc-bound residues (Fig. 6A ▶). In contrast, the charged aspartate side-chain in the E60D mutant is shorter than glutamate, and therefore, this mutation is expected to effect the local charge distribution in the catalytic pocket. This may explain the lower Ka value obtained for this mutant, indicating a stronger binding of the positively charged cofactor. Retaining the tetrahedral coordination of the zinc ion in the E60D-TbADH mutant was accomplished only by dislocating the zinc ion toward the Asp-60 residue (Fig. 6B ▶). Previously, we constructed a model structure for the complex of wild-type TbADH + NADP+ + DMSO (Kleifeld et al. 2000a). In TbADH, the tetrahedral coordination of the zinc ion is distorted, and three out of the four ligands are almost coplanar. Such configuration allows the accommodation of DMSO as fifth ligand on the zinc ion to create a bipyramidal geometry. Specifically, we showed that DMSO binds the catalytic zinc ion opposite to the Glu-60 residue (Kleifeld et al. 2000a). Therefore, applying this model for the binding of DMSO to the catalytic zinc ion in E60A-TbADH and E60D-TbADH reveals that the mutation of Glu-60 does not affect the binding of DMSO, as demonstrated by our EXAFS results.
Figure 6.


Proposed active site configuration of E60A-TbADH and E60D-TbADH. (A) Schematic view in stereo of the active site configuration in E60A-TbADH mutant. Elimination of the Glu-60 residue creates an open space, which can be filled by adjacent water molecule. (B) E60D-TbADH mutant. Substitution of Glu-60 with Asp-60 resulted in a shift in the zinc ion position in order to retain the tetrahedral coordination of the metal ion.
Altogether, these results indicate that the E60A and E60D mutations in TbADH caused only a slight structural change in the active site topography. These mild structural distortions in the first coordination shell of the catalytic zinc ion are also reflected in the kinetic behavior of these enzymes.
Discussion
A great deal of experimental and theoretical work has been devoted to elucidating the reaction mechanism of ADHs (Eklund et al. 1974; Dunn et al. 1982; Makinen et al. 1983; Zeppezauer 1983; Formicka et al. 1992; Meijers et al. 2001). These enzymes have been the focus of intensive studies involving kinetics, biophysical, spectroscopical, and structural studies. However, the actual detailed reaction mechanism is still not clear. The major research objectives have been the role the catalytic zinc ion plays in catalysis, the mode of substrate binding, and the role of the catalytic metal-bound water during catalysis (Eklund et al. 1974; Makinen et al. 1983; Meijers et al. 2001). Two main types of structural mechanisms have been proposed that differ specifically in the hypothesized coordination of the zinc ion during catalysis. In both mechanisms, the coordination number of the catalytic zinc ion and the role of the coordinated water molecule (in HLADH) seems to be important for catalysis. However, whether the zinc ion changes its original tetrahedral coordination during turnover, and whether the water stays bound to the catalytic zinc ion during catalysis remained controversial issues (Meijers et al. 2001).
Unlike most ADHs, the crystal structures of TbADH and CbADH (Korkhin et al. 1998) indicate that the catalytic zinc ion in these bacterial ADHs is coordinated to three protein residues (Asp-150, His-59, and Cys-37) and to the conserved Glu-60, instead of the water/OH− molecule. Interestingly, the coordination of Glu-60 to the catalytic metal ion in both holo-CbADH and holo-TbADH is not conclusive, owing to its relatively long intraatomic distance from the catalytic zinc ion observed in these structures. However, no other protein residue or water molecule was found to complement the coordination of the Glu-60 residue with the zinc ion. These studies raised the possibilities that the reaction mechanism of TbADH may differ from other ADHs, and that the conserved Glu-60 residue in TbADH has critical role in catalysis. These hypotheses provided the basis for our structure-function studies reported in this work.
Kinetic studies of TbADH and its highly homologous secondary ADH from Thermoanaerobacter ethanolicus show that site-directed mutagenesis of the zinc-bound residues (Asp-150, His-59, and Cys-37) abolish the enzymatic activity and dramatically lower the zinc content of the mutated enzymes (Bogin et al. 1997; Burdette et al. 1997). These effects were dependent on the type of amino acid substitution. Nonchelating residues totally abolish the activity and the zinc content, and chelating residues exhibited moderate effect, which was mainly owing to lowering the zinc content in the enzyme (Bogin et al. 1997). Strikingly, our site-directed mutagenesis studies on Glu-60 reported here reveal a different scheme. Replacement of the conserved Glu-60 in TbADH with a nonchelating (alanine) and charged (aspartate) residues resulted in a moderate reduction in the kinetic behavior relative to the wild-type enzyme (Table 1). Previously reported site-directed mutagenesis of the conserved Glu-68 residue in YADH (equivalent to Glu-60 in TbADH) to glutamine resulted in a significant decrease in enzyme efficiency (∼100-fold decrease than wild-type-YADH) and the affinity to cofactor and substrate.
Glutamine is the same size as glutamate and can form hydrogen bonding. However, it lacks the charged carboxyl group. On the basis of these results, it was proposed that the net reduction in enzyme activity is owing to local electrostatic and structural changes in the active site (Ganzhorn and Plapp 1988). Hence, the charge on the Glu-68 residue in YADH is critical in mediating catalysis. In contrast, mutating the conserved Glu-60 residue in TbADH did not have such a dramatic effect on the enzymatic activity. The Ka and Kb values of the E60A-TbADH mutant indicate a moderate decrease in binding affinities of both cofactor and substrate (twofold). Interestingly, the less-active E60D-TbADH mutant exhibits better binding affinity for the cofactor than for the wild-type enzyme. The stronger binding of the cofactor hinders its release from the catalytic site and therefore slows down the rate of the reaction. This may result from shortening the amino acid side-chain by one carbon, which presumably alters the charge distribution in the catalytic pocket.
Our edge and EXAFS curve-fitting analysis of apo-E60A-TbADH and holo-E60A-TbADH + DMSO shows a similar structural organization at the catalytic zinc ion with the analogous complexes of wild-type TbADH (Kleifeld et al. 2000a). Specifically, a distinct Zn-O contribution was detected, in the absence of a potentially zinc-binding protein residue in E60A-TbADH complexes. In addition, this mutant exhibits similar near-UV-CD spectral changes, as was observed for the wild-type enzyme, during its interactions with cofactor and DMSO (Fig. 5 ▶). Therefore, we ruled out the possibility that another active site protein residue contributed the additional Zn-O bond distance. Modeling of the E60A mutant indicates that the replacement of Glu-60 by alanine creates an open space near the catalytic zinc ion (Fig. 6A ▶). This free space can be easily filled by a water/OH− molecule that coordinates to the zinc ion, as indicated by our kinetic and curve-fitting EXAFS analyses (Figs. 2–4 ▶ ▶ ▶; Table 2). Hence, the Zn-O bond distance contribution may be assigned to a water/OH− molecule that is bound to the catalytic zinc ion.
The E60A mutation eliminates the ionic bond between the Glu-60 and Lys-346 and, therefore, gives rise to accumulation of positive charge in the catalytic pocket that needs to be neutralized. This may be the reason to the lower binding affinities of both substrate and cofactor detected for this mutant (Table 1). In addition, this charge neutralization process may alter the position of aromatic residues in the catalytic pocket, which may explain the near-UV CD changes observed at the tyrosine level. These results indicate that the reaction mechanism of E60A-TbADH is mediated via a water/OH− molecule.
The substitution of Glu-60 with aspartate limited the flexibility of this residue, owing to its shorter side-chain; however, these residues have the same charge. Interestingly, this mutation resulted in a greater reduction in the enzyme activity (Table 1). Yet, the edge and EXAFS data analysis indicate that the coordination of the nearest shell ligands to the catalytic zinc site in E60D-TbADH is similar to the E60A-TbADH mutant and wild-type TbADH (Table 2). Specifically, a tetrahedral coordination is detected in the apo-form of the mutant around the zinc ion. This configuration is altered to penta-coordination structure on the binding of cofactor and DMSO. Our modeling studies indicate that the E60D mutation forces the zinc ion to coordinate to the Asp-60 carboxyl group while withdrawing it from its original position (Fig. 6B ▶). However, we cannot rule out the possibility of a water molecule bound between Asp-60 and the zinc ion.
All together, our structure-function studies demonstrate that the conserved Glu-60 in TbADH is not essential for catalysis. Therefore, we suggest that this residue may be replaced by a water molecule during catalysis. The involvement of water molecule is further supported by site-directed mutagenesis (cysteine 295 to alanine) and kinetic studies of the highly homologous ADH from T. ethanolicus, which show that water plays a key role in the enzyme catalysis (Heiss et al. 2001a,b).
Although not completely analogous to our system, theoretical studies by Ryde (1995b) demonstrated that the conserved Glu-68 residue in HLADH plays a dynamic role in the reaction mechanism. Specifically, it was proposed that Glu-68 might intermittently coordinate to the zinc ion via an energetically favored process. Such coordination may facilitate the exchange of ligands in the active site. Additional experimental evidence of the dynamic role of the conserved glutamate residue in ADHs was provided by the crystal structures of FDH, in which Glu-67 exchanges the zinc-bound water molecule in the ternary complex of the enzyme (Sanghani et al. 2002).
The involvement of a coordinated water/OH− molecule, which forms a penta-coordinated zinc ion in the productive complex of HLADH, was proposed previously, on the basis of spectroscopic studies (Makinen et al. 1983). In addition, recent high-resolution crystallographic studies of HLADH demonstrate a direct binding of a water/OH− molecule to the zinc ion (in the holo-enzyme), which coordinates to the nicotinamide ring of the cofactor (Meijers et al. 2001). Importantly, these studies propose a refined reaction mechanism for HLADH, which include a penta-coordinated zinc ion as an intermediate complex in which both water and substrate are bound to the metal ion. Previously reported results on TbADH catalysis (Kleifeld et al. 2000a, 2001a) and the data presented here strongly support the formation of penta-coordinated intermediate states during TbADH turnover.
To conclude, our results show the conserved Glu-60 in TbADH is not essential for catalysis. Integration of previously reported results on the role of the conserved glutamate residue in ADHs with the results reported here indicates that the Glu-60 in TbADH may possess a pivotal role during catalysis. Specifically, its ability to intermittently coordinate to the catalytic zinc ion may facilitate the coordination of water/OH− or substrates to the catalytic zinc ion during substrate turnover.
Materials and methods
Materials
All chemicals were of highest purity grade and were obtained from Merck (Darmstadt, Germany), except when otherwise noted. Trisma base, PMSF, β-nicotinamide adenine dinucleotide phosphate (potassium salt) (NADP+), d,l-dithiotheitol (DTT), ampicillin, and benzamidine were purchased from Sigma. DEAE-Sepharose and red-Sepharose CL-6B were purchased from Pharmacia. Ethylenediaminetetraacetic acid (EDTA) and 2-butanol were obtained from Fluka Chemical AG. Protein assay kit was from Bio-Rad Laboratories.
Site-directed mutagenesis and purification of recombinant enzymes
Wild-type TbADH containing plasmid was obtained from the laboratory of Prof. Yigal Burstein from the department of Organic Chemistry in the Weizmann Institute of Science. This plasmid was used as a template to generate the E60A-TbADH and E60D-TbADH mutations. The mutations were created by the Kunkel method (Kunkel 1985) and verified by DNA sequencing. Recombinant enzymes were purified according to a modification of the procedure described by Bogin et. al. (1997). Briefly, plasmids with wild-type or mutated TbADH were transformed into E. coli TG-1 strain. The TG-1 cells were grown aerobically for 16 h at 37°C in 2YT medium with ampicillin (100 μg/mL). Pelleted cells were resuspended in 25 mM Tris-HCl, 0.1 mM DTT, 0.1 mM EDTA, 1 mM benzamidine, and 0.02% sodium azide (pH 7.3; buffer A). Cells were disrupted for 5 min by pulsed sonication (Branson Sonifier 450) within ice, followed by centrifugation at 23,000g for 15 min. The supernatant was heat treated for 10 min at 65°C and then centrifuged again at 23,000g for 20 min. The supernatant was applied to a DEAE-Sepharose column (7 × 3 cm), preequilibrated with buffer A containing protease inhibitors at 4°C, and was extensively washed with buffer A until no protein was eluted. The recombinant protein was eluted from the column with a solution of 0.1 M NaCl in buffer A and applied to a red-Sepharose CL-6B column (13 × 3 cm), which was preequilibrated with buffer A containing protease inhibitors at 4°C. The recombinant protein was eluted from the red-Sepharose column with 400 mL NaCl linear gradient (0.1 to 1 M). Active fractions were collected and concentrated by ultra-filtration, using Amicon YM-30 from Millipore. The concentrated protein solution was collected and dialyzed extensively against buffer A for 12 h.
Enzyme activity assay
Enzymatic activity of the TbADH mutants (assayed in the direction of 2-propanol oxidation) was measured at 40°C by following the absorption increase at 340 nm (e340 = 6.2 mM−1 cm−1) for the formation of NADPH from NADP+. The standard assay mixture contained 150 mM 2-butanol, 0.5 mM NADP+, and 100 mM Tris-HCl (pH 9.0) in a total volume of 1 mL. One unit of enzyme activity is defined as the amount of enzyme that catalyzes the formation of 1μmole of NADPH per minute under initial velocity at the above-mentioned conditions.
Enzymatic activity and kinetic parameters were measured and calculated by using a Beckman DU-7500 spectrophotometer equipped with a multicomponent/SCA/Kinetics Plus software package and thermostat water-circulating bath. Reaction mixtures contained in a total volume of 1 mL in 0.15 M Tris-HCl buffer (pH 9.0). The 2-propanol concentrations were 0.05, 0.1, 0.2, 0.5, 0.7, and 1.0 mM for E60D-TbADH and wild-type TbADH; the data for E60A-TbADH were also measured with 2.0 mM 2-propanol. The NADP+ concentrations were 5, 10, 20, 30, 50, and 70 μM for E60D-TbADH; 5, 10, 20, 50, 70, 100, and 150 μM for wild-type TbADH; and 10, 20, 50, 70, 100, and 150 μM for E60A-TbADH. Therefore, the total number of points for analysis was 35, 36, and 42 for E60A-TbADH, E60D-TbADH, and wild-type TbADH, respectively.
Initial velocities were fitted with Equation 1 (as described in al-Kassim et al. 1990) by nonlinear regression using the Solver tool implemented in Microsoft Excel:
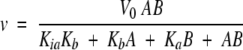 |
(1) |
ICP-AES
The metal content in TbADH mutants was analyzed by using an ICP-AES model Spectroflame from Spectro. Zinc content was determined, after extensive dialysis of protein samples, against three changes of X100 volumes of 25 mM HEPES-HCl, 100 mM NaCl (pH 8.3). Samples were digested in nitric acid, and the volume was adjusted to 6 mL. Analysis was conducted on portions of this solution versus certified standards. The measurement was repeated three times by using protein samples from different batches.
Protein concentrations
Protein concentrations were determined by both Bradford assay (Bradford 1976), with bovine serum albumin as standard, and amino acid analysis.
Circular dichroism
CD measurements were made by using the Aviv spectrophotometer model 202. The data collection was made in quartz cells with a light path of 10 mm. Samples for near-UV scan contain 10 μM enzyme, 25 μM NADP+, and 0.75 mM DMSO in 150 mM Tris-HCl buffer (pH 9.0). Samples for far-UV scans contain 10μM enzyme, 50μM NADP+, and 1 mM DMSO in 50 mM phosphate buffer (pH 8.0).
X-ray Absorption
Sample preparation
Enzyme samples were extensively dialyzed against three changes of X100 volumes of 25 mM HEPES and 100 mM NaCl (pH 8.3). A HEPES buffer was used to avoid drastic pH changes in cryogenic temperature. TbADH enzymatic activity was checked in HEPES buffer for all samples. After dialysis, ultra-filtration, using Millipore Centricon-30 apparatus, concentrated the enzyme to a final concentration of 1 mM (∼40 mg/mL). Samples were loaded into copper sample holders (in the size of 10 × 5 × 0.5 mm) covered with Mylar tape and were immediately frozen in liquid nitrogen. Frozen samples were mounted into a Displex closed-cycle helium cryostat, and the temperature of the samples was maintained at 30 K to minimize the thermal disorder in the XAS data.
Data collection
XAS data collection was performed at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory (BNL), beam line X9B. The spectra were recorded at the zinc K-edge in fluorescence geometry at low temperature (30 K). The beam energy was defined using a flat silicon (111) monochromator crystal. The incident beam intensity (I0) was recorded by using an ionization chamber. The fluorescence intensity was recorded using a 13-element Germanium detector. The transmission signal from a zinc foil was measured with a reference ion chamber, simultaneously with fluorescence, for beam energy calibration purposes. Several scans of each sample were collected for a total statistic of 1 million counts across the edge. The samples were checked for burn marks after each scan, and the beam position on the sample was changed before each scan to minimize radiation damages. Enzyme activity was checked after exposure to X-ray, and the enzyme was found to be fully active.
Data processing and analysis
The average zinc K-edge absorption coefficient μ(E) was obtained after several independent XAS measurements, for each sample, were aligned in absolute energy, using the reference zinc metal foil XAS data as absolute energy calibrant. Then, the absorption coefficient for different samples was shifted in X-ray energy, until the first inflection points were aligned at the same energy (9659 eV). This alignment assured a good approximation of the ability of the same X-ray energy, E0 = 9659 eV being chosen as the photoelectron energy origin in all data sets.
The smooth atomic background was removed with the AUTOBK program of the UWXAFS data analysis package, developed at the University of Washington, Seattle (Stern et al. 1995). The same energy, E0 = 9659 eV, was chosen for the background removal purpose as the photoelectron energy origin. The R-space region for minimizing the signal below the first shell was chosen between 0 and 1 Å. After the background removal, the useful k-range in the resultant k2 weighted is between χ(k) and 10 −10.5 Å−1. Model data for the fitting procedure were constructed by extracting the catalytic zinc site coordinates (in a radius of 8 Å) from crystallographic coordinates of CbADH, because of its better structure resolution over the crystal structure of TbADH (Korkhin et al. 1998). By using the computer code FEFF7 (Rehr et al. 1991; Zabinsky et al. 1995), we calculated the theoretical photoelectron-scattering amplitudes and phase shifts. Total theoretical χ(k) was constructed by adding the most important partial χ(k), which contributes to the r-range of interest.
The k2 weighting factor and the Hanning window function, defined between 2 and 10−10.5 Å−1, were used in Fourier transformations for all data sets. During the fitting procedure, the various parameters—including the corrections to the energy origin (ΔE0), bond distances (ΔR), and mean square disorders of the distances, Debye-Waller factors (σ2)—were varied until the best fit was achieved. The number of relevant independent data points Nidp in the data was calculated by using Equation 2 (Stern 1993):
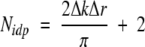 |
(2) |
Where Δk and Δr are the data ranges in k and r spaces, respectively. Equation 2 implies that the number of fit variables should be smaller than Nidp. To reduce the number of fit variables, the many-body factor S20was optimized and fixed at 0.8. The starting point for analysis of each data set was the model used for fitting of wild-type TbADH (Kleifeld et al. 2000a). Theoretical XAFS signal was fitted to experimental data, by using the nonlinear least-squares method implemented in the program FEFFIT (Stern et al. 1995) in R-space, by Fourier-transforming both theory and data. Data and theory were weighted by k2 and multiplied by a Hanning window function in Fourier transforms.
Structural analysis and modeling studies
The crystal structures of HLADH, TbADH, and CbADH were analyzed by using Insight II software from Acclerys. The model of TbADH-NADP+-DMSO complex was constructed on the base of experimental X-ray structures of CbADH (PDB 1PED, 1KEV) and HLADH-DMSO complex (PDB 6ADH).
Acknowledgments
We thank Prof. Yigal Burstein, Dr. Moshe Perez, Hava Gil-Henn, and Dr. Oren Bogin for their useful advice and technical assistance. This work was supported by the USA-Israel Binational Science Foundation project no. 6602.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ADH, alcohol dehydrogenase
CbADH, Clostridium beijerinckii alcohol dehydrogenase
TbADH, Thermoanaerobacter brockii alcohol dehydrogenase
EXAFS, extended X-ray absorption fine structure
XAS, X-ray absorption spectroscopy
CD, circular dichroism
NADP+, oxidized form of β-nicotinamide adenine dinucleotide phosphate
apo-TbADH, recombinant TbADH
holo-TbADH, binary complex of TbADH and NADP+
DMSO, dimethyl sulfoxide
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0221603.
References
- al-Kassim, L.S. and Tsai, C.S. 1990. Studies of NADP(+)-preferred secondary alcohol dehydrogenase from Thermoanaerobium brockii. Biochem. Cell Biol. 68 907–913. [DOI] [PubMed] [Google Scholar]
- Bertini, I., Luchinat, C., Maret, W., and Zeppezauer, M. 1986. Zinc enzymes. Birkhauser Inc., Boston.
- Bogin, O., Peretz, M., and Burstein, Y. 1997. Thermoanaerobacter brockii alcohol dehydrogenase: Characterization of the active site metal and its ligand amino acids. Protein Sci. 6 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Burdette, D.S., Secundo, F., Phillips, R.S., Dong, J., Scott, R.A., and Zeikus, J.G. 1997. Biophysical and mutagenic analysis of Thermoanaerobacter ethanolicus secondary-alcohol dehydrogenase activity and specificity. Biochem. J. 326 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna-Cesari, F., Perahia, D., Karplus, M., Eklund, H., Braden, C.I., and Tapia, O. 1986. Interdomain motion in liver alcohol dehydrogenase: Structural and energetic analysis of the hinge bending mode. J. Biol. Chem. 261 15273–15280. [PubMed] [Google Scholar]
- Dunn, M.F., Biellmann, J.F., and Branlant, G. 1975. Roles of zinc ion and reduced coenzyme in horse liver alcohol dehydrogenase catalysis: The mechanism of aldehyde activation. Biochemistry 14 3176–3182. [DOI] [PubMed] [Google Scholar]
- Dunn, M.F., Dietrich, H., MacGibbon, A.K., Koerber, S.C., and Zeppezauer, M. 1982. Investigation of intermediates and transition states in the catalytic mechanisms of active site substituted cobalt(II), nickel(II), zinc(II), and cadmium(II) horse liver alcohol dehydrogenase. Biochemistry 21 354–363. [DOI] [PubMed] [Google Scholar]
- Dworschack, R.T. and Plapp, B.V. 1977. Kinetics of native and activated isozymes of horse liver alcohol dehydrogenase. Biochemistry 16 111–116. [DOI] [PubMed] [Google Scholar]
- Eklund, H., Nordstrom, B., Zeppezauer, E., Soderlund, G., Ohlsson, I., Boiwe, T., and Branden, C.I. 1974. The structure of horse liver alcohol dehydrogenase. FEBS Lett. 44 200–204. [DOI] [PubMed] [Google Scholar]
- Eklund, H., Plapp, B.V., Samama, J.P., and Branden, C.I. 1982. Binding of substrate in a ternary complex of horse liver alcohol dehydrogenase. J. Biol. Chem. 257 14349–14358. [PubMed] [Google Scholar]
- Esposito, L., Sica, F., Raia, C.A., Giordano, A., Rossi, M., Mazzarella, L., and Zagari, A. 2002. Crystal structure of the alcohol dehydrogenase from the hyperthermophilic archaeon Sulfolobus solfataricus at 1.85 Å resolution. J. Mol. Biol. 318 463–477. [DOI] [PubMed] [Google Scholar]
- Formicka, G., Zeppezauer, M., Fey, F., and Huttermann, J. 1992. Copper(II)-substituted horse liver alcohol dehydrogenase: Structure of the minor species. FEBS Lett. 309 92–96. [DOI] [PubMed] [Google Scholar]
- Ganzhorn, A.J. and Plapp, B.V. 1988. Carboxyl groups near the active site zinc contribute to catalysis in yeast alcohol dehydrogenase. J. Biol. Chem. 263 5446–5454. [PubMed] [Google Scholar]
- Heiss, C., Laivenieks, M., Zeikus, J.G., and Phillips, R.S. 2001a. Mutation of cysteine-295 to alanine in secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus affects the enantioselectivity and substrate specificity of ketone reductions. Bioorg. Med. Chem. 9 1659–1666. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. The stereospecificity of secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus is partially determined by active site water. J. Am. Chem. Soc. 123 345–346. [DOI] [PubMed] [Google Scholar]
- Johansson, K., El-Ahmad, M., Kaiser, C., Jornvall, H., Eklund, H., Hoog, J., and Ramaswamy, S. 2001. Crystal structure of sorbitol dehydrogenase. Chem. Biol. Interact. 130–132 351–358. [DOI] [PubMed] [Google Scholar]
- Kleifeld, O., Frenkel, A., Bogin, O., Eisenstein, M., Brumfeld, V., Burstein, Y., and Sagi, I. 2000a. Spectroscopic studies of inhibited alcohol dehydrogenase from Thermoanaerobacter brockii: Proposed structure for the catalytic intermediate state. Biochemistry 39 7702–7711. [DOI] [PubMed] [Google Scholar]
- Kleifeld, O., Van den Steen, P.E., Frenkel, A., Cheng, F., Jiang, H.L., Opdenakker, G., and Sagi, I. 2000b. Structural characterization of the catalytic active site in the latent and active natural gelatinase B from human neutrophils. J. Biol. Chem. 275 34335–34343. [DOI] [PubMed] [Google Scholar]
- Kleifeld, O., Frenkel, A., and Sagi, I. 2001a. Time-dependent XAS studies of trapped enzyme-substrate complexes of alcohol dehydrogenase from Thermoanaerobacter brockii. J. Synchrotron Radiat. 8 978–980. [DOI] [PubMed] [Google Scholar]
- Kleifeld, O., Kotra, L.P., Gervasi, D.C., Brown, S., Bernardo, M.M., Fridman, R., Mobashery, S., and Sagi, I. 2001b. X-ray absorption studies of human matrix metalloproteinase-2 (MMP-2) bound to a highly selective mechanism-based inhibitor. comparison with the latent and active forms of the enzyme. J. Biol. Chem. 276 17125–17131. [DOI] [PubMed] [Google Scholar]
- Korkhin, Y., Kalb, A.J., Peretz, M., Bogin, O., Burstein, Y., and Frolow, F. 1998. NADP-dependent bacterial alcohol dehydrogenases: Crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278 967–981. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lamed, R.J. and Zeikus, J.G. 1981. Novel NADP-linked alcohol–aldehyde/ketone oxidoreductase in thermophilic ethanologenic bacteria. Biochem. J. 195 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen, M.W., Maret, W., and Yim, M.B. 1983. Neutral metal-bound water is the base catalyst in liver alcohol dehydrogenase. Proc. Natl. Acad. Sci. 80 2584–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijers, R., Morris, R.J., Adolph, H.W., Merli, A., Lamzin, V.S., and Cedergren-Zeppezauer, E.S. 2001. On the enzymatic activation of NADH. J. Biol. Chem. 276 9316–9321. [DOI] [PubMed] [Google Scholar]
- Peretz, M., Bogin, O., Keinan, E., and Burstein, Y. 1993. Stereospecificity of hydrogen transfer by the NADP-linked alcohol dehydrogenase from the thermophilic bacterium Thermoanaerobium brockii. Int. J. Pept. Protein Res. 42 490–495. [DOI] [PubMed] [Google Scholar]
- Perlman, R.L. and Wolff, J. 1968. Dimethyl sulfoxide: An inhibitor of liver alcohol dehydrogenase. Science 160 317–319. [DOI] [PubMed] [Google Scholar]
- Rehr, J.J., Mustre de Leon, J., Zabinsky, S.I., and Albers, R.C. 1991. Theoretical X-ray absorption fine structure standards. J. Am. Chem. Soc. 113 5135–5140. [Google Scholar]
- Ryde, U. 1995a. Molecular dynamics simulations of alcohol dehydrogenase with a four- or five-coordinate catalytic zinc ion. Proteins 21 40–56. [DOI] [PubMed] [Google Scholar]
- ———. 1995b. On the role of Glu-68 in alcohol dehydrogenase. Protein Sci. 4 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanghani, P.C., Stone, C.L., Ray, B.D., Pindel, E.V., Hurley, T.D., and Bosron, W.F. 2000. Kinetic mechanism of human glutathione-dependent formaldehyde dehydrogenase. Biochemistry 39 10720–10729. [DOI] [PubMed] [Google Scholar]
- Sanghani, P.C., Robinson, H., Bosron, W.F., and Hurley, T.D. 2002. Human glutathione-dependent formaldehyde dehydrogenase: Structures of apo, binary, and inhibitory ternary complexes. Biochemistry 41 10778–10786. [DOI] [PubMed] [Google Scholar]
- Scott, R.A. 1985. Measurement of metal-ligand distances by EXAFS. Meth. Enzymol. 117 414–459. [Google Scholar]
- Stern, E.A. 1993. Number of relevant independent points in X-ray-absorption fine-structure spectra. Phys. Rev. B 48 9825–9827. [DOI] [PubMed] [Google Scholar]
- Stern, E.A., Newville, M., Ravel, B., Yacoby, Y., and Haskel, D. 1995. The UWXAFS analysis package: Philosophy and details. Physica B (Amsterdam) 208/209 117–120. [Google Scholar]
- Strickland, E.H. 1974. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit. Rev. Biochem. 2 113–175. [DOI] [PubMed] [Google Scholar]
- Sun, H.W. and Plapp, B.V. 1992. Progressive sequence alignment and molecular evolution of the Zn-containing alcohol dehydrogenase family. J. Mol. Evol. 34 522–535. [DOI] [PubMed] [Google Scholar]
- Wilkinson, G. 1987. Comprehensive coordination chemistry: The synthesis, reactions, properties and applications of coordination compounds. Pergamon Press, London.
- Woody, R.W. 1995. Circular dichroism. Meth. Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]
- Zabinsky, S.I., Rehr, J.J., Ankudinov, A., Albers, R.C., and Eller, M.J. 1995. Multiple scattering calculations of X-ray absorption spectra. Phys. Rev. B 52 2995–3009. [DOI] [PubMed] [Google Scholar]
- Zeppezauer, M. 1983. Coordination properties and mechanistic aspects of liver alcohol dehydrogenase. NATO Adv. Study Inst. Ser. C 100 99–122. [Google Scholar]