

Abstract
Depression and sub-syndromal depressive symptoms are important predictors of morbidity and mortality after acute coronary syndrome (ACS). Prior trials of depression treatment in post-ACS patients have demonstrated no improvement for event-free survival, and only modest improvement in depression symptoms. These trials have raised a number of important issues regarding timing of depression intervention, acceptability of depression treatment to ACS patients, and safety for subsets of the treated population. This article describes Project COPES (Coronary Psychosocial Evaluation Studies), a multi-center Phase-I randomized clinical trial. Project COPES uses a patient preference depression treatment that has previously been found acceptable to medical patients, and a 3-month pre-randomization observation period to insure depression status. The study sample will include 200 post-ACS patients. The primary outcome is patient satisfaction with depression care. Secondary, exploratory aims include the acceptability of depression treatment, reduction in depressive symptoms, and the effects of treatment on two key pathways - medication adherence and inflammation - hypothesized to link depression to post-ACS prognosis. These analyses will provide important data to inform subsequent clinical trials with this population.
INTRODUCTION
Cardiovascular diseases remain the leading cause of death and a major contributor to medical morbidity and disability in the United States. Despite improved prognosis associated with the identification of clinical risk factors and the development of sophisticated interventions, there continues to be a 2.40-fold (1.80-3.20) mortality risk associated with clinical depression and a 1.76-fold (1.30-2.40) mortality risk associated with depression symptoms (c.f., 1) for patients after acute coronary syndrome (ACS), which is defined to include acute myocardial infarction (AMI) and unstable angina (UAP). In an effort to reduce this risk, two large randomized clinical trials, M-HART (2) and ENRICHD (3), were initiated in the 1990’s. The success of prior trials that targeted psychosocial factors that have either a direct or indirect relationship to depression, such as anger, hostility, type A behavior pattern, and general distress (c.f., 4-6), provided support for the overall notion of intervening on an emotion related factor to improve cardiovascular outcomes. In addition, a rich literature demonstrated the efficacy for both cognitive behavioral and pharmacologic therapies in the treatment of depression (c.f., 7). Despite the logic of treating depression to improve cardiac event-free survival - and of the approaches chosen for such treatment - neither trial was successful at improving these outcomes, though statistically modest improvements were realized in the level of depressive symptoms and/or depression status for patients who received active treatment (e.g., for ENRICHD, a mean decrease in BDI score of 49% vs. 33%, intervention vs. usual care, p<.001; 3).
Several reasons have been discussed for the failure of ENRICHD and M-HART to affect medical outcomes (c.f., 3). Among these is that while statistically significant, the modest improvements in depression for patients randomized to intervention may not have been sufficient to affect the pathophysiological pathway(s) (e.g., inflammation, medication adherence) tying depression to post-ACS prognosis. At the same time, patients randomized to usual cardiologic care often demonstrated early and substantial improvement in depression, suggesting that many of the patients randomized into the trial were experiencing a short-lived adjustment reaction rather than real depression, and hence did not carry the associated risk to survival that had been identified in observational studies (1, 8). Under such a scenario, the power to detect an effect would have been greatly reduced. This could have been a particular issue for the ENRICHD trial, which enrolled patients within days after their index ACS event.
Another reason discussed for the modest depression treatment effects and hence, lack of effect on event-free survival concerns the degree to which patients in these trials received a “full dose” of therapy. Analyses from ENRICHD found that because of missed appointments and treatment dropout, only 54% of patients randomized to intervention received the complete course of protocol defined individual treatment (3), while only 31% received the group therapy that was an augmentation to this primary, individual therapy (9). The underlying issue may have been the satisfaction with, and acceptability of the Trial intervention for those randomized to receive treatment. Psychiatric depression trials typically employ reactive recruitment methods, enrolling those who are highly motivated treatment seekers. In contrast, ENRICHD and M-HART employed proactive recruitment methods, approaching all patients in hospital with an admission diagnosis of AMI, screening them to ascertain whether they met depression criteria, and then enrolling them if they did. Hence, because the patients did not “self-present” for depression treatment, many randomized to treatment may have been less than fully motivated to engage in therapy, particularly the approach offered by the Trial.
Concerns regarding the cardiovascular safety of the psychosocial treatments used by M-HART and ENRICHD have also been raised. For example, there was a non-significant but elevated trend to harm (death, p=.06) in the post-ACS female sub-sample randomized to treatment in the M-HART study (11). An exploratory, planned subgroup analysis of the ENRICHD cohort revealed a similar, non-significant but elevated trend to harm (death, p=.12) in post-ACS women randomized to the intervention condition of ENRICHD (3). In both cases excess deaths were primarily cardiovascular and none were from suicide. Speculation for these findings - while post-hoc - includes that the women enrolled and randomized into the treatment condition(s) found the type of treatment provided neither welcome nor particularly helpful to them. As a result, they may have experienced increased distress rather than decreased distress. While this is a post hoc suggestion, it is supported by analyses from M-HART demonstrating a lower event rate among women whose distress symptoms abated as a function of treatment, and a higher event rate among those whose symptoms either increased or did not decrease (12).
Thus, although depression as a clinical syndrome and symptom constellation are each detrimental to event-free survival for post-ACS patients, key questions remain regarding how best to safely and effectively treat depression and its symptoms in post-ACS patients and whether and how this treatment improves event-free survival. These key questions, obstacles to the mounting of any future clinical trials of depression treatment in ACS patients for which event-free survival is the primary outcome, concern: 1) the timing of treatment delivery to capture patients whose depression symptoms are neither transient nor merely reflective of adjustment issues, but rather indicative of depression related risk; 2) the selection of a treatment that is acceptable to post-ACS patients with depressive symptoms, and efficacious for these symptoms; and, 3) the safety of the delivered intervention with regard to medical morbidity and mortality. In addition, the effect of the treatment on hypothesized pathways tying depression to post-ACS survival – such as inflammation and medication adherence - is an important, intermediate issue, as these pathways can serve as surrogate endpoints in smaller, exploratory trials that are not powered for event-free survival. For example, since depression is associated with increased levels of circulating inflammatory markers and ACS is a disease of inflammation, an intervention that reduces both depression and inflammation would be seen as more promising to test in a large trial than an intervention that only decreases depression. Similarly, since depression is associated with reduced medical adherence and reduced medical adherence is in turn associated with reduced post-ACS event-free survival, an intervention that decreases depression and improves medication adherence would be seen as more promising to test in a large trial.
By its design and clinical approach, the Project COPES randomized clinical trial is designed to specifically address these questions. The Trial utilizes a 3-month observational “run-in” phase to insure that the depressive symptoms of immediate post-ACS patients are not merely reflective of transient adjustment issues and persist at 3-months. In addition, the treatment is based on a 6-month patient preference, stepped-care approach that has previously been found acceptable to medical patients with depression (10). Adverse events are monitored to assess cardiovascular safety, and depressive symptoms are assessed prior to, and immediately after the 6-month treatment window to provide estimates of the effect size on depression symptoms that the intervention yields. Information concerning effect size will then contribute to power calculations for future, larger trials should this trial be demonstrated safe for, and acceptable to the patient population. Lastly, questions regarding the ability of this intervention to effect underlying pathophysiology are explored by monitoring of two key pathways – inflammation and medication adherence - hypothesized as tying depression to post-ACS prognosis.
TREATMENT RATIONALE AND DESCRIPTION
For Project COPES the selection of a treatment was based on the need for an approach that would be acceptable to and successful with depressed medical patients, that was amenable to individual tailoring, and that was appropriate for a heterogeneous population comprised of many minority and elderly patients. Based on these concerns the intervention that was selected follows the protocol developed for the IMPACT Trial (10). The IMPACT intervention is defined by: 1) a collaborative care approach, with treatment delivered by a clinical nurse specialist, psychologist, social worker, and/or psychiatrist; 2) a patient choice of psychotherapy and/or pharmacotherapy for the treatment; 3) the use of a form of psychotherapy called Problem Solving Therapy (PST) that is brief, problem-focused and designed to augment the patient’s own skill repertoire; and, 4) the use of a “stepped-care” approach by which symptom severity is reviewed at regular intervals, and treatment is augmented - according to pre-determined decision rules - to promote improvement.
PST (10) is a protocol driven and problem-focused form of cognitive behavioral therapy that comprises one element of the overall IMPACT intervention being utilized by Project COPES. It is delivered by a clinical nurse specialist, social worker, or clinical psychologist trained to criterion in the approach by IMPACT Trial developers and trainers. PST teaches patients how to systematically solve the psychosocial problems that can promote depression onset and maintenance. PST has been found particularly effective for treating depression in patients with comorbid major medical conditions, such as diabetes and musculo-skeletal disease, and it is well accepted by these populations (10, 15). The structure of PST provides for the tailoring of treatment to the specific problems or psychosocial vulnerabilities faced by the individual patients. PST can be delivered by a broad range of mental health providers, making it particularly applicable in medical settings. The treatment model is taught in entirety in the initial session, and reinforced in subsequent sessions, using individual problems that the patient chooses to discuss in these sessions. Through repeated use and reinforcement, problem-solving strategies become an essential element in the patient’s overall behavioral repertoire, thereby contributing to maintenance of treatment gains. The frequency of treatment visits is determined by a combination of treatment progress and patient preference for treatment, utilizing consultation between the patient and the member(s) of the treatment team the patient is working with. PST treatment visits initially occur weekly, with each visit lasting approximately 30-45 minutes. As progress is realized (based on reduction of depressive symptoms), visit frequency is decreased to every 2 weeks, and if progress continues, less frequently, following the IMPACT guidelines. If progress is not being realized, visits can occur more frequently.
Pharmacotherapy treatment utilizes Sertraline, Escitalopram, Venlafaxine, Bupropion, and Mirtazipine, thereby providing for substantial treatment flexibility and tailoring according to the patient’s own medication history and side-effect experiences. Furthermore, these classes of agents have shown a benign side-effect profile for patient with cardiovascular disease in that their use has not been associated with the adverse cardiovascular outcomes seen with the use of other classes of antidepressant medication (16). Patients who choose pharmacotherapy are interviewed by the study psychiatrist to establish the appropriate medication and dosage, based on the patient’s prior medication history and current symptoms. Dosing follows standard clinical practice, with patients placed on what has been identified as the minimally effective dose when treatment is initiated (e.g., for Sertraline, 50mg), though some patients are started on lower doses if medically indicated (e.g., frailty). Patients are initially seen at 1-2 week intervals for dose titration, and thereafter every 3-5 weeks as needed. The study treatment nurse and/or psychiatrist conduct these visits. The focus of these treatment contacts is on change in depressive symptoms and identification of any untoward medication side effects. If a patient choosing pharmacotherapy is already prescribed an antidepressant, their treatment in the study is coordinated with the prescribing physician.
Stepped-care decisions for all patients randomized to intervention are guided by scores on the 9-item Patient Health Questionnaire (PHQ-9; 17), which is administered at each treatment visit. For the purpose of stepped care decisions, these scores are evaluated at 2-month intervals. Patients are maintained on their initial therapy choice as long as they show sufficient improvement in their PHQ-9 score. For these purposes, sufficient improvement is defined as a function of PHQ-9 score at the previous 2-month assessment. If that score was between 5-10, a 30% improvement is required; if 11-20, a 50% improvement is required; if greater than 20, a 60% improvement is required. Patients who do not show sufficient improvement are offered treatment augmentation. For example, patients who initially chose PST could be offered augmentation with pharmacotherapy and/or an increase in the frequency of therapy sessions. Those who initially selected pharmacotherapy could be offered augmenting treatment with PST, a change in their medication, or the addition of a second medication. In this way, level of treatment is matched to the level of depressive symptoms, while patient preferences for treatment guide the treatment decision process.
Successful treatment is also defined by a score on the PHQ-9, in this case by a score ≤ 3 for 2 consecutive therapy sessions; this is independent of the process involving stepped-care decisions that is conducted at 2-month intervals. When a patient achieves this criterion score for 2 consecutive therapy sessions, the patient moves to a monitoring phase of monthly phone contacts, which is maintained for as long as the PHQ-9 remains ≤ 3. If however, PHQ-9 score falls above this threshold during a phone contact, treatment is reinitiated. Patients receiving pharmacotherapy continue to receive their final dosage for up to 12 months after randomization.
UCC
The control condition for the RCT is usual cardiologic care (UCC), as defined by the patient’s treating physician(s). Patients assigned to UCC are referred back to their physicians, where they receive the routine cardiologic care that their physician provides, and any depression or other treatment that the physician feels is warranted; receipt of any depression care out of study is monitored at follow-up assessments. In this way, the effects of the trial intervention can be evaluated against standard of care delivered to patients after ACS. As with patients assigned to active treatment, depression symptoms of patients assigned to UCC are evaluated at 2-month intervals for 6-months.
METHODS
Overview and Objectives
The Project COPES RCT will randomize 200 post-ACS patients meeting depression symptom eligibility criteria (see below) over a 24-month period from 4 tertiary care medical facilities. The recruitment period began in January 2005 and will run to December 2008. The time of patient study participation is 9-months, including the initial 3-month observation period and subsequent 6-month treatment period (see Figure 1).
Figure 1.
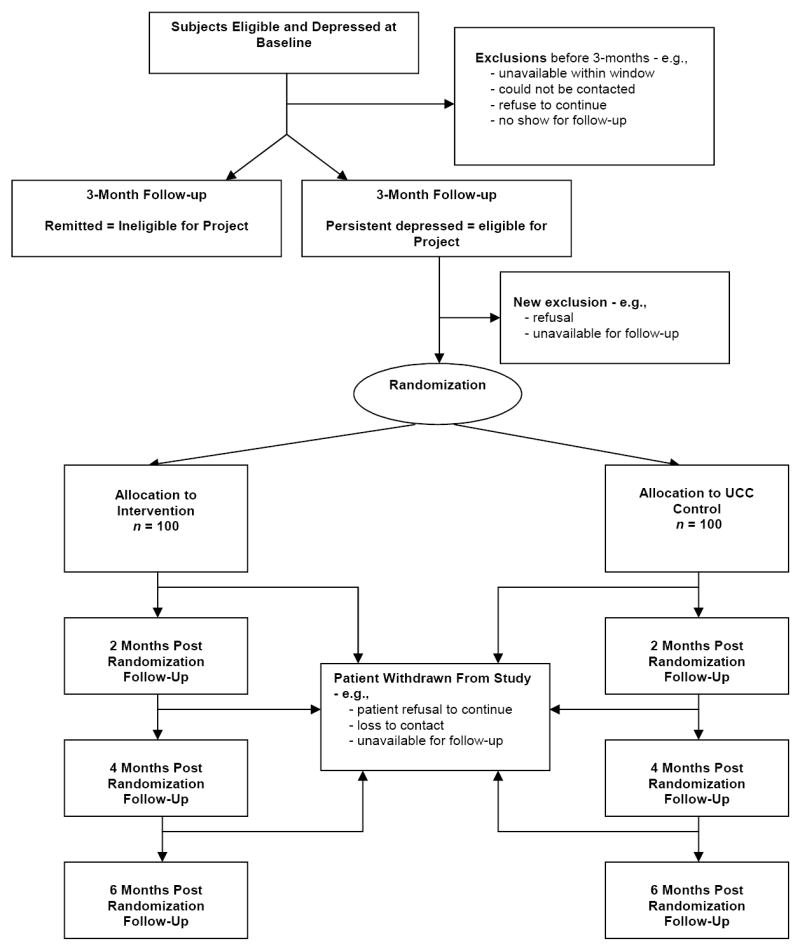
Project COPES RCT Design
The primary hypothesis is that a higher percentage of patients randomized to active treatment (INT) will endorse ‘excellent/very good’ satisfaction with care than will those randomized to UCC. A secondary, exploratory hypothesis is that patients in the INT and UCC arms will have comparable levels of adverse event rates - a combined endpoint that includes all-cause mortality, and cardiac (ACS events - UA, AMI, cardiac hospitalization) and medical morbidity. Side effects associated with any anti-depressant medications used in the trial will also be examined between the two groups. The treatment effect size on depressive symptom reduction on the BDI (13) will be explored, and this information will be used to inform the design of future, post-ACS depression treatment trials.
An additional feature of the COPES RCT is an exploration of the effect that treatment has on patient medication adherence and levels of circulating inflammatory markers, two processes hypothesized as links between depression and post-ACS event-free survival. The focus on pathophysiological pathways has been suggested by the research community as an intermediate step prior to the conduct of further large-scale post-ACS depression trials aimed at medical event-free survival (c.f., 3). For purposes of this study, adherence to the patients prescribed dose of aspirin is assessed, using MEMSCAP technology. The inflammatory markers assessed include C-Reactive Protein (CRP), Interleukin-6 (IL-6), and Intercellular Adhesion Molecule (ICAM).
Eligibility Criteria
Study participants are men and women recruited during a hospitalization with a verified diagnosis of AMI or UA. AMI is defined as at least 2 of the following: ischemic chest pain lasting ≥20 minutes, acute rise in serum troponin-I ≥1.0 μg/L, and new pathologic ST segments in ≥2 contiguous ECG leads. UA is defined as new-onset angina within the past 2 months, exacerbation of previous angina with pain at rest or with minimal exertion, prolonged chest pain (lasting ≥ 20 minutes), or angina within 2 weeks following discharge for myocardial infarction in patients with documented coronary artery disease (defined as ischemic ECG ST-T segment changes, previously documented MI, positive nuclear treadmill test result, or coronary angiographic evidence of blockage of 50% stenosis in ≥1 major coronary artery).
Participants must also meet criteria for symptoms of depression that are evident within 1-week of hospitalization for the index ACS event (baseline) and persist for a 3-month observation period. For this study, the depression symptom criterion is based on a score ≥10 (indicative of at least mild depression) on the BDI, a well-validated assessment tool that is used to screen patients for such purposes (13-14). The original form of the BDI is used so that findings of this study can be directly compared to findings of previous studies with depressed post-ACS patients. The criterion score of ≥10 is based these prior studies, which have shown that this score independently predicts post-ACS event-free survival (c.f., 1).
Patients are excluded if they have a medical condition that is likely to be terminal within the time frame of their participation in the study; if their ACS diagnosis is secondary to a medical procedure; if they have a history of, or are currently demonstrating psychosis, a history of bipolar disorder, or if they meet criteria for current alcohol or other substance abuse disorder; if they demonstrate moderate to severe cognitive impairment on Mini-Mental Status Exam; if there are logistical barriers that preclude their participation in treatment should they be so randomized; if they are unable to communicate in English or Spanish. In addition, patients are excluded if at baseline or 3-month follow-up their symptoms are consistent with melancholic depression, if their BDI score is ≥45, if their score on the BDI suicidality item is ≥2, and/or if they indicate suicidal ideation with intent during clinical interview. In cases where patients meet BDI exclusion criteria (or if patients indicate suicidal ideation with intent on interview) standard risk protocols are initiated (see Safety Protocol, below).
Recruitment, Enrollment, and Informed Consent
The Project COPES protocol was approved by the Institutional Review Board at each recruitment hospital prior to the initiation of screening and recruitment and by an independent Data Safety and Monitoring Board (DSMB).
Prospective study participants are identified through monitoring of hospital admissions for ACS diagnoses, and by subsequent examination of medical records after patients are admitted to coronary care units. After receiving permission from the admitting physician, recruitment staff approach patients meeting ACS medical eligibility criteria at hospital bedside. The study is described, and informed consent and HIPAA (Health Insurance Portability Authorization Act) authorization obtained. Patients are then briefly interviewed to ascertain whether they have any exclusionary criteria; for this purpose, the Mini-Mental Status Exam, the Alcohol Audit, and the psychosis, bipolar disorder, and personality disorder screening items from the SCID are administered. If a participant does not have any exclusion, the BDI is administered. Those who score ≥ 10 on the BDI, whose score is not exclusionary, and who meet full eligibility are then offered participation in the study. Recruitment into the study must be completed within 7-days of hospital admission for the index ACS event.
After informed consent is obtained, patients complete a battery of psychosocial measures, including the SF-36 (18), the Hospital Anxiety and Depression Scale (19), and the ENRICHD Social Support Instrument (20); these scales do not bear directly on the primary and secondary aims of the study. They then provide a blood sample for measurement of inflammatory markers (CRP, IL-6, ICAM). In addition, immediately after discharge, patients are provided with a 3-month supply of their discharge aspirin prescription in a bottle that allows for day/time monitoring of adherence (MEMSCAP). Aspirin was selected for measurement of medication adherence since it is standard of care for post-ACS patients to be prescribed aspirin upon discharge from hospital.
Three months after this baseline assessment, patients complete a follow-up assessment during which depressive symptom status is re-assessed by the BDI. Patients with a persistent score ≥ 10 on the BDI are then invited to participate in the Phase-I RCT, and informed consent acquired. Patients undergo a clinical interview for the assessment of major depression under DSM-IV criteria (21), using the Diagnostic Interview and Structured Hamilton protocol (22). A second blood sample is collected for inflammatory markers and information regarding adherence with aspirin is downloaded from the MEMSCAP bottle, which is then refilled and given back to the patient for monitoring during the treatment phase. Information regarding medical procedures performed and/or hospitalizations experienced since the last contact is also collected.
Key elements of the patient-preference, stepped-care approach to treatment are explained to the patient as part of the informed consent process. To ensure equipoise, the description of the study to patients and their physicians emphasizes that at this time, no data indicate that treating depression and/or its symptoms is of survival benefit to patients after ACS.
Randomization
Eligible patients who consent to the RCT at the 3-month visit are randomized to either the INT or UCC condition, with condition assignment being determined through use of an algorithm that is computer generated and managed. The patient is informed of the condition to which they have been assigned, and questions are solicited and answered. All patients are scheduled for their follow-up assessment visits and letters to their physicians are sent informing the physician of their patient’s participation in the trial and of their randomization condition; the letter also informs the physician if their patient meets diagnostic criteria for major depressive disorder (MDD). Patients assigned to UCC are dismissed with the schedule of their follow-up assessment visits. Patients assigned to the INT condition are given an information brochure describing the intervention. This description includes an overview of the two elements of treatment (PST, pharmacotherapy), the collaborative and patient-preference nature of the treatment, and the stepped care aspect of treatment. Furthermore, this brochure states that there is no evidence of greater efficacy with post-ACS patients for either form of depression treatment, with studies showing that both can be effective. The brochure goes on to state that the choice on their part will be made in concert with members of the research team, who will discuss the patient’s prior treatment experience. After reading the brochure and discussing any questions they have with members of the research team, the patient is asked to indicate their treatment preference, and an appointment is made with the appropriate member of the treatment team. If the patient is not ready to choose a treatment, they are sent home with the information brochure and contacted within 2-3 days by a treatment specialist from the study. Patients are allowed to choose both PST and pharmacotherapy.
Psychosocial interventions cannot be fully blinded. Throughout the process of randomization, selection of treatment element, and subsequent treatment participation, full effort is put forth to ensure that those staff responsible for the collection of treatment outcome data are kept blinded to patient treatment assignment.
Endpoint Follow-up and Measures
Follow-up assessment is conducted 2-, 4-, and 6-months after randomization, and along with baseline assessment includes interview and questionnaire based measures of patient satisfaction with care (see Table 1). At each follow-up visit, patients are asked the following questions regarding satisfaction with care: 1) Over the last 2 months how would you rate the quality of care you have received for your heart, and 2) Over the last 2 months how would you rate the quality of professional care you have received for your symptoms of distress or depression. Patients respond on a 5-point likert scale (1-excellent, 5-poor). For the distress/depression question, patients can also indicate that they have received no care for these symptoms. In addition to satisfaction with care, patients are asked questions regarding any changes in their medical status. Ascertainment of medical status includes the occurrence of any hospitalizations and the reasons, the occurrence/frequency of symptoms associated with coronary disease, and the occurrence/frequency of symptoms consistent with the side-effect profiles of the medications being used in the trial. This information is recorded and checked against the medical record. In addition, receipt of any out-of-study treatment for depression is ascertained, and patients are queried regarding their current medications, and any life-style modifications they have engaged in since their last assessment visit (e.g., exercise program/cardiac rehabilitation, smoking cessation). An ECG is performed at the time of randomization and 6-months later, for ascertainment of silent ACS events and other safety related indices. Level of depressive symptoms is assessed by the PHQ-9 and BDI. Safety of the intervention is determined by a combined endpoint of all-cause mortality, and cardiac (ACS events - UA, AMI, cardiac hospitalization) and medical morbidity (including the above mentioned side effects). Acceptability of and satisfaction with treatment is determined by both the patient response to the question concerning satisfaction with depression care, and by the percent of therapy appointments attended by those randomized to INT. Impact of treatment on pathophysiological processes is determined by changes from baseline in levels of inflammatory markers, and in degree of adherence with aspirin medication among INT patients compared to UCC patients. Percent adherence to antidepressant medication is determined by MEMSCAP.
Table 1.
Study measures and schedule of administration
| Months | ||||
|---|---|---|---|---|
| Measures | Baseline | 3 | 2 & 4 Post-Randomization | 6 Post-Randomization |
| Medical Eligibility | X | |||
| Mini Mental Status Exam | X | X | ||
| Alcohol/Substance Use AUDIT | X | X | ||
| Beck Depression Inventory | X | X | X* | X* |
| SF-36 | X | X* | X* | |
| Hospital Anxiety & Depression Scale | X | X* | X* | |
| ENRICHD Social Support Instrument | X | X* | X* | |
| Diagnostic Interview and Structured Hamilton (DISH) | X* | X* | ||
| PHQ-9 | X* | X* | X* | |
| Adverse Event Checklist | X | X* | X* | |
| Satisfaction with Care items | X* | X* | X* | |
| Blood Sample | X | X* | X* | |
| MEMSCAP | X | X* | X* | |
Indicates items that are administered if the patient meets eligibility for, and is randomized into the RCT
Safety Protocol
Information regarding adverse events, which includes any change in medical status, is reported to hospital IRBs within 48-hours, and to the DSMB every two months. The DSMB meets twice annually.
At any time during their participation in the study, patients who present with suicidal ideation (e.g., BDI item #9 score ≥1) are questioned further regarding specifics of ideation, prior history of such, and all elements of safety (e.g., intent, plan, availability of plan elements, mitigating factors) by a research associate. The associate then contacts senior research investigators by phone to discuss this information, using a “contact tree” until they reach one of these investigators. If warranted, the investigator speaks with the patient, and if the patient is deemed at risk, full efforts are mobilized to mitigate the risk (e.g., transport to emergency room).
Therapy Quality Assurance Procedures
All PST therapy sessions are audiotaped, and local supervisors who have been trained by IMPACT personnel provide 1-hour of weekly supervision that utilizes these audiotapes to insure that therapists are adhering to the protocol. In addition, a random selection of 10% of tapes from each therapist is reviewed by IMPACT personnel to insure adherence to protocol.
Sample Size, Power Calculations and Data Analysis
Primary Hypothesis – Satisfaction With Care, Acceptability of Care
A total of 200 people will be equally randomized to INT and UCC in Project COPES. The IMPACT Trial served as the primary source for determination of power estimates for the primary outcome. In that trial, the odds ratio for patients endorsing a high level of satisfaction with care (care as either excellent or very good) in INT vs. UCC conditions was 3.26 (10). The population in Project COPES differs from that of IMPACT in that we are studying post-ACS patients, in contrast to the primary medical care patients of IMPACT. Hence, we conservatively applied the findings from IMPACT to the current study and anticipate an only moderate effect size (vs. the large effect size for IMPACT), with power at 0.80 for the sample of 200. Analyses of the effects of treatment condition on satisfaction with care will utilize a 2 (INT and UCC) × 4 (time of assessment) repeated measure design; out of study treatment will be included as a covariate. The analysis for satisfaction with care will entail the use of intention to treat principles. Secondary analyses concerning acceptability of treatment will examine the percent of therapy appointments kept by participants randomized to INT.
Exploratory Hypotheses
The dependent variables for exploratory analyses are, 1) BDI score, 2) number of adverse events, 3) percent adherence with aspirin, and 4) levels of inflammatory markers. The approach to exploratory analyses regarding the effects of INT on BDI score will utilize the same 2 (INT and UCC) × 4 (time of assessment) repeated measure design (with out of study treatment included as a covariate) as for satisfaction with care and again, will entail the use of intention to treat principles. For exploratory analyses concerning number of adverse events, we will again employ a 2 (INT and UCC) × 4 (time of assessment) repeated measures design, and the use of antidepressant medications as a covariate; this analysis will be repeated for major adverse cardiac events, and for medication side-effects, separately. This same design will be employed for exploratory analyses concerning percent adherence with aspirin and levels of inflammatory markers, however in these cases, there are only 2 assessments, in contrast to the 4 assessments involved for the other dependent variables.
Organizational Structure
The Project Coordinating Center is in the Behavioral Cardiovascular Health and Hypertension Program, Section of General Medicine, Columbia University School of Medicine. Three clinical centers, Mt. Sinai School of Medicine, Columbia University medical Center, and Yale University School of Medicine participate in recruitment, with the latter center including Yale-New Haven Hospital, Hospital of St. Raphael, and VA Connecticut-West Haven. The Behavioral Medicine Research Group of the National Heart, Lung and Blood Institute is the project office.
Summary
The Project COPES RCT is a multi-site, Phase-I trial of depression treatment in patients after ACS. The RCT intervention utilizes the approach taken by the IMPACT Trial, which was found both highly effective and well accepted by depressed patients followed in medical primary care settings. Overall, COPES is designed to address critical questions raised by prior depression trials among post-ACS patients, including issues regarding satisfaction with/acceptability of treatment, timing of treatment, and patient safety. In addition, questions regarding the effect of treatment on pathophysiological pathways proposed as linking depression to post-ACS event-free survival will be explored. In this way, the Project COPES RCT stands to address key obstacles to future, large-scale clinical trials of depression treatment in this population.
Acknowledgments
This work was supported by grants HC-25197 and HL-04458 from the National Heart, Lung and Blood Institute to Dr. Davidson. The COPES Phase-I Clinical Trial is registered at clinicaltrials.gov
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med. 2004;66:802–13. doi: 10.1097/01.psy.0000146332.53619.b2. [DOI] [PubMed] [Google Scholar]
- 2.Frasure-Smith N. The Montreal Heart Attack Readjustment Trial. J Cardiopulm Rehabil. 1995;15:103–6. doi: 10.1097/00008483-199503000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Berkman LF, Blumenthal J, Burg M, Carney RM, Catellier D, Cowan MJ, Czajkowski SM, DeBusk R, Hosking J, Jaffe A, Kaufmann PG, Mitchell P, Norman J, Powell LH, Raczynski JM, Schneiderman N ENRICHD Investigators. The enhancing recovery in coronary heart disease patients (ENRICHD) study: The effects of treating depression and low social support on clinical events after myocardial infarction. JAMA. 2003;289:3106–16. doi: 10.1001/jama.289.23.3106. [DOI] [PubMed] [Google Scholar]
- 4.Friedman M, Thoresen CE, Gill J, Ulmer D, Powell LH, Price VA, Brown B, Thompson L, Rabin DD, Breall WS, Bourg W, Levy R, Dixon T. Alteration of type A behavior and its effect on cardiac recurrences in post-myocardial infarction patients: summary results of the recurrent coronary prevention project. Am Heart J. 1986;112:653–65. doi: 10.1016/0002-8703(86)90458-8. [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal JA, Jiang W, Babyak MA, Krantz DS, Frid DJ, Coleman RE, Waugh R, Hanson M, Appelbaum M, O’Connor C, Morris JJ. Stress management and exercise training in cardiac patients with myocardial ischemia. Effects on prognosis and evaluation of mechanisms. Arch Intern Med. 1997;157:2213–23. [PubMed] [Google Scholar]
- 6.Linden W, Stossel C, Maurice J. Psychosocial interventions for patients with coronary artery disease: a meta-analysis. Arch Intern Med. 1996;156:745–52. [PubMed] [Google Scholar]
- 7.Davidson KW, Kupfer DJ, Bigger JT, Califf RM, Carney RM, Coyne JC, Czajkowski SM, Frank E, Frasure-Smith N, Freedland KE, Froelicher ES, Glassman AH, Katon WJ, Kaufmann PG, Kessler RC, Kraemer HC, Krishnan RR, Lespérance F, Rieckmann N, Sheps DS, Suls JM. Assessment and treatment of depression in patients with cardiovascular disease: National Heart, Lung, and Blood Institute Working Group report. Psychosom Med. 2006;68:645–50. doi: 10.1097/01.psy.0000233233.48738.22. [DOI] [PubMed] [Google Scholar]
- 8.Lesperance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation. 2002;105:1049–53. doi: 10.1161/hc0902.104707. [DOI] [PubMed] [Google Scholar]
- 9.Saab PG, Bang H, Williams RB, Powell LH, Schneiderman N, Thoresen C, Burg MM, Keefe K. The impact of cognitive behavioral group therapy on event-free survival in patients with new myocardial infarction: The ENRICHD experience. doi: 10.1016/j.jpsychores.2009.01.015. Under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Unützer J, Katon W, Callahan CW, Williams JW, Hunkeler E, Harpole L, Hoffing M, Della Penna RD, Noel PH, Lin EH, Arean PA, Hegel MT, Tang L, Belin TR, Oishi S, Langston C the IMPACT Investigators. Collaborative Care Management of Late-Life Depression in the Primary Care Setting: A Randomized Controlled Trial. JAMA. 2002;288:2836–45. doi: 10.1001/jama.288.22.2836. [DOI] [PubMed] [Google Scholar]
- 11.Frasure-Smith N, Lespérance F, Prince RH, Verrier P, Garber RA, Juneau M, Wolfson C, Bourassa MG. Randomised trial of home based psychosocial nursing intervention for patients recovering from myocardial infarction. Lancet. 1997;350:473–9. doi: 10.1016/S0140-6736(97)02142-9. [DOI] [PubMed] [Google Scholar]
- 12.Cosette S, Frasure-Smith N, Lesperance F. Clinical implications of a reduction in psychological distress on cardiac prognosis in patients participating in a psychosocial intervention program. Psychosom Med. 2001;63:257–66. doi: 10.1097/00006842-200103000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatr. 1961;4:561–71. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 14.Beck AT, Steer RA. Manual for the Beck Depression Inventory. San Antonio, TX: Psychological Corporation; 1993. [Google Scholar]
- 15.Katon WJ, Von Korff M, Lin EH, Simon G, Ludman E, Russo J, Ciechanowski P, Walker E, Bush T. The Pathways Study: a randomized trial of collaborative care in patients with diabetes and depression. Arch Gen Psychiatry. 2004;61:1042–9. doi: 10.1001/archpsyc.61.10.1042. [DOI] [PubMed] [Google Scholar]
- 16.Glassman AH, O’Connor CM, Califf RM, Swedberg K, Schwartz P, Bigger JT, Jr, Krishnan KR, van Zyl LT, Swenson JR, Finkel MS, Landau C, Shapiro PA, Pepine CJ, Mardekian J, Harrison WM, Barton D, McIvor M Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA. 2002;288(6):701–9. doi: 10.1001/jama.288.6.701. [DOI] [PubMed] [Google Scholar]
- 17.Kroenke K, Spitzer RL, Williams JB. The PHQ–9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606–13. doi: 10.1046/j.1525-1497.2001.016009606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ware J, Sherbourne R. The MOS 36-item short-form health survey (SF36): I. Conceptual framework and item selection. Med Care. 1992;30:473–83. [PubMed] [Google Scholar]
- 19.Herrmann C, Buss C, Snaiht RP, editors. HADS-D Hospital Anxiety and Depression Scale. Bern, Goettingen, Toronto, Seattle: Hans Huber; 1995. [Google Scholar]
- 20.Mitchell PH, Powell L, Blumenthal JA, Norten J, Ironson G, Pitula CR, Froelicher ES, Czajkowski S, Youngblood M, Huber M, Berkman LF. A short social support measure for patients recovering from myocardial infarction: The ENRICHD Social Support Inventory. J Cardiopulm Rehab. 2003;23:398–403. doi: 10.1097/00008483-200311000-00001. [DOI] [PubMed] [Google Scholar]
- 21.DSM-IV. Diagnostic and statistical manual of mental disorders. 4. Washington DC: American Psychiatric Association; 1994. [Google Scholar]
- 22.The depression interview and structured Hamilton (DISH): Rationale, development, characteristics and clinical validity. Psychosom Med. 2002;64:897–905. doi: 10.1097/01.psy.0000028826.64279.29. [DOI] [PubMed] [Google Scholar]