

Abstract
The chemotherapeutic agent cisplatin is widely used in treatment of solid tumors. In breast cancer cells, cisplatin produces early and marked changes in cell morphology and the actin cytoskeleton. These changes manifest as loss of lamellipodia/filopodia and appearance of membrane ruffles. Furthermore, cisplatin induces dephosphorylation of the actin-binding protein ezrin, and its relocation from membrane protrusions to the cytosol. Because cisplatin activates acid sphingomyelinase (ASMase), we investigate here the role of the ASMase/ceramide (Cer) pathway in mediating these morphological changes. We find that cisplatin induces a transient elevation in ASMase activity and its redistribution to the plasma membrane. This translocation is blocked upon overexpression of a dominant-negative (DN) ASMaseS508A mutant and by a DN PKCδ. Importantly; knockdown of ASMase protects MCF-7 cells from cisplatin-induced cytoskeletal changes including ezrin dephosphorylation. Reciprocally, exogenous delivery of D-e-C16-Cer, but not dihydro-C16-Cer, recapitulates the morphotropic effects of cisplatin. Collectively, these results highlight a novel tumor suppressor property for Cer and a function for ASMase in cisplatin-induced cytoskeletal remodeling.
Introduction
Ceramides, ubiquitous sphingolipids in biological membranes, and their metabolites are receiving increasing attention in cancer research (Ogretmen and Hannun, 2004). Besides established roles in apoptosis and growth arrest, novel tumor suppressor properties of ceramide have emerged, including effects on cellular cytoskeleton and motility. Exogenous short chain ceramides (C2- and C6-ceramides) were shown to induce rearrangement of the F-actin network, cell rounding, and detachment in various cell lines, including neuroepitheliomas, breast and cervical cancers (Panigone et al., 2001; Di Bartolomeo and Spinedi, 2002; Hu et al., 2005). These morphological changes precede and appear to be independent of the apoptotic signaling triggered by ceramide. Recent studies demonstrated that ceramide impedes surface availability and trafficking of integrin receptors; thus, interfering with cellular adhesion and adhesion-related survival signaling (Panigone et al., 2001; Hu et al., 2005). Although these studies begin to shed light on the morphotropic effects of ceramide, the role of endogenous ceramide in these processes and the mechanisms by which ceramide influences the actin cytoskeleton are still unclear.
The actin cytoskeleton and its associated proteins are essential players in cell motility, adhesion, and morphogenesis. Ezrin, a member of the ERM family, regulates cytoskeletal dynamics by cross-linking actin filaments to the plasma membrane (Bretscher et al., 2002; Fievet et al., 2007). Two cellular forms of ezrin have been studied: an inactive cytoplasmic form and an active membrane-bound form. In its inactive conformation, the N-terminal membrane association domain of ezrin exerts intramolecular masking of the actin-binding domain located at the C terminus. Sequential binding of the phosphoinositide PIP2 at the N terminus followed by phosphorylation of the C-terminal threonine 567 residue results in unmasking of the functional protein binding sites, thus allowing ezrin to associate simultaneously with the cytoplasmic tail of various transmembrane proteins and with F-actin (Fievet et al., 2004). In its active form, ezrin promotes formation of specialized cellular protrusions such as lamellipodia, filopodia, and microvilli, essential structures in motility and contact with other cell types.
Effectors of ceramide signaling have been implicated in regulating cellular motility. In particular several studies have correlated the ceramide-activated serine/threonine phosphatase PP2A with decreased tumor metastasis (Metz et al., 1996; Meisinger et al., 1997). Xu and Deng (2006) recently reported that loss of PP2A increases migration and invasion of lung cancer cells. Treatment with C2-ceramide, a potent PP2A agonist, suppressed motility of cancer cells (Xu and Deng, 2006). However, the underlying mechanisms by which PP2A affects tumor cell motility remain unknown.
To understand the molecular mechanisms by which sphingolipids influence cellular architecture, we investigated the effects of stress-induced endogenous ceramide generation in MCF-7 breast cancer cells, using cisplatin, a chemotherapeutic agent of major clinical utility for treatment of breast cancer.
We demonstrate here that ceramide production plays an important role in stress-induced cytoskeletal remodeling. Cisplatin induced marked changes in the actin cytoskeleton, including dephosphorylation and cytosolic translocation of ezrin. Mechanistically, activation of acid sphingomyelinase (ASMase) is shown to be required upstream of the observed morphological changes. Further investigation revealed a novel association between ceramide-activated PP2A and ezrin at the plasma membrane. These findings demonstrate that the ASMase/ceramide pathway is instrumental for cytoskeletal remodeling induced by chemotherapeutic interventions.
Results
Cisplatin treatment induces acute cytoskeletal remodeling in MCF-7 cells
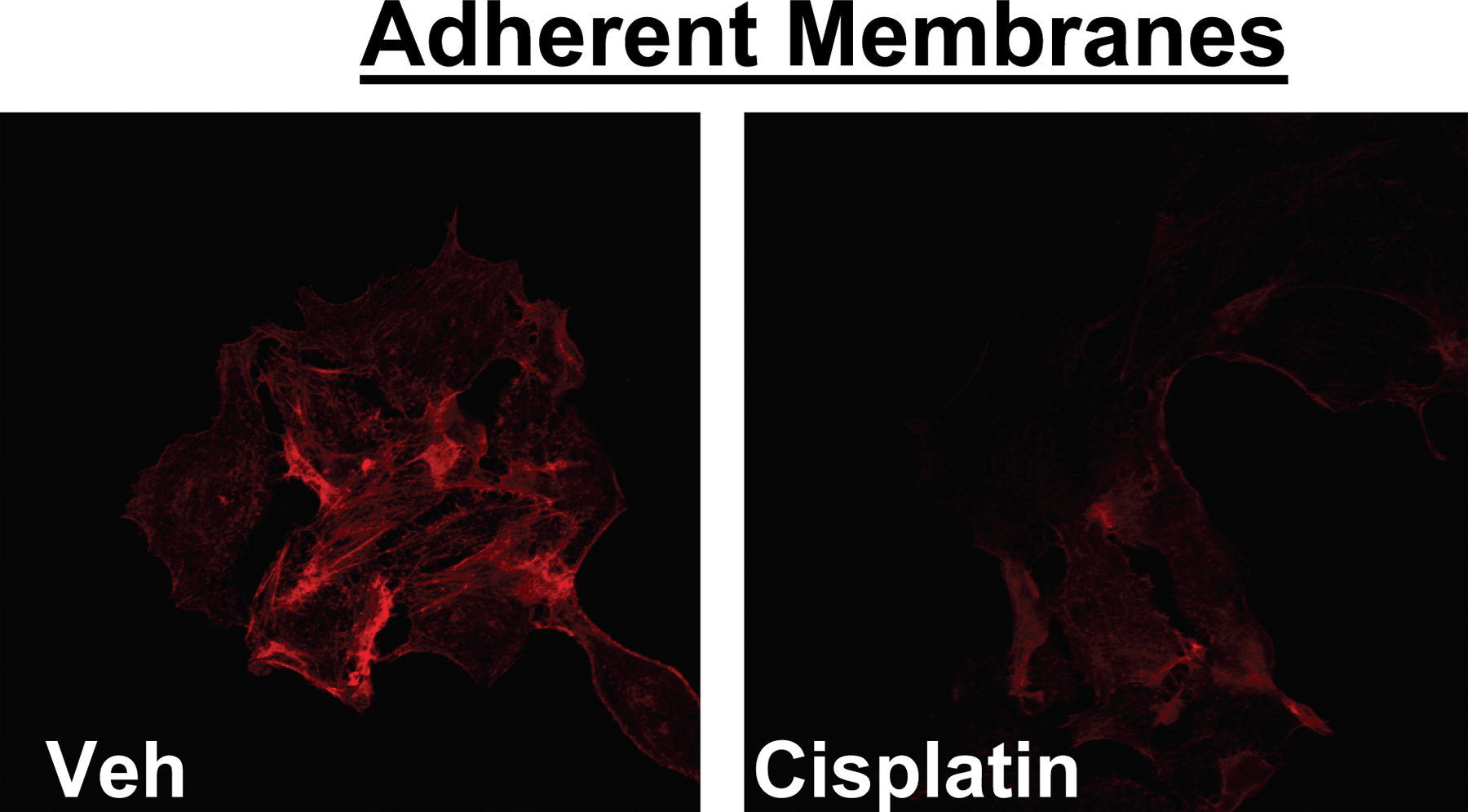
Several studies have previously shown that anticancer drugs induce changes in cellular morphology (Kruidering et al., 1998; Bijman et al., 2006). As a model, we used the human mammary carcinoma cells, MCF-7, originally obtained from pleural effusion of a patient with breast cancer. Treatment with cisplatin (5 μg/ml) caused marked morphological changes within 2 h. As shown in Fig. 1 A, the morphological changes can be readily observed by light microscopy as major reduction in cellular processes, consistent with filopodia and lamellipodia. However, cisplatin did not display major cytotoxic effects on MCF-7 cells even when kept for 12 h (unpublished data). Because the actin cytoskeleton has a major role in formation of cellular processes, the effects of cisplatin on the actin network were evaluated next. Using phalloidin, which specifically stains F-actin, remodeling of actin network was observed after cisplatin treatment. As shown in Fig. 1 B, cisplatin disrupted the filamentous pattern of phalloidin staining and caused the appearance of cortical stress fibers. Importantly, phalloidin staining of adherent membranes revealed a decreased membrane-bound F-actin after treatment with cisplatin (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200705060/DC1). Intriguingly, cisplatin treatment did not alter filamentous (F) to globular (G) actin ratio (Fig. 1 C). Therefore, these results suggest that cisplatin destabilizes membrane anchoring of actin filaments, leading to rearrangement of filamentous actin network and overall loss of cellular processes.
Figure 1.

Effects of cisplatin on MCF-7 cellular cytoskeleton. (A) MCF-7 cells were grown on 10-cm dishes up to 60% confluency. Then cells were treated with either cisplatin (5 μg/ml) or DMSO for 2 h. Cell morphology was examined under an inverted light microscope equipped with a CCD camera using 20x objectives. Representative areas are enlarged in the insets. (B) Cells treated as in A were fixed and stained with rhodamine-phalloidin. (C) Cells treated with vehicle (DMSO), phalloidin, cytochalasin D, or cisplatin (60 and 120 min) were analyzed for filamentous (F) and globular (G) actin by Western blotting. Results were quantitated by densitometry using NIH ImageJ. Figures are representative of at least three independent experiments.
Relocation of ezrin after cisplatin treatment
Because the actin-binding protein ezrin plays a major role in stabilizing the association of actin filaments with the plasma membrane, the effect of cisplatin treatment on the distribution of ezrin was evaluated. As expected, under basal conditions, ezrin mainly localized to cellular filopodia with minor cytosolic staining. Interestingly, treatment with cisplatin induced major relocation of ezrin from the cell periphery to a more cytosolic distribution (Fig. 2 A). This observation was further corroborated by a cellular fractionation approach in which the membrane fraction (100,000 g) was separated from the cytosolic fraction. As shown in Fig. 2 B, Western blotting for ezrin showed a marked drop in membrane-associated ezrin. (Fig. 2 B). Collectively, these results suggest that the morphological changes induced by cisplatin are associated with rearrangement of the actin cytoskeleton and redistribution of the actin-binding protein, ezrin.
Figure 2.

Treatment with cisplatin causes relocation of ezrin. MCF-7 cells were treated with DMSO or cisplatin (5 μg/ml) for 2 h. (A) Cells were fixed and stained with ezrin-specific polyclonal antibody (green channel) before laser-scanning confocal microscopy. Nuclei were visualized using DRAQ5 nuclear dye (red channel). Shown are representative images of multiple experiments. Representative areas are enlarged in the insets. (B) Cells were homogenized and fractionated into cytosolic and membrane fractions by centrifugation at 100,000 g. Amount of ezrin in total, cytosolic, and membrane fractions (concentrated eightfold) was detected by Western blotting. (C) MCF-7 cells (5 x 105 cells/10-cm plate) were treated with DMSO or cisplatin (5 μg/ml) for the indicated time points up to 2 h. Levels of p-ezrin and total ezrin were detected by specific rabbit polyclonal antibodies. (D) Cells treated with DMSO or cisplatin (5 μg/ml) for 2 h were stained for p-ezrin (green channel) and phalloidin (red channel). Blots from three independent experiments were quantitated using NIH ImageJ (*, P < 0.05). Images are representative of three independent experiments. Bars, 20 μm.
In addition to PIP2 binding, phosphorylation of the C-terminal threonine residue (T567) is a hallmark of ezrin activation. In its active form, ezrin adopts a peripheral localization that allows it to cross-link the plasma membrane to the cellular cytoskeleton. The observation that cisplatin induced cytosolic redistribution of ezrin prompted us to examine the phosphorylation status of ezrin. Using a phospho (T567) specific antibody, the levels of active ezrin were evaluated by Western blotting after a time course of cisplatin treatment. The results showed that cisplatin caused a time-dependent dephosphorylation of ezrin seen after 1 h and persisted up to 2 h (Fig. 2 C). It is worth indicating, however, that total protein levels of ezrin remained unchanged. Dephosphorylation of ezrin at T567 was further confirmed by immunofluorescence after cisplatin treatment (Fig. 2 D). Therefore, these findings indicate that the cytoskeletal changes induced by cisplatin are associated with inactivation of the actin-binding protein, ezrin.
Cisplatin activates the ASMase/ceramide pathway
The effects of cisplatin on dephosphorylation of ezrin raised the possibility of activation of a serine/threonine phosphatase. Importantly, previous studies have shown that the bioactive lipid ceramide activates phosphatases in vitro and in cells (Pettus et al., 2002). Moreover, the ASMase/ceramide pathway has been previously shown to be activated by cisplatin (Levade and Jaffrezou, 1999; Lacour et al., 2004). Therefore, it became important to evaluate whether the observed changes with cisplatin are due to modulation of sphingolipid metabolism. To that end, MCF-7 cells were treated with cisplatin (5 μg/ml) over a time course of 3 h, and sphingolipids were extracted by the Bligh Dyer technique. Mass spectrometric measurements revealed a transient elevation in total ceramide (twofold) within 30 min of cisplatin treatment (Fig. 3 A). Further analysis indicated that this change was mainly accounted for by an increase in ceramide species carrying C16 and C24:1 acyl chains (Fig. 3 C). Importantly, the observed changes in ceramide levels were paralleled by a transient drop in sphingomyelin (SM) levels (Fig. 3 B). In particular, a major change in C16 SM and a minor one in C24:1 SM were detected by mass spectrometry after 15 min of cisplatin treatment (Fig. 3 D). Together, this sphingolipidomic profile suggests the operation of a sphingomyelinase.
Figure 3.

LC/MS analysis of cisplatin effects on ceramide and sphingomyelin. Cells were treated with cisplatin (5 μg/ml) for 15, 30, 60, or 180 min. After Bligh Dyer extraction of lipids, the different treatment groups were subjected to mass spectrometric analysis as indicated in Materials and methods. Sphingolipids measurements were normalized to total phospholipids. Shown are the normalized results for (A) total ceramide, (B) total SM, (C) C16 and C24:1-ceramide, and (D) C16 and C24:1-SM. Ceramide and SM results represent averages ± S.E. from three independent experiments.
To investigate this possibility, in vitro sphingomyelinase assays were conducted using radiolabeled sphingomyelin. As shown in Fig. 4 A, a significant elevation in ASMase activity was observed within 15 min and reached up to twofold over baseline after 30 min of cisplatin addition. However, neutral sphingomyelinase (NSMase) and secretory sphingomyelinase activities (S-SMase) were not significantly changed along this time frame of cisplatin treatment.
Figure 4.

Activation of ASMase after treatment with cisplatin. (A) MCF-7 cells were treated with either DMSO or cisplatin (5 μg/ml) for 15, 30, 60, or 180 min. Total lysates (100 μg/sample) were subjected to SMase assays (ASMase, NSMase; neutral sphingomyelinase and S-SMase; secretory sphingomyelinase) as described under Materials and methods. Results shown are averaged from three enzymatic assays ± S.E. (*, P < 0.05). (B and C) Cells plated on 2-cm confocal dishes (5 × 105 cells/plate) were transfected with 1 μg of the V5-ASMase plasmid. After 24 h, cells were incubated with either DMSO or cisplatin (5 μg/ml) for 30 min. Localization of ASMase was detected using a V5 monoclonal antibody (green channel) in permeabilized (B) or non-permeabilized cells (C). Sphingomyelin was visualized indirectly using the SM-binding protein, lysenin (red channel). Immunofluorescence was performed using a polyclonal lysenin antibody. Nuclei (NUC) were labeled using DRAQ5 nuclear dye. Shown are representative images of three independent experiments. Bars, 5 μm.
Importantly, confocal microscopy analysis revealed translocation of ASMase from its endolysosomal compartment to the cell periphery in response to cisplatin (Fig. 4 B). In particular, imaging of unpermeabilized cells not only confirmed this translocation process, but it also suggested localization of ASMase to the outer cell surface. Notably, the exoplasmic leaflet has been previously shown to be a major site of SM enrichment (Calderon and DeVries, 1997; van Helvoort et al., 1997). Lysenin, a hemolytic protein from the earthworm Eisenia foetida, functions as a high affinity SM-binding protein (Hanada et al., 1998). Therefore, inactivated lysenin was used to evaluate the effects of cisplatin on sphingomyelin. A clear drop in fluorescence intensity of lysenin staining was seen after cisplatin treatment (Fig. 4 C); however, lysenin staining retained a uniform plasma membrane distribution under these experimental conditions. Moreover, cisplatin induced significant colocalization of ASMase with lysenin; however, it was noted that ASMase did not show a uniform staining in contrast to lysenin (Fig. 4 C, inset). Collectively, these results provide biochemical and cell biological evidence that cisplatin modulates the sphingolipid pathway through activating ASMase.
ASMase is upstream of ezrin dephosphorylation and the cytoskeletal changes
Given the above results, it became important to investigate whether changes in sphingolipid metabolites mediate the action of cisplatin. Therefore, knockdown of ASMase was attempted using RNAi technology. Two different 21-bp oligonucleotides targeting ASMase or a control (scrambled) sequence were transfected into MCF-7 cells. Western blotting confirmed loss of the ASMase protein (>70%) after 48 h of RNAi transfection (Fig. 5 A). Importantly, knockdown of ASMase blocked cisplatin-induced ezrin dephosphorylation as determined by Western blotting (Fig. 5 B).
Figure 5.

ASMase is upstream of cisplatin-induced ezrin dephosphorylation. (A) MCF-7 cells were transfected with 10 nM of either SCR or ASMase RNAi oligonucleotides. After 48 h, levels of ASMase and β-actin were determined by Western blotting and quantitated by densitometry. (B) Cells were treated with either DMSO or cisplatin (5 μg/ml) for 60 min. Lysates (30 μg) from each treatment group were used to detect levels of phosphorylated ezrin (p-ezrin) and ezrin by Western blotting. Blots from three independent experiments were quantitated using NIH ImageJ (**, P < 0.01; n.s, non significant). (C) Cells plated on 2-cm confocal dishes were transfected with 20 nM of SCR or two ASMase RNAi oligonucleotides (ASMase RNAi [1] and [2]). After 48 h, cells were treated with either DMSO or cisplatin for 60 min. Then cells were fixed and stained using ezrin-specific polyclonal antibody. Images are representative of three independent experiments. Bars, 10 μm.
This result prompted us to define the role of ASMase in cisplatin-induced cytoskeletal changes. To this end, cells were transfected with either ASMase RNAi or SCR oligonucleotides for 48 h. Confocal microscopy imaging revealed that cisplatin treatment caused an overall rounding, loss of cellular filopodia, and relocation of ezrin to a more cytosolic distribution in the control group (SCR). It is worth noting that a normal nuclear morphology was maintained (red channel), suggesting that the observed changes are not secondary to cell death. Importantly, cells transfected with ASMase RNAi were remarkably protected from the morphological effects of cisplatin. This was indicated by retention of not only cellular filopodia but also peripheral ezrin localization after cisplatin treatment (Fig. 5 C). Therefore, these findings suggest a critical role for ASMase in mediating the cytoskeletal effects of cisplatin.
Exogenous ceramide recapitulates the cytoskeletal effects induced by cisplatin
Because the above results suggested that ceramide production is necessary for cytoskeletal changes induced by cisplatin, we next evaluated whether ceramide is sufficient for these changes. C16-ceramide was chosen for these experiments, as it was abundant in MCF-7 cells and almost doubled in response to cisplatin (Fig. 3 C). Microscopically, treatment with C16-ceramide recapitulated the effects of cisplatin, including loss of filopodia and redistribution of ezrin to the cytosol (Fig. 6 A). After separation of cytosolic and membrane pools by cell fractionation, Western blot analysis revealed reduction of membrane-associated ezrin after ceramide treatment (Fig. 6 B). In line with these results, treatment of HL-60 cells with short chain C2-ceramide was earlier reported to decrease the levels of ERM proteins in the membrane fraction (Kondo et al., 1997), suggesting that the above findings are neither specific to a particular cell line nor a ceramide species. Additionally, similar to cisplatin, administration of exogenous C16-ceramide caused a time-dependent dephosphorylation of ezrin, although dephosphorylation occurred in a more acute manner (30 min) (Fig. 6 C). Of note, treatment with the ceramide analogue, dihydro-C16-ceramide, failed to influence the levels of p-ezrin, suggesting a ceramide-specific response. Thus, ceramide is not only necessary but also sufficient for stress-induced cytoskeletal responses.
Figure 6.

Exogenous ceramide mimics cytoskeletal effects of cisplatin. MCF-7 cells were treated with D-e-C16-ceramide (5 μM) or vehicle (2% dodecane/98% ethanol) for 2 h. (A) Cells were fixed and stained with ezrin-specific polyclonal antibody (green channel). Nuclei were visualized using DRAQ5 nuclear dye. (B) Cells were homogenized and fractionated into a cytosolic and membrane fractions by centrifugation at 100,000 g. Amount of ezrin in total, cytosolic, and membrane fractions (concentrated eightfold) was detected by Western blotting. (C) Cells were treated with D-e-C16-ceramide (5 μM), dh-C16-ceramide (5 μM), or vehicle (2% dodecane/98% ethanol) up to 3 h. Levels of p-ezrin and total ezrin were detected by Western blotting. (D) Cells treated with bSMase (100 mU/ml) for 0, 15, 30, and 60 min were subjected to Western blot analysis for ezrin and p-ezrin levels. (E) Subcellular distribution of ezrin in control cells and cells treated with bSMase (100 mU/ml) for 60 min. (F) Cells treated as in D were stained for p-ezrin (green channel) and phalloidin (red channel). Results are representative of three independent experiments. Blots from three independent experiments were quantitated using NIH ImageJ (*, P < 0.05). Bars, 20 μm.
The above hypothesis was further evaluated by studying the effects of ceramide, endogenously generated through the action of bacterial sphingomyelinase (bSMase). Importantly, addition of bSMase (100 mU/ml) induced a time-dependent dephosphorylation of ezrin (Fig. 6 D), and subcellular redistribution of ezrin from cell periphery to the cytosol was observed within 60 min of bSMase addition (Fig. 6 E). This was accompanied by a parallel loss of p-ezrin staining from cellular filopodia (Fig. 6 F). Collectively, these data suggest that ceramide is a key lipid regulator of ezrin function and distribution.
PP2A mediates ezrin dephosphorylation
Ceramide-activated protein phosphatases (CAPPs) are well established effectors of ceramide generating pathways of which PP2A and PP1 remain the best studied thus far (Ruvolo, 2003). Therefore, it became important to determine whether the observed dephosphorylation of ezrin is mediated by CAPPs. In previous studies, it was demonstrated that okadaic acid potently inhibits PP2A when used at low doses (Chalfant et al., 1999, 2001). Cells were pretreated with either okadaic acid (10 nM) or vehicle (DMSO) 1 h before cisplatin addition. Western blot analysis revealed that inhibition of PP2A with okadaic acid restored levels of p-ezrin (Fig. 7 A). We also entertained the potential involvement of another well-established ceramide activated phosphatase, PP1. However, treatment with tautomycin (10 nM), a specific PP1 inhibitor, had little effect on cisplatin-induced ezrin dephosphorylation (Fig. 7 B). These results were further corroborated using RNAi technology. For these experiments, MCF-7 cells were transfected with 20 nM of either control (SCR) or PP2A-specific 21-bp oligonucleotides. After 48 h, marked knockdown (>70%) of PP2A protein was observed by Western blotting (Fig. 7 C). Importantly, loss of PP2A conferred protection from cisplatin-induced ezrin dephosphorylation (Fig. 7 C). These findings provide strong pharmacologic and genetic evidence for PP2A in ezrin dephosphorylation.
Figure 7.

PP2A mediates dephosphorylation of ezrin by cisplatin. Cells plated on 10-cm dishes (5 × 105 cells/dish) were pretreated with 10 nM of either okadaic acid (A) or tautomycin (B). After 1 h, cells were incubated with DMSO or cisplatin (5 μg/ml) for 1 h. Phospho-ezrin levels were evaluated by Western blotting using a specific polyclonal antibody. (C) Cells were transfected with 20 nM of either SCR or PP2A (catalytic subunit) specific oligonucleotides. After 48 h, cells were subjected to DMSO or cisplatin treatments for 1 h. Lysates (30 μg) were analyzed by Western blotting for levels of p-ezrin and PP2A. Blots shown are representative of at least three independent experiments. Densitometric analysis was performed using NIH Image software. (D) MCF-7 cells expressing YFP-ezrin and CFP-PP2A (catalytic subunit) were subjected to sensitized emission FRET analysis. After 24 h of plasmid transfection, cells were treated with DMSO or cisplatin for 30 and 60 min. A representative FRET image of at least 30 cells imaged in three experiments is shown. FRET efficiencies are encoded by using the color bar scale shown on the left. Colors range between blue (lowest FRET) and red (highest FRET).
Because p-ezrin localizes primarily at (or close to) the plasma membrane, it became important to determine the effects of cisplatin on PP2A localization and possible association of PP2A and ezrin. For these studies, a FRET microscopy approach was used whereby CFP-PP2A served as the donor and YFP-ezrin as the acceptor. As shown in Fig. 7 D, within 30 min, cisplatin treatment induced redistribution of PP2A from the cytosol to the cell periphery. Importantly, this redistribution of PP2A increased FRET efficiency to 40%, suggesting that PP2A was in close proximity to ezrin (nm). Intracellular relocation of overexpressed YFP-ezrin was observed after 60 min of cisplatin treatment along with a drop of FRET efficiency to baseline level. Together, these results suggest that PP2A mediates ezrin dephosphorylation during stress-induced ceramide.
PKCδ mediates activation of ASMase by cisplatin
Recently, we defined a mechanism for ASMase activation in which PKCδ serves as a key upstream kinase (Zeidan and Hannun, 2007). Therefore, we investigated whether cisplatin treatment modulated PKC activity in MCF-7 cells. In vitro kinase assays revealed a significant elevation (1.5-fold) in PKCδ activity by 30 min and further elevation (3-fold) after 1 h of cisplatin treatment (Fig. 8 A). Because this time frame of PKCδ induction is consistent with the kinetics of ASMase activation by cisplatin, the role of PKCδ in this process was evaluated. Therefore, cells were infected with either control adenoviruses (LacZ) or viruses encoding a dominant-negative construct of PKCδ (DN-PKCδ) before stimulation with cisplatin. In comparison with the control group, cells infected with DN-PKCδ showed a significant reduction in ASMase activity after cisplatin treatment (Fig. 8 B). Additionally, the effect of DN-PKCδ on cisplatin-induced ASMase relocation was evaluated. The results revealed a 40% drop (65% inhibition of the response) in cells showing plasma membrane translocation of ASMase after cisplatin treatment (Fig. 8 C).
Figure 8.

PKCδ is upstream of ASMase activation by cisplatin. (A) MCF-7 cells (5 × 105 cells/plate) were treated with DMSO or cisplatin (5 μg/ml) for 5, 30, 60, or 180 min. PKCδ was immunoprecipitated from lysates of each treatment group and subjected to in vitro kinase assay as described in Materials and methods.32P-MBP was visualized by autoradiography. Average densitometric analysis + S.E. of three independent experiments are plotted. (B) Cells were infected with control LacZ or DN-PKCδ adenoviruses (multiplicity of infection of 50). After 24 h, cells were treated with DMSO or cisplatin for 30 min. Samples of total lysates (100 μg) were then subjected to in vitro ASMase assay. (C and D) Cells overexpressing ASMase (or ASMaseS508A) were infected with LacZ or DN-PKCδ adenoviruses as in B before treatment with either DMSO or cisplatin for 30 min. Localization of ASMase was observed by confocal microscopy using a V5 monoclonal antibody staining in permeabilized cells. Percentage of cells showing plasma membrane translocation of ASMase was quantitated from at least 10 random visual fields each containing 15–20 cells per field. (*, P < 0.05). ns, not significant.
Moreover, because we had identified phosphorylation of S508 residue in ASMase as an essential step for ASMase translocation, we next studied the role of this residue in cisplatin-induced ASMase translocation. To this end, cells overexpressing V5-tagged WT-ASMase or ASMaseS508A were treated with cisplatin for 30 min. As shown in Fig. 8 D, although ASMase translocation could be detected in 75 ± 10% of WT-ASMase cells, this response could be seen in only 22 ± 9% of ASMaseS508A cells. Collectively, these results strongly suggest that activation of ASMase by cisplatin is mediated by PKCδ.
ASMase mediates the effect of cisplatin on cell migration
Recent studies have indicated a major role for ezrin in regulating cellular motility (Fievet et al., 2007; Prag et al., 2007). In particular, the active phosphorylated form of ezrin is tumor promoting as it enhances cellular migration and invasion. Therefore, we hypothesized that inactivation of ezrin by cisplatin treatment influences cellular motility. This hypothesis was tested using a wound-healing assay applied to confluent monolayers of MCF-7 cells stably overexpressing WT ASMase or ASMaseS508A. As shown in Fig. 9 A, under basal conditions both ASMase and ASMaseS508A stables were able to reduce the wound to 27% of its original size after 48 h. Importantly, the wound-healing ability of cells incubated with cisplatin (5 μg/ml) was markedly reduced to only 70% of original wound size, indicating reduced migration in the presence of cisplatin. On the contrary, cells overexpressing ASMaseS508A were significantly refractory to the effects of cisplatin, as they were still able to reduce wound size to 40%. Importantly, ASMaseS508A cells were also refractory to ezrin dephosphorylation and cytosolic translocation after cisplatin treatment as compared with WT-ASMase cells (Fig. S2, A and B; available at http://www.jcb.org/cgi/content/full/jcb.200705060/DC1). Together, these findings strongly suggest that suppression of cellular motility by cisplatin requires activation of ASMase.
Figure 9.

Differential migratory response of WT-ASMase vs. ASMaseS508A cells to cisplatin treatment. MCF-7 cells stably overexpressing WT-ASMase or ASMaseS508A were treated with either cisplatin or DMSO. (A) Cell migration was analyzed by wound-healing assay as described under Materials and methods. Wound size was measured at 0, 24, and 48 h after scratching a confluent monolayer of cells. (B) Cells overexpressing ezrin T567D or a control (pCB6) plasmid were subjected to the wound-healing migration assay. Cell migration was assessed by measuring wound size at 0, 24, and 48 h. Results shown are representative of three independent experiments (*, P < 0.05).
Previous studies have shown that the T567D mutant form of ezrin mimics structural and functional properties of phosphorylated ezrin (Gautreau et al., 2000; Bretscher et al., 2002). To investigate the role of ezrin in the cisplatin response discussed above, we studied the migratory response of cells expressing ezrin T567D or a control plasmid. The results showed that cells expressing ezrin T567D were resistant to cisplatin as they rescued the wound size to 40% of its original size within 48 h (Fig. 9 B). In contrast, cells transfected with the control plasmid retained 75% of the wound size, suggesting impaired migration in presence of cisplatin (Fig. 9 B). Therefore, regulation of ezrin appears to be critical for the effect of cisplatin on cellular migration.
Discussion
The results in this study demonstrate that the ASMase/ceramide pathway is instrumental for remodeling of cellular architecture during stress responses. Treatment with cisplatin caused loss of filopodia and rearrangement of the actin cytoskeleton. These morphological changes were associated with dephosphorylation of ezrin and its further relocation from the plasma membrane to the cytosol. Knockdown of ASMase rescued cells from cisplatin-induced ezrin inactivation and overall morphological change, demonstrating a critical role for ASMase. Moreover, addition of C16-ceramide recapitulated the effects of cisplatin on ezrin and on the cytoskeleton, indicating that activation of the ASMase/ceramide pathway is sufficient for induction of these responses. Mechanistically, the results demonstrate a critical role for PKCδ upstream of ASMase and for PP2A downstream of ceramide in the dephosphorylation of ezrin. Functionally, dephosphorylation/inactivation of ezrin correlated with reduced mobility of MCF-7 cells after cisplatin treatment in an ASMase-dependent manner. Collectively, these results identify a novel pathway involving ASMase/ceramide in mediating stress responses on the actin cytoskeleton.
A major conclusion from this study emanates from defining a novel molecular/biochemical mechanism for ezrin inactivation. Previous studies demonstrated that binding of the phosphoinositide PIP2 at the N terminus of ezrin facilitates its conformational change (Fievet et al., 2004). However, reciprocal regulation (inactivation) of ezrin by other bioactive lipids and by other pathways remains largely unknown. The data presented here suggest that the ASMase/ceramide pathway mediates stress-induced ezrin dephosphorylation and consequent retraction of filopodia (Fig. 10, model). This model is supported by several lines of evidence. First, cisplatin activated ASMase and induced ceramide in a time course consistent with a role upstream of ezrin dephosphorylation. Second, genetic knockdown of ASMase blocked ezrin dephosphorylation and restored cellular morphology after cisplatin treatment. Third, cells overexpressing DN ASMaseS508A maintained levels of p-ezrin after cisplatin treatment and, more importantly, preserved their migratory response in a wound-healing model. Finally, addition of exogenous ceramide mimicked the morphotropic effects of cisplatin including dephosphorylation of ezrin.
Figure 10.

Model of cisplatin-induced cytoskeletal remodeling. The model depicts activation of the ASMase/ceramide pathway by cisplatin as a key step upstream of actin rearrangement. The serine/threonine phosphatase PP2A links ceramide generation to ezrin dephosphorylation and cytosolic relocation. Inactivation of ezrin results in dissociation of actin filaments from the plasma membrane and resorption of cellular filopodia.
These findings hold major biological implications to tumor progression and therapy. Mounting evidence indicates a role for ezrin not only in tumor survival but also in tumor migration and invasion (Curto and McClatchey, 2004). Aberrant overexpression of ezrin has been documented in a myriad of cancers, some of which hold fatal clinical prognosis such as pancreatic tumors. Additionally, ezrin has been linked to regulation of P-glycoprotein, a pump that is involved in multiple drug resistance (Luciani et al., 2002). As such, ezrin has emerged as a candidate target for cancer therapy. Based on the current study, one could speculate that cisplatin and/or ceramide analogues could be used to combat tumors of high ezrin levels. Intriguingly, using in vivo mouse models, Stover et al. (2005) showed that intravenous delivery of liposomal ceramide reduced the dissemination of ovarian and breast cancers. However, the underlying mechanisms were not investigated (Stover et al., 2005).
The results from this study also significantly extend the spectrum of stress responses mediated by ceramide. Modulation of sphingolipid metabolism is emerging as a shared cellular stress response to chemotherapy (Bezombes et al., 2003; Marchesini and Hannun, 2004). In particular, the ASMase/ceramide pathway has been suggested as a target for different classes of cytotoxic drugs including: DNA damaging agents (doxorubicin [Herr et al., 1997]), histone deacetylase inhibitors (Rahmani et al., 2005), fenretinide (Lovat et al., 2004), and microtubule-targeting agents (paclitaxel [Prinetti et al., 2006]). Activation of ASMase by these agents has been associated with apoptotic cell death. Although the actions of chemotherapies are not limited to cell killing, the role of ASMase in responses other than cytotoxicity have not been well studied. For the first time, the results presented in this study demonstrate that ASMase hinders the mobility of mammalian cells through remodeling of the cellular cytoskeleton.
The present study established that ceramide signaling mediates the cytoskeletal rearrangement produced by cisplatin. Recently, identification of the morphotropic properties of ceramide and other bioactive sphingolipids has emerged as an important goal. Sphingosine-1-phosphate (S1P), a catabolite of ceramide, elicits profound cytoskeletal effects mainly through regulating Rho GTPases (Toman et al., 2004; Donati and Bruni, 2006). Although an increase in ceramide and a drop in S1P may be detrimental for cell survival (Maceyka et al., 2002), the current study suggests that the bioactive lipid ceramide also governs cellular morphology and its associated biological processes. For the first time, we show that ceramide modulates the actin cytoskeleton by inducing dephosphorylation of ezrin. These changes seem to be specific to ceramide, as the inactive dihydroceramide analogue failed to dephosphorylate ezrin. Using the same cell line used in this study, Panigone et al. (2001) observed that up-regulation of prosaposin, an activator of ASMase, or treatment with C2-ceramide, causes defective cell spreading and impaired adhesion. Such cytoskeletal responses elicited by ceramide do not seem to be specific for MCF-7 cells, as similar observations have been made by other investigators in different cell lines. In a recent study, Hu et al. (2005) demonstrated that exogenous C6-ceramide induced loss of filopodia in HeLa cells. The divergence of the morphological effects of ceramide from apoptosis was demonstrated by Spinedi and coworkers, as caspase inhibitors blocked only cell death but not detachment (Di Bartolomeo and Spinedi, 2002). With this in mind, one can propose that ceramide derives its tumor suppressor properties from being not only a pro-apoptotic lipid, but also a morphotropic one.
The results from this study also significantly extend our understanding of signaling mechanisms both upstream and downstream of ASMase. Upstream, the results implicate PKCδ in this pathway. Although activation of ASMase was reported to be a critical step for cisplatin-induced ceramide generation (Lacour et al., 2004), the molecular mechanism underlying this process is not known. A critical role for PKCδ in mediating activation of ASMase by cisplatin is supported by several lines of evidence. In vitro enzymatic assays revealed similar kinetics for ASMase and PKCδ activation after cisplatin treatment. Additionally, infection with DN-PKCδ adenovirus impaired not only activation but also membrane translocation of ASMase in response to cisplatin. These results are in concert with our recently reported mechanism of ASMase regulation (Zeidan and Hannun, 2007) in which activation of ASMase proceeds through PKCδ-mediated phosphorylation of the enzyme at S508. The validity of this model is further corroborated by the finding that the ASMaseS508A mutant failed to translocate to the plasma membrane after cisplatin treatment.
As to downstream mechanisms, the identification of ezrin as a target for PP2A-mediated dephosphorylation extends the current understanding of PP2A as a regulator of cellular motility. Previous work from our laboratory and others established PP2A as a lipid-binding phosphatase that is activated upon ceramide binding to its catalytic domain (Chalfant et al., 1999, 2004). Although the catalytic activity of PP2A has been tightly associated with reduced cellular motility in various tumors, there is little molecular understanding of this process. The present study proposes regulation of ezrin as a key mechanism by which PP2A limits cellular motility. First, targeting PP2A using pharmacologic (okadaic acid) and genetic approaches (RNAi) blocked ezrin dephosphorylation. Second, FRET experiments uncovered an association of PP2A and ezrin at the cell membrane in response to cisplatin. Furthermore, dephosphorylation of ezrin at Thr 526 seemed to be specifically carried by PP2A but not PP1, another ceramide-activated phosphatase, as inhibition of PP1 with tautomycin did not rescue levels of p-ezrin. It is worth noting that dephosphorylation of ERM proteins has been previously reported in response to chemokines such as SDF (Brown et al., 2003), FMLP, and GM-CSF (Yoshinaga-Ohara et al., 2002), but the identities of the phosphatases involved were not established.
Based on the current findings, ASMase emerges as a novel extranuclear target for cisplatin action. Although classically regarded as a nuclear DNA damaging agent, recent studies support a more promiscuous mode of cisplatin action. Safaei et al. (2005) demonstrated that fluorescently labeled cisplatin concentrates within lysosomes. Additional evidence suggests that cisplatin targets lysosomal proteases such as cathepsin D (Emert-Sedlak et al., 2005). Interestingly, cathepsin D has been previously shown as a downstream protease of ASMase activation and a key ceramide-binding protein (Heinrich et al., 1999, 2000, 2004). Although we did not directly address whether activation of ASMase by cisplatin is mediated by a nuclear signal, a recent study by Charruyer et al. (2005) demonstrating activation of ASMase in platelets after UV radiation supports a model of ASMase that is independent of nuclear signaling. Of note, other reports have shown intact cytotoxic effects of cisplatin in enucleated cells (cytoplasts) (Mandic et al., 2003).
In conclusion, the results in this study demonstrate that the ASMase/ceramide pathway conveys stress signals to the actin cytoskeleton machinery. The study unveils a novel sphingolipid-mediated mechanism of ezrin regulation. Based on these findings, a hypothetical model of stress-induced cytoskeletal remodeling is illustrated in Fig. 10.
Materials and methods
Materials
Cell culture material including RPMI media, fetal bovine serum (FBS), and the antibiotic blasticidin S were from Invitrogen. Bovine sphingomyelin and phosphatidylserine were from Avanti Polar Lipids, Inc. [Choline-methyl-14C] sphingomyelin, C16-ceramide, and dhC16-ceramide were provided by Dr. Alicja Bielawska (The Lipidomics Core at the Medical University of South Carolina, Charleston, SC). Antibodies against PKCδ (polyclonal) and HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology, Inc. Monoclonal antibodies against the V5 epitope were from Invitrogen. PP2A (catalytic subunit α) antibody was from BD Biosciences. ASMase polyclonal antibody has been previously described (Zeidan and Hannun, 2007). Polyclonal ezrin antibody (antibody 1; Millipore) was used for Western blotting. A second ezrin polyclonal antibody (antibody 2; Cell Signaling Technology) was used for immunofluorescence. Anti p-ERM antibody and the second anti-ezrin antibody were purchased from Cell Signaling Technology. AlexaFluor 488, 633, and 546 were from Invitrogen. Okadaic acid and tautomycin were purchased from Calbiochem. Lysenin, cisplatin, bacterial sphingomyelinase derived from Bacillus cereus, and all other materials were from Sigma-Aldrich.
Cell lines and culture conditions
MCF-7 cells were originally purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 supplemented with 10% FBS at 37°C in a 5% CO2 incubator. Where indicated, cells were shifted to a phosphate-free medium. Testing for the presence of mycoplasma infections was performed routinely on a monthly basis.
Immunoprecipitation and Western blotting
Cells were treated as indicated in figure legends. Cells were then lysed in a buffer containing 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% NP-40, 2 mM EDTA, 5 mM sodium fluoride, 1.75 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerol phosphate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 μM microcystin. Homogenates were centrifuged at 3,000 g for 10 min. The supernatants were used for immunoprecipitation using specific monoclonal antibodies (1 μg/ml) overnight. Immune complexes were isolated by incubation with protein G–Agarose beads for 3 h. Western blot analysis was carried using similar procedures as described previously (Zeidan et al., 2006).
SMase assays
In vitro enzymatic assays for acid and neutral sphingomyelinases were performed using [choline-methyl-14C] sphingomyelin. The assays were performed as previously reported (Marchesini et al., 2003; Zeidan et al., 2006).
RNA interference
Gene silencing of human ASMase and PP2A (catalytic subunit) was performed essentially according to a standard protocol (Zeidan et al., 2006). Sequence-specific ASMase siRNA reagents were purchased from QIAGEN. The sequences of ASMase RNAi duplexes were sequence [1]: sense CUC CUU UGG AUG GGC CUG G antisense CCA GGC CCA UCC AAA GGA G, and sequence [2]: sense GGU CUA UUC ACC GCC AUC A antisense UGA UGG CGG UGA AUA GAC C. The PP2A (catalytic subunit α) RNAi was purchased from Santa Cruz Biotechnology, Inc. The specificity of all RNAi sequences was verified by sequence comparison with the human genome database using the NIH Blast program.
Plasmid constructs and subcloning procedures
The V5-ASMase plasmid has been previously described (Zeidan and Hannun, 2007). Full-length ezrin cDNA and ezrin T567D were a gift of Dr. Monique Arpin (INSERM, Paris, France). The full-length cDNAs of ezrin and PP2Aα (catalytic subunit) were subcloned into pECFP-C1 and p-EYFP-N1 plasmids, respectively. The CFP tag is situated at the N terminus of PP2A, and the YFP tag is situated at the C terminus of ezrin. EcoRI and BamH1 restriction enzymes were used to place the 929-bp PP2A sequence in frame. EcoRI and SalI restriction enzymes were used to place human ezrin 1760 bp in frame. The integrity of the newly developed plasmids was verified by DNA sequencing. For transient overexpression experiments, endotoxin-free plasmids were transfected into MCF-7 cells using Effectene (QIAGEN) according to the manufacturer's recommendations. LacZ and DN-PKCδ adenoviruses were provided by Dr V. Natarajan (The Johns Hopkins University, Baltimore, MD).
In vitro PKCδ kinase assay
PKCδ immune complexes were washed four times with ice-cold kinase buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgAc, 0.5 mM EGTA, and 0.1 mM DTT). Immunoprecipitates were incubated with the assay buffer consisting of 20 μM ATP, 5 μCi 32P-ATP, 0.2 mg/ml MBP in the presence of 0.2 mg/ml phosphatidylserine and 20 μg/ml DAG. The reaction was carried at 30°C for 15 min and terminated by addition of SDS-PAGE sample buffer. Proteins were separated by electrophoresis, and autoradiography was performed. Samples from each treatment group were tested for equal levels of PKCδ after immunoprecipitation by Western blotting.
Confocal microscopy
Approximately 5 × 104 cells were seeded into 2-cm poly-l-lysine coated confocal plates (MatTek Corp). After treatment, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with methanol for 5 min. Blocking was performed in 2.5% FBS solution. Primary antibodies were diluted in a solution of 1.5% FBS/0.15% saponin and incubated for 3 h (1:200 dilution). The samples were then washed with 1.5% FBS solution three times. This was followed by incubation with secondary antibodies for 1 h at room temperature. Samples were stored at 4°C until image acquisition. Images were acquired using a laser-scanning confocal microscope (LSM 510; Carl Zeiss, Inc.). Excitation wavelengths 488, 543, and 633 were used. Images were acquired at the equatorial plane of monolayer cells guided by DRAQ5 (Qbiogene) nuclear stain and processed using LSM 5 Image Browser (Carl Zeiss, Inc.).
Fluorescence resonance energy transfer (FRET) was used to evaluate protein–protein interaction of PP2A and ezrin in the presence or absence of cisplatin. The applied FRET parameters were: excitation for CFP: 458 nm (argon laser), emission: BP (band pass) 475–525 nm, and excitation for YFP: 514 nm (argon laser) emission LP (long pass) 530. MCF-7 cells were co-transfected with CFP-PP2A (catalytic subunit α) and YFP-ezrin using standard protocols and incubated for 24 h. Images were acquired using a laser-scanning microscope (LSM 510-META; Carl Zeiss, Inc.) under sensitized emission settings. Each image consisted of three channels: donor (CFP) channel; acceptor (YFP) channel; and FRET channel (excitation wavelength 458 nm and emission filters LP 530). For each experiment, appropriate controls transfected with either donor alone (CFP-PP2A) or acceptor alone (YFP-ezrin) were prepared. Cells showing similar fluorescence intensity for the donor (CFP) and acceptor (YFP) were selected for analysis. FRET efficiency was determined using the LSM Image Examiner software (Carl Zeiss, Inc.). FRET pictures were normalized for the FRET efficiencies in the respective setting as indicated by the accompanying rainbow scale (FRET efficiency ranges from blue/0 to red/100%).
Preparation of cytosolic/membrane fractions
Cell pellets from the different treatment groups were lysed by sonication in a buffer consisting of 10 mM Hepes, 150 mM NaCl, 1 mM EDTA, pH 7.4, 5 mM sodium fluoride, 1.75 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerol phosphate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 μM microcystin. Homogenates were cleared of debris by centrifugation at 500 g at 4°C. Samples of the postnuclear supernatants were supplied with sample buffer and corresponded to total cell lysate fractions (T). The membrane fraction (Mb) was obtained by centrifugation at 100,000 g at 4°C for 1 h. The resultant supernatant was denoted as the cytosolic fraction (C).
Mass spectroscopy for ceramide and sphingomyelin
Sphingolipid analysis was performed using Electrospray Ionization/Tandem Mass Spectrometry (ESI-MS/MS) on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple reaction monitoring positive ionization mode. This method has been recently described (Bielawski et al., 2006).
Cell wounding and migration assay
5 × 105 MCF-7 cells stably transfected with V5-ASMase or V5-ASMaseS508A were plated in 6-well culture plates in RPMI medium plus 10% FBS. When cultures were 80% confluent, wounding was performed with a 200-μl Eppendorf micropipette tip. Cells were then washed twice with PBS followed by addition of fresh medium supplemented with mitomycin C (0.5 μg/ml) to control for proliferation. Immediately after wounding, cells were inspected under the microscope (Eclipse TE 200; Nikon) (t = 0). Then at 24 and 48 h, cell migration was analyzed by measuring the maximum migration distance of the cell front into the wounded area. Experiments were performed in triplicate and the distance was measured in three fields of each well.
Preparation of adherent plasma membranes
MCF-7 cells were grown on poly-l-lysine coated confocal dishes 2 cm in diameter. Using a syringe with 25-gauge needle, cells were subjected to repeated streams of 5 ml buffer A: pH 7.4, 10 mM Hepes, 50 mM KCl, and 2 mM MgCl2. Membranes were checked by light microscopy, fixed with 4% formaldehyde, and stained with rhodamine-phalloidin.
F-actin/G-actin in vivo assay
The ratio of filamentous actin (F-actin) to globular actin (G-actin) in MCF7 cells was analyzed using an F-actin/G-actin in vivo assay kit (Cytoskeleton, Inc.) based on the manufacturer's protocol. In brief, following indicated treatments cells were lysed with a cell lysis/F-actin stabilization buffer containing 1 mM ATP and protease inhibitor cocktail. Cells were homogenized by 10 passages through a 28.5-gauge insulin syringe and incubated for 10 min at 37°C. Positive control lysates were incubated with 1 μM phalloidin, and negative control lysates were incubated with 10 μM cytochalasin D. The cell lysates were cleared of unbroken cells and cellular debris by 5 min centrifugation at 2,000 rpm (37°C) followed by centrifugation of the supernatant at 100,000 g for 60 min (37°C) to separate F-actin (pellet) from G-actin (supernatant). The supernatants were separated from the pellets and were immediately placed on ice. The pellets were resuspended to the same volume as the supernatants using ice cold Milli-Q water containing 10 μM cytochalasin D and were incubated on ice for 60 min. Samples were then diluted 10-fold in cold Milli-Q water, and equal volumes (20 μl per lane) were resolved by SDS-PAGE and analyzed by Western blotting with an anti-actin antibody provided with the kit. Western blotting was performed as described above.
Statistical analysis
Mann-Whitney or student's t tests were performed between control and treated states and/or between treatment and treatment plus RNAi-mediated inhibition states on a minimum of three independent experiments. A P-value of 0.05 or less is considered as statistically significant and indicated with an asterisk.
Online supplemental material
Fig. S1 demonstrates the effect of cisplatin on membrane-bound F-actin. Fig. S2 shows resistance of ASMaseS508A cells to cisplatin. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200705060/DC1.
Supplemental Material
Acknowledgments
We thank the Lipidomics Core Facility (Drs. Jacek Bielawski and Alicja Bielawska) at the Medical University of South Carolina. The authors are grateful to Dr. Monique Arpin (INSERM, Paris) for sharing ezrin cDNA, Patrick Roddy for subcloning experiments, and Dr. Chiara Luberto for careful reading of this manuscript.
This work was supported by the National Cancer Institute grant NCI P01-CA97132 (to Y.A. Hannun). Work at the Lipidomics Core is supported by National Institutes of Health grant C06 RR018823. Y.H. Zeidan is supported in part by the Cystic Fibrosis Research, Inc. New Horizons Fund. R.W. Jenkins is supported by Medical Scientist Training Program grant GM08716.
Abbreviations used in this paper: ASMase, acid sphingomyelinase; CN, control; DN, dominant negative; p-ezrin, phosphorylated ezrin; SCR, scrambled RNAi; WT, wild type.
References
- Bezombes, C., G. Laurent, and J.P. Jaffrezou. 2003. Implication of raft microdomains in drug induced apoptosis. Curr. Med. Chem. Anti-cancer. Agents. 3:263–270. [DOI] [PubMed] [Google Scholar]
- Bielawski, J., Z.M. Szulc, Y.A. Hannun, and A. Bielawska. 2006. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 39:8 [DOI] [PubMed] [Google Scholar]
- Bijman, M.N., G.P. van Nieuw Amerongen, N. Laurens, V.W. van Hinsbergh, and E. Boven. 2006. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol. Cancer Ther. 5:2348–2357. [DOI] [PubMed] [Google Scholar]
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Brown, M.J., R. Nijhara, J.A. Hallam, M. Gignac, K.M. Yamada, S.L. Erlandsen, J. Delon, M. Kruhlak, and S. Shaw. 2003. Chemokine stimulation of human peripheral blood T lymphocytes induces rapid dephosphorylation of ERM proteins, which facilitates loss of microvilli and polarization. Blood. 102:3890–3899. [DOI] [PubMed] [Google Scholar]
- Calderon, R.O., and G.H. DeVries. 1997. Lipid composition and phospholipid asymmetry of membranes from a Schwann cell line. J. Neurosci. Res. 49:372–380. [PubMed] [Google Scholar]
- Chalfant, C.E., K. Kishikawa, M.C. Mumby, C. Kamibayashi, A. Bielawska, and Y.A. Hannun. 1999. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274:20313–20317. [DOI] [PubMed] [Google Scholar]
- Chalfant, C.E., B. Ogretmen, S. Galadari, B.J. Kroesen, B.J. Pettus, and Y.A. Hannun. 2001. FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J. Biol. Chem. 276:44848–44855. [DOI] [PubMed] [Google Scholar]
- Chalfant, C.E., Z. Szulc, P. Roddy, A. Bielawska, and Y.A. Hannun. 2004. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 45:496–506. [DOI] [PubMed] [Google Scholar]
- Charruyer, A., S. Grazide, C. Bezombes, S. Muller, G. Laurent, and J.P. Jaffrezou. 2005. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem. 280:19196–19204. [DOI] [PubMed] [Google Scholar]
- Curto, M., and A.I. McClatchey. 2004. Ezrin … a metastatic detERMinant? Cancer Cell. 5:113–114. [DOI] [PubMed] [Google Scholar]
- Di Bartolomeo, S., and A. Spinedi. 2002. Ordering ceramide-induced cell detachment and apoptosis in human neuroepithelioma. Neurosci. Lett. 334:149–152. [DOI] [PubMed] [Google Scholar]
- Donati, C., and P. Bruni. 2006. Sphingosine 1-phosphate regulates cytoskeleton dynamics: implications in its biological response. Biochim. Biophys. Acta. 1758:2037–2048. [DOI] [PubMed] [Google Scholar]
- Emert-Sedlak, L., S. Shangary, A. Rabinovitz, M.B. Miranda, S.M. Delach, and D.E. Johnson. 2005. Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol. Cancer Ther. 4:733–742. [DOI] [PubMed] [Google Scholar]
- Fievet, B.T., A. Gautreau, C. Roy, L. Del Maestro, P. Mangeat, D. Louvard, and M. Arpin. 2004. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J. Cell Biol. 164:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet, B., D. Louvard, and M. Arpin. 2007. ERM proteins in epithelial cell organization and functions. Biochim. Biophys. Acta. 1773:653–660. [DOI] [PubMed] [Google Scholar]
- Gautreau, A., D. Louvard, and M. Arpin. 2000. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J. Cell Biol. 150:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, K., T. Hara, M. Fukasawa, A. Yamaji, M. Umeda, and M. Nishijima. 1998. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 273:33787–33794. [DOI] [PubMed] [Google Scholar]
- Heinrich, M., M. Wickel, W. Schneider-Brachert, C. Sandberg, J. Gahr, R. Schwandner, T. Weber, P. Saftig, C. Peters, J. Brunner, et al. 1999. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 18:5252–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich, M., M. Wickel, S. Winoto-Morbach, W. Schneider-Brachert, T. Weber, J. Brunner, P. Saftig, C. Peters, M. Kronke, and S. Schutze. 2000. Ceramide as an activator lipid of cathepsin D. Adv. Exp. Med. Biol. 477:305–315. [DOI] [PubMed] [Google Scholar]
- Heinrich, M., J. Neumeyer, M. Jakob, C. Hallas, V. Tchikov, S. Winoto-Morbach, M. Wickel, W. Schneider-Brachert, A. Trauzold, A. Hethke, and S. Schutze. 2004. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 11:550–563. [DOI] [PubMed] [Google Scholar]
- Herr, I., D. Wilhelm, T. Bohler, P. Angel, and K.M. Debatin. 1997. Activation of CD95 (APO-1/Fas) signaling by ceramide mediates cancer therapy-induced apoptosis. EMBO J. 16:6200–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W., R. Xu, G. Zhang, J. Jin, Z.M. Szulc, J. Bielawski, Y.A. Hannun, L.M. Obeid, and C. Mao. 2005. Golgi fragmentation is associated with ceramide-induced cellular effects. Mol. Biol. Cell. 16:1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, T., K. Takeuchi, Y. Doi, S. Yonemura, S. Nagata, and S. Tsukita. 1997. ERM (ezrin/radixin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J. Cell Biol. 139:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidering, M., B. van de Water, Y. Zhan, J.J. Baelde, E. Heer, G.J. Mulder, J.L. Stevens, and J.F. Nagelkerke. 1998. Cisplatin effects on F-actin and matrix proteins precede renal tubular cell detachment and apoptosis in vitro. Cell Death Differ. 5:601–614. [DOI] [PubMed] [Google Scholar]
- Lacour, S., A. Hammann, S. Grazide, D. Lagadic-Gossmann, A. Athias, O. Sergent, G. Laurent, P. Gambert, E. Solary, and M.T. Dimanche-Boitrel. 2004. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 64:3593–3598. [DOI] [PubMed] [Google Scholar]
- Levade, T., and J.P. Jaffrezou. 1999. Signalling sphingomyelinases: which, where, how and why? Biochim. Biophys. Acta. 1438:1–17. [DOI] [PubMed] [Google Scholar]
- Lovat, P.E., F. Di Sano, M. Corazzari, B. Fazi, R.P. Donnorso, A.D. Pearson, A.G. Hall, C.P. Redfern, and M. Piacentini. 2004. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J. Natl. Cancer Inst. 96:1288–1299. [DOI] [PubMed] [Google Scholar]
- Luciani, F., A. Molinari, F. Lozupone, A. Calcabrini, L. Lugini, A. Stringaro, P. Puddu, G. Arancia, M. Cianfriglia, and S. Fais. 2002. P-glycoprotein-actin association through ERM family proteins: a role in P-glycoprotein function in human cells of lymphoid origin. Blood. 99:641–648. [DOI] [PubMed] [Google Scholar]
- Maceyka, M., S.G. Payne, S. Milstien, and S. Spiegel. 2002. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta. 1585:193–201. [DOI] [PubMed] [Google Scholar]
- Mandic, A., J. Hansson, S. Linder, and M.C. Shoshan. 2003. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 278:9100–9106. [DOI] [PubMed] [Google Scholar]
- Marchesini, N., and Y.A. Hannun. 2004. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem. Cell Biol. 82:27–44. [DOI] [PubMed] [Google Scholar]
- Marchesini, N., C. Luberto, and Y.A. Hannun. 2003. Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J. Biol. Chem. 278:13775–13783. [DOI] [PubMed] [Google Scholar]
- Meisinger, J., S. Patel, K. Vellody, R. Bergstrom, J. Benefield, Y. Lozano, and M.R. Young. 1997. Protein phosphatase-2A association with microtubules and its role in restricting the invasiveness of human head and neck squamous cell carcinoma cells. Cancer Lett. 111:87–95. [DOI] [PubMed] [Google Scholar]
- Metz, R.J., K. Vellody, S. Patel, R. Bergstrom, J. Meisinger, J. Jackson, M.A. Wright, and M.R. Young. 1996. Vitamin D3 and ceramide reduce the invasion of tumor cells through extracellular matrix components by elevating protein phosphatase-2A. Invasion Metastasis. 16:280–290. [PubMed] [Google Scholar]
- Ogretmen, B., and Y.A. Hannun. 2004. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer. 4:604–616. [DOI] [PubMed] [Google Scholar]
- Panigone, S., R. Bergomas, E. Fontanella, A. Prinetti, K. Sandhoff, G.A. Grabowski, and D. Delia. 2001. Up-regulation of prosaposin by the retinoid HPR and its effect on ceramide production and integrin receptors. FASEB J. 15:1475–1477. [DOI] [PubMed] [Google Scholar]
- Pettus, B.J., C.E. Chalfant, and Y.A. Hannun. 2002. Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta. 1585:114–125. [DOI] [PubMed] [Google Scholar]
- Prag, S., M. Parsons, M.D. Keppler, S.M. Ameer-Beg, P. Barber, J. Hunt, A.J. Beavil, R. Calvert, M. Arpin, B. Vojnovic, and T. Ng. 2007. Activated ezrin promotes cell migration through recruitment of the GEF Dbl to lipid rafts and preferential downstream activation of Cdc42. Mol. Biol. Cell. 18:2935–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinetti, A., D. Millimaggi, S. D'Ascenzo, M. Clarkson, A. Bettiga, V. Chigorno, S. Sonnino, A. Pavan, and V. Dolo. 2006. Lack of ceramide generation and altered sphingolipid composition are associated with drug resistance in human ovarian carcinoma cells. Biochem. J. 395:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani, M., E. Reese, Y. Dai, C. Bauer, S.G. Payne, P. Dent, S. Spiegel, and S. Grant. 2005. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res. 65:2422–2432. [DOI] [PubMed] [Google Scholar]
- Ruvolo, P.P. 2003. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 47:383–392. [DOI] [PubMed] [Google Scholar]
- Safaei, R., K. Katano, B.J. Larson, G. Samimi, A.K. Holzer, W. Naerdemann, M. Tomioka, M. Goodman, and S.B. Howell. 2005. Intracellular localization and trafficking of fluorescein-labeled cisplatin in human ovarian carcinoma cells. Clin. Cancer Res. 11:756–767. [PubMed] [Google Scholar]
- Stover, T.C., A. Sharma, G.P. Robertson, and M. Kester. 2005. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin. Cancer Res. 11:3465–3474. [DOI] [PubMed] [Google Scholar]
- Toman, R.E., S.G. Payne, K.R. Watterson, M. Maceyka, N.H. Lee, S. Milstien, J.W. Bigbee, and S. Spiegel. 2004. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 166:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvoort, A., M.L. Giudici, M. Thielemans, and G. van Meer. 1997. Transport of sphingomyelin to the cell surface is inhibited by brefeldin A and in mitosis, where C6-NBD-sphingomyelin is translocated across the plasma membrane by a multidrug transporter activity. J. Cell Sci. 110:75–83. [DOI] [PubMed] [Google Scholar]
- Xu, L., and X. Deng. 2006. Suppression of cancer cell migration and invasion by protein phosphatase 2A through dephosphorylation of mu- and m-calpains. J. Biol. Chem. 281:35567–35575. [DOI] [PubMed] [Google Scholar]
- Yoshinaga-Ohara, N., A. Takahashi, T. Uchiyama, and M. Sasada. 2002. Spatiotemporal regulation of moesin phosphorylation and rear release by Rho and serine/threonine phosphatase during neutrophil migration. Exp. Cell Res. 278:112–122. [DOI] [PubMed] [Google Scholar]
- Zeidan, Y.H., and Y.A. Hannun. 2007. Activation of acid sphingomyelinase by protein kinase C{delta }-mediated phosphorylation. J. Biol. Chem. 282:11549–11561. [DOI] [PubMed] [Google Scholar]
- Zeidan, Y.H., B.J. Pettus, S. Elojeimy, T. Taha, L.M. Obeid, T. Kawamori, J.S. Norris, and Y.A. Hannun. 2006. Acid ceramidase but not acid sphingomyelinase is required for tumor necrosis factor-{alpha }-induced PGE2 production. J. Biol. Chem. 281:24695–24703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}