

Abstract
Antioxidant action is an important component of the complex neuroprotective action of estrogens. Combining theoretical prediction and subsequent experimental confirmation by chemical and in vitro paradigms, this study focused on the mechanistic aspects of hydroxyl-radical scavenging by 17β-butoxy-1,3,5(10)-estratrien-3-ol, a synthetic derivative of 17β-estradiol with increased potency to inhibit lipid peroxidation and reduced affinity to estrogen-receptors compared to the endogenous hormone. In the process that acts as a “chemical shield,” the phenolic A-ring turns into 10β-hydroxy-17β-butoxy-1,3,5(10)-estratrien-3-one, a non-aromatic para-quinol, upon capturing hydroxyl-radicals, which results in the complete loss of estrogen-receptor affinity and antioxidant activity. However, the parent compound is apparently recovered in brain tissue from this para-quinol via enzyme-catalyzed NAD(P)H-dependent reductive aromatization without causing oxidative stress. Taken together, our report argues for a previously unrecognized antioxidant cycle for synthetic estrogen-derived compounds.
Keywords: 17β-Butoxy-1,3,5(10)-estratrien-3-ol; Antioxidant neuroprotection; Chemical shield; Steroidal para-quinol; Hydroxyl radical; Oxidative stress
1. Introduction
Oxidative damage by reactive oxygen species (ROS) to neuronal cells, accumulation of iron in specific brain areas, and inflammatory processes are considered major pathological aspects of aging and neurodegenerative diseases such as Parkinson’s and Alzheimer's diseases (PD and AD, respectively) [1–3]. ROS are free radicals and molecules, e.g., hydrogen peroxide (H2O2), that have high propensity to produce free radicals under specific conditions. Free radicals have unpaired electron(s) and in order to reach a more stable energy level, they easily pick up electron(s) (or hydrogen atom) from another molecule that, in turn, converts those molecules into free radicals, thus, setting up a chain reaction. Once the process is started, it can cascade, finally resulting in tissue and cell damage eventually leading to cell death and diseases [4]. Therefore, antioxidants that mainly work as chemical electron sinks and quench the free-radical chain reaction has been implicated as potential therapeutic agents to fight against the detrimental effects of brain aging and neurodegenerative diseases [5,6].
It has been well-established that estrogens influence memory and cognition [7, 8], decrease the risk and/or delay the onset of neurological diseases (e.g., AD and PD), and attenuate the extent of cell death that results from a variety of insults involving ROS-induced oxidative stress implicated among others in stroke or neurotrauma [9,10]. Various in vitro and in vivo models show that estrogens are capable to protect neurons in a concentration range of 0.1 nM to 50 µM suggesting the involvement of different modes of action [11–15]. The complex events by which estrogens orchestrate neuronal survival have not been clearly understood [16]. While “classical,” nuclear estrogen-receptor (ER) dependent genomic actions seem to be plausible, it has also been established that the involvement of ERs is not necessary for neuroprotection [17]. There has been mounting evidence that estrogens enhance cell survival by suppressing the neurotoxic stimuli partly via their antioxidant activity [18,19]. Free-radical scavenging is believed to be a significant contributor to the rapid ER-independent neuroprotective actions of estrogens and related compounds [20–22]. The antioxidant property of these compounds is due to the free phenolic hydroxyl group on the A- ring of the steroid [23]. Structure-activity relationship studies have clearly shown that this group is the only structural requirement to achieve cell survival upon oxidative insults [17,24]. Blocking of the phenolic hydroxyl group, e.g., by ether formation completely eliminates neuroprotection [22,25]. The direct oxylradical-scavenging by phenolic antioxidants has been thought to cause interruption of free-radical chain reactions such as lipid peroxidation by rapidly donating the hydrogen atom of the phenolic OH to radicals. Reduction of phenoxyl radicals by intracellular reductants (e.g., ascorbate, thiols) and by enzymes recycles the phenolic antioxidants [26,27]. Alternatively, additional non-radical products may also form [28].
Hormonal effects, including feminization and the potential stimulation of estrogen-sensitive malignancies, have blemished convincing in vitro and in vivo neuroprotection data on estrogens. Estrogen-derived synthetic neuroprotectants that retain the positive effects of estrogens but diminish undesirable side effects have been explored to overcome this problem [17]. 17-Alkyl ethers of 17β-estradiol [25] such as 17β-butoxyestra-1,3,5(10)trien-3-ol (17OBu-E2, 1, Fig. 1) is a representative compound in this regard, because of its significantly reduced ER-binding affinity and increased potency compared to 17β-estradiol (E2) to protect cultured HT-22 neurons against glutamate-induced oxidative stress principally by non-genomic mechanisms [25]. The selection of, e.g., 17OBu-E2 (1) as an estrogen derivative for in vitro neuroprotection experiments involving HT-22 cells and glutamate-induced oxidative stress is also advantageous, since it permits the studies to focus on non-genomic mechanisms such as the antioxidant action without interference from nuclear ER-dependent genomic mechanisms. In addition, the introduction of this D-ring substitution is expected to have weak, if any, influence on the chemistry of the phenolic A-ring of the steroid, which has been presumed to play an important role in the neuroprotective activity of estrogens by exerting an antioxidant action [13, 17]. Here we report the theoretical prediction and the subsequent experimental confirmation of (bio)chemical processes by which 1 may soak up the most toxic ROS, the hydroxyl-radical (•OH) produced by Fenton reaction [29] and argue for the existence of a previously unrecognized antioxidant cycle that maintains a “chemical shield” raised by lipophilic, neuroprotectant E2 derivatives.
Fig. 1.
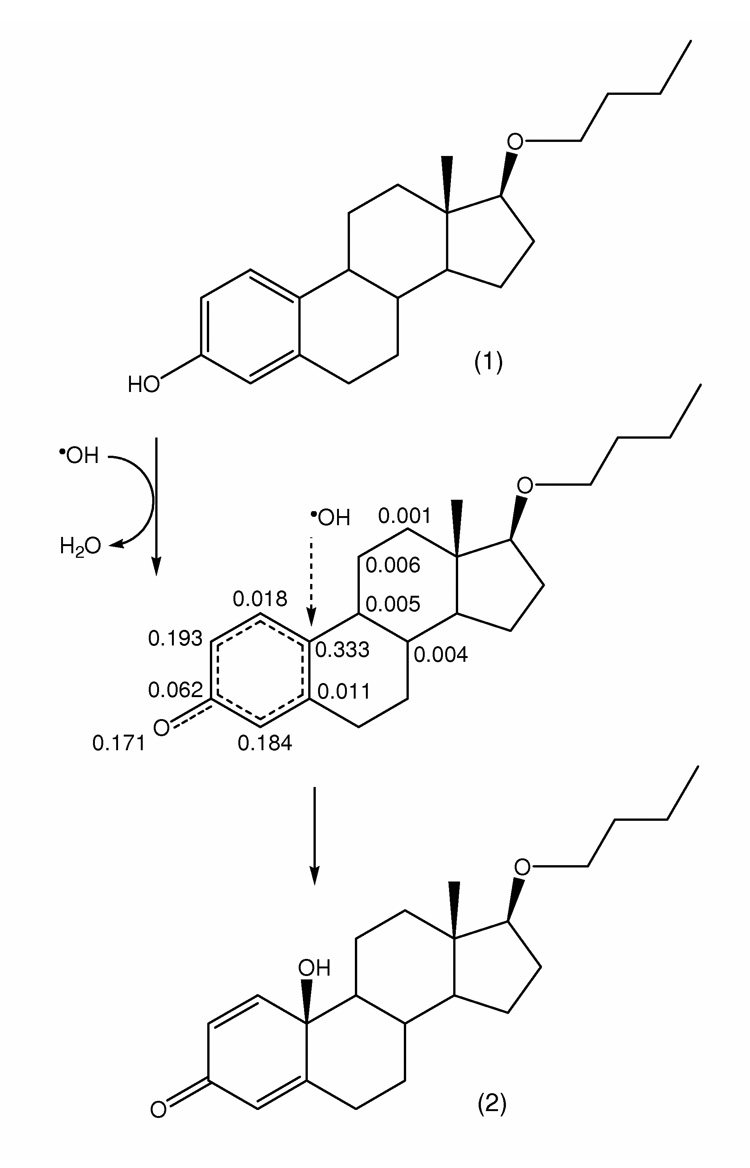
A predicted mechanism of •OH capture leading to the formation of 17OBu-E2-quinol (2) from 17OBu-E2 (1). The structure in the middle displays radical frontier densities on heavy atoms (C and O) for the intermediate obtained by semi-empirical quantum-chemical calculation (PM5, CAChe 6.1.12.34, Fujitsu), which indicates a preferential capture of •OH by the C-10 of the steroid. (For atoms with radical frontier densities <0.001, values are not shown.)
2. Materials and methods
2.1. Instruments and materials
Chemicals were purchased from TCI America (Oregon, USA). A CEM (Matthews, NC) Discover monomode microwave apparatus operating at a frequency of 2.45 GHz with a continuous irradiation power from 0 to 300 W was used for microwave-assisted organic synthesis (MAOS). Temperature was measured by infrared detection with continuous feedback temperature control, and maintained at a constant value by power modulation. The closed-vessel reaction was performed under controlled pressure in a glass tube (capacity 10 mL) sealed with a septum. After the irradiation period, the reaction vessel was cooled rapidly to ambient temperature by compressed air. NMR spectral data were recorded in CDCl3 on a Varian Mercury-400BB (Palo Alto, CA) instrument at 400 MHz instrument using tetramethylsilane as an internal standard. Mass spectra (MS) were obtained by using liquid chromatography (LC) coupled with atmospheric-pressure chemical ionization (APCI) on a quadrupole ion trap instrument (LCQ, Thermo Fisher, San Jose, CA). Melting point (uncorrected) was determined on a Fisher-Johns apparatus.
A brain-homogenate was prepared from ovariectomized (OVX) female rats (220–250 g body weight) purchased from Charles River Laboratories (Wilmington, MA). After killing by cervical dislocation, the brain was immediately removed, weighed and transferred to a 15-ml glass Potter-Elvehjem tissue grinder (Wheaton, Millville, NJ) placed in an ice bath. After addition of an ice-cold phosphate-buffered saline solution (PBS, volume in ml equivalent to four times of the weight of the brain in grams), brain homogenate was prepared by running the ball-shaped teflon pestle of the grinder, with the steel shaft connected to a motor-driven overhead stirrer (Wheaton), for 80 s at 1500 rpm. The brain homogenate was used immediately for the in vitro studies
2.2. Computational studies
Calculations were done after performing full geometry optimization (RMS gradient <0.1 kcal/[Å · mol]) and energy minimization by using the PM5 semi-empirical method with open-shell wave functions [unrestricted Hartree-Fock (UHF) approach], implemented in the BioMedCAChe (Fujitsu, Beaverton, OR) molecular modeling program (version 6.1.12.34).
2.3. Synthesis
17β-Butoxy-1,3,5(10)-estratrien-3-ol (1, 17OBu-E2, Fig. 1) was prepared from 3-O-benzyl-17β-estradiol with sodium hydride/butyl bromide, followed by the removal of the O-benzyl protecting group via catalytic transfer hydrogenation as reported earlier [25]. 10β-Hydroxy-17β-butoxy-1,4-estradiene-3-one (17OBu-E2-quinol, 2, Fig. 1) was obtained by MAOS [30]. Briefly, to 100 mg (300 µmol) of 1 dissolved in 3 ml acetic acid, 200 mg (450 µmol) lead(IV) acetate was added [31]. The reaction vessel was irradiated at 40 °C, with an irradiation power of 150 W, for 20 min. Then the solution was concentrated under a nitrogen stream and the residue was dissolved in 3 ml methanol and CH3OK (15 %, w/v) was added to adjust the pH to 9. This solution was irradiated again for 5 min at 40 °C to hydrolyze the acetyl quinol. The solution was concentrated again and glacial acetic acid was added to adjust the pH to 5. Upon adding ice cold water, the precipitate obtained was filtered off and washed thoroughly with water and purified by column chromatography [silica gel, hexane:ethyl acetate 4:1, (v/v) eluent]. 17OBu-E2-quinol (2), a white solid, was obtained with 54% yield, m.p. 52–56 °C; Rf = 0.65 (hexane : ethyl acetate 4:1, v/v); APCI-MS: m/z [M+H]+ 345; 1H-NMR δ (ppm) having diagnostic value: 7.08 (d, J = 10.2 Hz, 1H, H1); 6.17 (dd, J = 10.2 & 1.6 Hz, 1H, H2); 5.99 (d, J = 1.7 Hz, 1H, H4); 3.41 (m, 2H, OCH2 of butyl); 3.27 (t, J = 8.4 Hz, 1H, 17α); 0.84 (t, J = 7.4 Hz, 3H, CH3 of butyl). 13C-NMR δ (ppm): 185.6; 165.7; 150.7, 128.1; 122, 9; 88.7; 70.4; 69.9; 54.5; 50.1; 43.2; 37.5; 35.0; 33.2; 32.3; 32.0; 28.2; 28.0; 23.8; 23.6; 22.5; 19.4. 19.2. Anal. Calc. C22H30O2 × 0.5 H2O: C, 78.76, H, 9.31; Found C, 78.63, H, 9.40.
2.4. Fenton reaction
17OBu-E2 (1, 100 µM) was incubated in an aqueous medium containing FeSO4 (1.8 mM dissolved in pH 3 sulfuric acid) and H2O2 (36 mM in water). Aliquots were taken at 2.5, 10, 30 and 60 min time points and the solutions were extracted with ethyl acetate (3 × 0.5 mL). The combined organic extract was washed acid free with water, dried over sodium sulfate, and the solvent was evaporated under a nitrogen stream. Samples were dissolved in LC mobile phase and analyzed by LC/APCI-MS.
2.5. In vitro metabolism studies
In vitro formation of 2 from 1 was determined upon incubating 1 (100 µM) at 37 °C in freshly prepared OVX rat brain homogenate (20%, w/v) in 0.1 M phosphate buffer at pH 7.4. To the aliquots taken at different time points, ice-cold glacial acetic acid (50 µL) and ethyl acetate (3 × 0.5 mL) were added, the mixture was vortexed for 1 min and centrifuged at 10,000 rpm for 5 min. The organic layer was removed and the solvent was evaporated at room temperature under a nitrogen stream. Each experiment was performed in triplicate. Tissue-free incubations were done by dissolving NAD(P)H (1 mM) and 2 (100 µM) in 0.1 M phosphate buffer. Control incubations were performed in 0.1 M phosphate buffer only. The sample residues were dissolved in the LC mobile phase and analyzed by LC/APCI-MS. Compound identification was based on retention time (RT), APCI and tandem mass spectra (MS/MS) with synthetic compound (2) as reference.
In vitro reductive regeneration of 1 from 2 was determined analogously NAD(P)H-dependent conversion of para-quinol 2 to the phenolic parent compound (1) was determined after incubating 100 µM para-quinol and 1 mM NAD(P)H in phosphate buffer/acetonitrile (50:50, v/v) at 37°C for 5 min.
2.6. LC-MS analyses
LC separation was done using a Supelco (Bellefonte, PA) 5 cm × 2.1 mm i.d. Discovery HS C-18 reversed-phase column with 0.25 ml/min flow rate using water/acetonitrile/acetic acid (20:79:1, v/v) as a mobile phase. The sample residues were dissolved in 40 µl of mobile phase, respectively, and 5 µL of the solution was injected for analysis. MS/MS product-ion scans were obtained after collision-induced dissociation with helium as the target gas. Compound identification was based on RT, APCI mass spectra and MS/MS with authentic compounds as references [32]. Quantitative analyses were done by LC/APCI-MS/MS and calibration with solutions of known concentrations of the analytes (1 or 2). As an internal standard, 3-methoxy-1,3,5(10)-estratrien-17β-ol (1 µM) [25] was added before each sample extraction.
2.7. Induction of hydrogen peroxide production
Prooxidant activity of 1 and 2 in OVX brain homogenate was evaluated according to a previously published method based on the production of H2O2 production at 37 °C [33]. Briefly, in brain homogenate (0.2% w/v in 0.1M PBS, pH 7.4, final volume of 100 µL) 100 µM test compounds 1, 2 or 3,4-ortho-quinone of estrone as positive control, NAD(P)H (200 µM) and sodium azide (20 µM) at 37°C for 30 minutes in a shaking water bath. Then, 2 mL of a color developing solution (100 µM xylenol orange, 250 µM ferrous ammonium sulfate, and 100 µM sorbitol in 25 mM sulfuric acid) was added, and the mixture was allowed to stand at room temperature for 45 min before spectrophotometric analysis. Absorbance was recorded at 560 nm on a Shimadzu UV250-1PC (Columbia, MD) instrument.
Quantification of H2O2 was done by calibrating with serially diluted H2O2 standard solutions. Rates of H2O2 production were expressed in nmol·h−1·mg protein−1 (mean ± standard error of the mean, SEM). The protein content of the brain homogenates was determined by a dye-binding assay [34] using bovine serum albumin as a reference.
2.8. Estrogen-receptor (ER) binding
Affinities to ERs were evaluated by using an enzyme fragment complementation (EFC) method described in the HitHunter® EFC Estrogen Chemiluminescence Assay kit (Fremont, CA). Compounds (10 pM to 10 µM) were incubated with 5 nM recombinant ERα or ERβ (Panvera, Madison, WI) and estradiol-conjugated enzyme donor for 1.5 hours. The enzyme acceptor was then added for another 1.5 hours after which the chemiluminescence substrate was added for another hour. Relative luminescence was determined using a Biotek FL600 plate reader (Biotek Instruments, Winooski, VT). Sigmoidal standard curves were created by GraphPad Prism (version 3.02 for Windows, GraphPad Software, San Diego, CA) using a four-parameter logarithmic curve to determine the concentration to reach 50% inhibition (IC50).
2.9. Inhibition of lipid peroxidation
The capacity of 1 to inhibit lipid peroxidation was studied in comparison to E2 according to published methods [35,36]. Briefly, incubation mixtures consisted of 2 mL of the compound (at varying concentrations), 2.05 mL of 2.5% (v/v) linoleic acid in ethanol, 4 mL of 0.05 M phosphate buffer (pH 7.0) and 1.95 mL water, in a vial with a screw cap. Control experiments were also conducted using 2.05 ml ethanol without a compound. The incubations were carried out in the dark at 40°C for nine days. Then, the samples were analyzed using both the ferric thiocyanate (FTC) [37] and the thiobarbituric acid reactive species (TBARS) methods [38].
For the FTC method, 0.1 mL incubation mixture and, then, 0.1 mL of 30% (w/v) aqueous ammonium thiocyanate were added into 9.7 mL of 75% (v/v) aqueous ethanol. Thirty seconds after the addition of the ammonium thiocyanate, 0.1 mL of 0.02 M FeCl2 in 3.5% (v/v) HCl was added and the absorbance of the solution was measured at 500 nm 3 min later. For the TBARS method, 0.4 mL 20% (w/v) trichloroacetic acid and 0.4 mL 0.67% (w/v) aqueous thiobarbituric acid were added to 0.2 mL of incubation mixture. After keeping at 90 °C for 10 minutes, the sample was allowed to cool and a supernatant was obtained by centrifugation at 10,000 rpm for 40 minutes. The absorbance of the supernatant was measured at 532 nm. The percentage of inhibition was calculated as (1 –A/A0) × 100; where A was the absorbance of the solution containing the inhibitor and A0 represented the absorbance of the control. IC50 values were determined by non-lineal fitting to sigmoid dose–response profiles with the Scientist for Windows program (version 2.0, MicroMath, Salt Lake City, UT).
2.10. In vitro neuroprotection studies
All studies were done on mouse clonal hippocampal HT-22 cells. The cells were cultured in Dulbecco’s Modified Eagle’s media supplemented with 10% fetal bovine serum under the usual conditions. Experiments were done in 96-well culture plate containing approximately 5,000 cells/well as determined by a Neubauer hemacytometer. The cells were incubated for 24 h before the test compounds were added. 17OBu-E2 (1) and its quinol (2) were dissolved in absolute ethanol and diluted with the culture media to a final concentration of 0.01, 0.1, 1.0, 10 and 100 µM in their respective wells. Sodium glutamate (20 mM) in a solution of phosphate buffer was added together with the test compounds (co-treatment) or 5 h later (pre-treatment). Then, the cells were incubated for 24 h. Cell survival was quantified by the robust Calcein AM assay [39] in a phosphate buffer solution.
2.11. Data analysis
Data are expressed as mean ± SEM. Statistical evaluations were done by one-way analysis of variance (ANOVA) followed by post hoc Dunnett’s test (comparison to a single control group). P < 0.05 was considered statistically significant.
3. Results
3.1. Computational studies
Theoretical calculations on the phenoxyl radical derived from 1 indicated that susceptibility for radical reactions, such as a radical exchange causing an interruption of free-radical chain reactions, was highest at the C-10 (tertiary) carbon. Based on this assessment, we predicted a hydroxyl radical (•OH) radical attack to occur according to the simplified mechanisms shown in Fig. 1. Calculated heats of formation (ΔHf) have also indicated that localizing the radical site formally at the C-10 tertiary carbon may be considered the most stable radical intermediate (−104.4 kcal·mol−1, the lowest among the calculated ΔHf values for possible isomeric radicals), while the C-2/C-4 radical forms obtained by a tautomeric H-shift are considerably less stable [40]. ΔHf values of 10β-hydroxy-17β-butoxy-1,4-estradien-3-one (2, Fig. 1) and the isomeric 10α-hydroxy-17β-butoxy-1,4-estradien-3-one were −155.7 kcal·mol−1 and −151.1 kcal·mol−1, respectively, indicating the preferential formation of the β-isomer. Reaction trajectory calculations from the radical intermediate for the recombination with •OH (ΔHf = 8.9 kcal·mol−1 [41]), an exothermic reaction (ΔH = −60.2 kcal·mol−1) based on the ΔHf values of the reactants and products according to Hess’s Law [42], revealed no transition states. The conclusions that could be drawn from a computational study for the formation of 10β, 17β-dihydroxy-1,4-estradien-3-one from E2 by the mechanism shown for 2 in Fig. 1 were essentially the same as described above in this section (However, the predicted heat of reaction was ΔH = −57.7 kcal·mol−1, which indicated that the radical recombination leading to the formation of the E2-derived para-quinol could be slightly less exothermic than the formation of 2 out of phenoxyl radical derived from 1 and •OH).
The logarithms of the n-octanol/water partition coefficients calculated by the atomic typing scheme of BioMedCAChe (ClogP) for 1 and 2 were 5.49 and 3.15, respectively. The latter lipophilicity-indicating measures were considerably higher than those of the corresponding parent steroids E2 and 10β, 17β-dihydroxy-1,4-estradien-3-one (ClogP of 4.01 and 1.67, respectively).
3.2. Microwave-assisted organic synthesis (MAOS)
The 17OBu-E2-derived para-quinol, 10β-hydroxy-17β-butoxy-1,4-estradiene-3-one (17OBu-E2-quinol, 2), as authentic reference compound, was synthesized by MAOS in acetic acid at 40 °C within 25 min. A rapid hydrolysis of the intermediate para-quinol acetate was also done under microwave irradiation. Our approach [30] significantly reduced the reaction time and the amount of lead(IV) acetate conventionally used [31] providing, therefore, an increased throughput and a “greener” chemistry to obtain synthetic estrogen-derived para-quinols.
3.3. Fenton reaction
When 17OBu-E2 (1) was exposed to •OH generated by chemical Fenton reaction, we found that 1 rapidly (with second-order rate constant of approx. 15 M−1· s−1) converted to one major product that showed an increase of 16 in molecular weight according to its APCI mass spectrum [m/z (M+H)+ 345] obtained after LC separation (Fig. 3), implicating incorporation of a single oxygen into 1 [(M+H)+: m/z 329]. Based on detailed LC/APCI-MS analyses and coelution/comparison (tR, MS and MS/MS) with the synthetic standard prepared from 1 (see previous subsection), this product was identified as 10β-hydroxy-17β-butoxyestra-1,4-diene-3-one (2, Fig. 1); the corresponding para-quinol derived from 1.
Fig. 3.
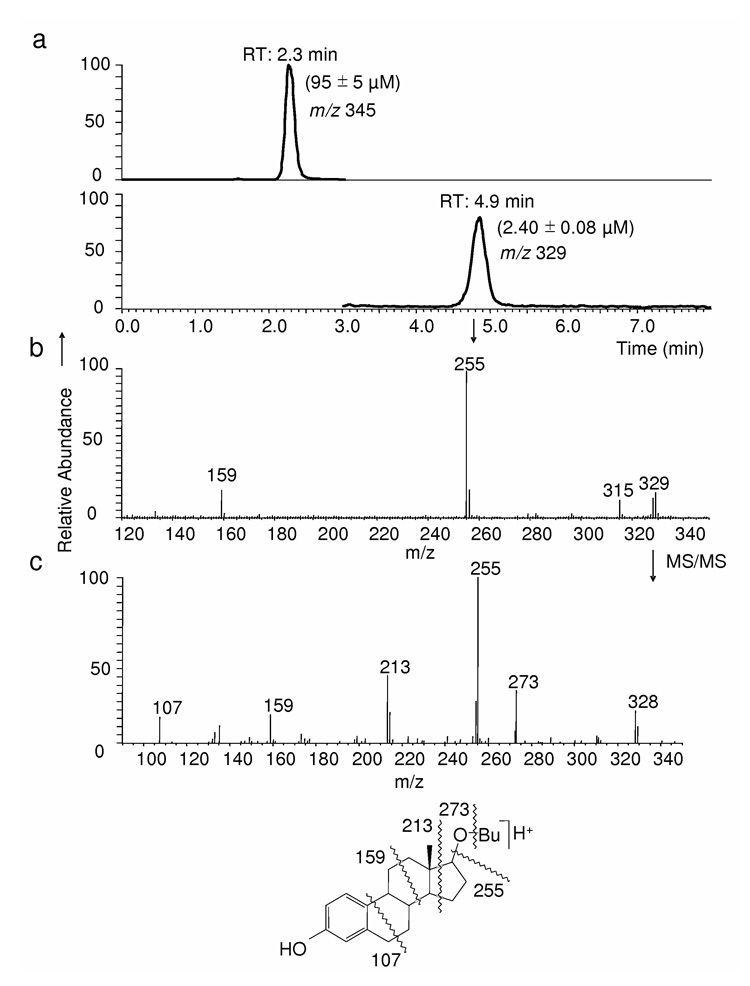
Reductive regeneration of 17OBu-E2 (1) from 17OBu-E2-quinol (2) upon incubation with NADPH. (a) The chromatographic traces displayed are the selected-ion monitoring (SIM) of m/z 345 for 2 and the SIM of m/z 329 for 1. (b) The peak at RT: 4.9 min is unequivocally identified, based on coelution with an authentic reference compound and identical APCI, MS/MS (shown in chart c together with the origin of the major fragments observed), as 1.
3.4. In vitro metabolism studies
To probe whether 2 also arises under biological Fenton reaction, we incubated 1 in brain homogenate of OVX rats for 5 min and confirmed the formation of 2 [at an approximate initial rate of 2.1 ± 0.4 nmol·min−1·(mg protein)−1] as the principal •OH-scavenging product by LC-APCI-MS analyses. Since para-quinols (similarly to cyclohexadienones) can be converted chemically to the corresponding phenols [43], we also studied whether biological reducing agents such as NAD(P)H would also regenerate the synthetic estrogen derivative (1) from its para-quinol (2) by reductive aromatization. We not only confirmed that 2, indeed, smoothly converted chemically to 1 upon treatment with zinc/acetic acid [31], but the formation of 1 (2.4 ± 0.08 µM) could also be detected by LC/APCI-MS (Fig. 3, tR = 4.9 min), when 2 (100 µM) was incubated in the presence of 1 mM NAD(P)H for 5 min. When OVX brain homogenate (20 % w/v) was also used, the 2→1 conversion was found to be approximately 8-times faster; [rate of formation ≈ 210 nmol·min−1 ·(mg protein)−1], implicating that enzymes in the brain further catalyze the recovery of 1 from its radical scavenging product.
3.5. Hydrogen peroxide production
It is imperative to probe whether the reductive recovery of 1 from 2 via an enzymecatalyzed NAD(P)H-dependent reduction in the brain would produce oxidative stress similarly to that of redox cycling between catechol estrogens-derived quinones and their corresponding semiquinones [44]. Such redox cycling is believed to be responsible, in part, for the carcinogenicity of estrogens [45]. The profoundly prooxidant estrone-3,4-quinone [33] was used as positive control in this experimental paradigm. The measured hydrogen peroxide production (given in nmol·h−1· mg protein−1) in OVX rat brain homogenate clearly showed that the para-quinol to phenol (2→1) conversion does not produce oxidative stress compared to that of the control, while estrone-3,4-quinone produced approximately a 5-fold increase in H2O2 production (Fig. 4).
Fig. 4.
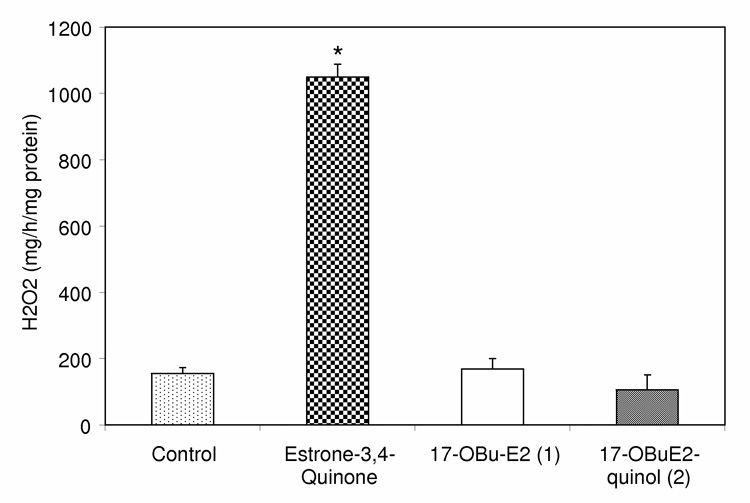
Effect of compounds on H2O2 production in rat brain homogenate. Like 17OBu-E2 (1, open bar), 17OBu-E2-quinol (2, shaded bar) does not induce oxidative stress. Estrone-3,4-quinone [estra-1,5(10)-dien-3,4,17-trione, checkered bar], a profound pro-oxidant, was used as a positive control. (n = 3, *: P<0.05).
3.6. Receptor binding and inhibition of lipid peroxidation
As shown in Table 1, we have confirmed that 2, as expected, had no intrinsic affinity to ERs (IC50 > 10 µM), while the parent phenolic A-ring compound (1) showed competitive binding to ER-α and ER-β (with IC50 values of 12 and 15 nM, respectively), albeit with significantly less affinity compared to E2 (whose IC50 values were 1.3 and 0.7 nM for ER-α and ER-β, respectively). The IC50 values for the inhibition of lipid peroxidation determined by both the FTC and TBAR methods predicted that 1 was more potent [4.1 ± 0.8 µM, (FTC) and 2.7 ± 0.7 µM, (TBARS), respectively] than those of the parent E2 [11.9 ± 1.7 µM, (FTC) and 3.9 ± 0.6 µM, (TBARS), respectively]. On the other hand, estrogen-derived para-quinols (including 2) did not directly inhibit lipid peroxidation neither in the FTC- nor in the TBARS-based ex vivo models used in our study [46].
Table 1.
ER-binding and inhibition of lipid peroxidation of the compounds.
| Compound | IC50 for ER Binding (nM) | IC50 for Inhibition of Lipid Peroxidation (µM) | ||
|---|---|---|---|---|
| ERα | ERβ | FTC Method | TBARS Method | |
| 1,3,5(10)-Estratriene-3,17β-diol (E2) | 1.3 | 0.7 | 11.9 | 3.9 |
| 17β-butoxy-1,3,5(10)-estratriene-3-ol (17OBu-E2, 1) | 12.3 | 15.5 | 4.1 | 2.7 |
| 10β-Hydroxy-17β-butoxy-1,4-estradiene-3-one (17OBu-E2Q, 2) | > 10,000 | > 10,000 | n.i. | n.i. |
3.7. In vitro neuroprotection studies
Reductive recovery of 1 from its non-phenolic para-quinol (2) was also indicated by in vitro neuronal survival data (Fig. 5). When cells were exposed to glutamate-induced oxidative stress in the presence of varying concentrations of the test compounds, 1 and 2 provided protection at a concentration of 10−7 M and ≥ 10−6 M, respectively. However, 5-h pretreatment of the cultured HT-22 cells with 2 (and, thus, “allowing” the cultured cells to convert the para-quinol precursor to the phenolic A-ring steroid 1 before applying the oxidative challenge) significantly improved cell survival compared to co-treatment, and the shift in the dose–response curve compared to the parent phenolic compound (1) diminished.
Fig. 5.
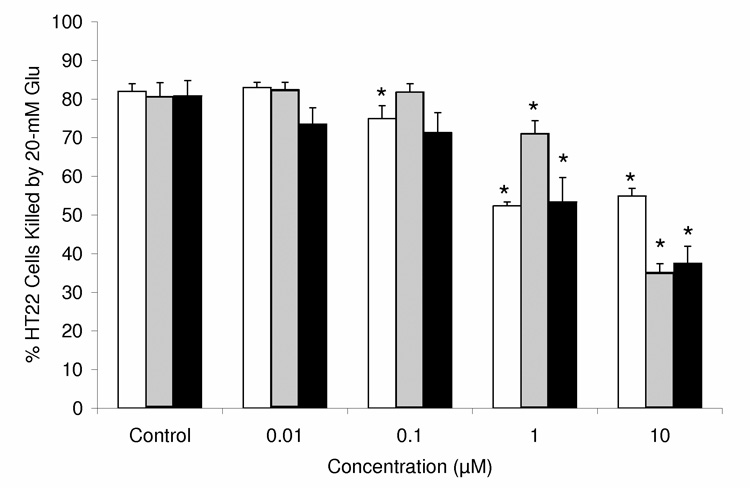
In vitro neuroprotection of hippocampus-derived HT-22 neuronal cells against glutamate-induced oxidative stress (with 20 mM Glu) by the non-phenolic 17OBu-E2-quinol (2, shaded bars), which is augmented upon pretreatment (closed bars), compared with that of the phenolic A-ring parent compound 17OBu-E2 (1, open bars) (n = 6–20, *: P<0.05).
4. Discussion
Considered the most toxic ROS, •OH is an indiscriminant oxidant that reacts at near-diffusion limited rates with a wide variety of biological macromolecules [47] and has been directly implicated in the development and/or progression of numerous neurodegenerative processes and neurotraumas [5]. Several compounds, including estrogens, have been shown to reduce oxidative stress on neurons [5, 6, 13, 15, 17]. In addition to binding to ERs in the cellular membrane, cytoplasm and nucleus [48], estrogens may also interact with lipid-transport proteins [49]. Because of their lipophilicity, they also concentrate in lipid-rich regions of cells such as cellular membranes and affecting and, thereby, have been shown to affect membrane fluidity [50]. Therefore, it is likely that estrogens act in vivo as highly localized antioxidants [28], and estrogen derivatives with enhanced lipophilicity (logP) are expected to increase antioxidant potency compared to the parent steroids. Here, we focused on the chemistry of antioxidant neuroprotection potentially provided by a lipophilic synthetic E2 derivative (1, with ClogP of 5.46 versus ClogP of 4.01 for E2) against •OH attack. This compound was chosen because of its reduced ERs-binding (Table 1), increased potency to inhibit lipid peroxidation (Table 1) and robust in vitro protection of HT-22 cells against glutamate insults [25] when compared to those of E2. Our theoretical calculations predicted that the capture of •OH by the phenoxyl-radical obtained from 1 (in a radical-exchange reaction such as chain-braking of lipid peroxidation [28]) results in a significant change in the aromatic A-ring to yield the principal molecular (non-radical) product. Specifically, the A-ring turns into a non-aromatic para-quinol bearing a hydroxyl group on the 10 (tertiary) position of the steroid (2, Fig. 1.). Because of the large steric congestion on the α-side of the steroid structure, formation of the 10β-isomer was found to be strongly favored thermodynamically. This was also reflected by the lower ΔHf of 10β-hydroxy-17β-butoxy-1,4-estradien-3-one (2, −155.7 kcal·mol−1) versus the isomeric 10α-hydroxy-17β-butoxy-1,4-estradien-3-one (−151.1 kcal·mol−1). Therefore, a central hypothesis of our study was that formation of 2 upon •OH exposure played an important role in the neuroprotective antioxidant action of 1. As an analytical standard and test compound for in vitro studies, 2 was prepared by MAOS [30] that has attracted considerable attention for the rapid synthesis of a variety of organic compounds [51].
An unequivocal experimental conformation of 2 as the principal non-radical product upon •OH attack was done by LC-APCI-MS analyses (Fig. 2). The significant chemical change in the A-ring of the steroid after the completion of this radical-scavenging process is also clearly expressed by the loss of ER-binding affinities and lack of antioxidant potency of 2 when compared to those of 1 (Table 1).
Fig. 2.
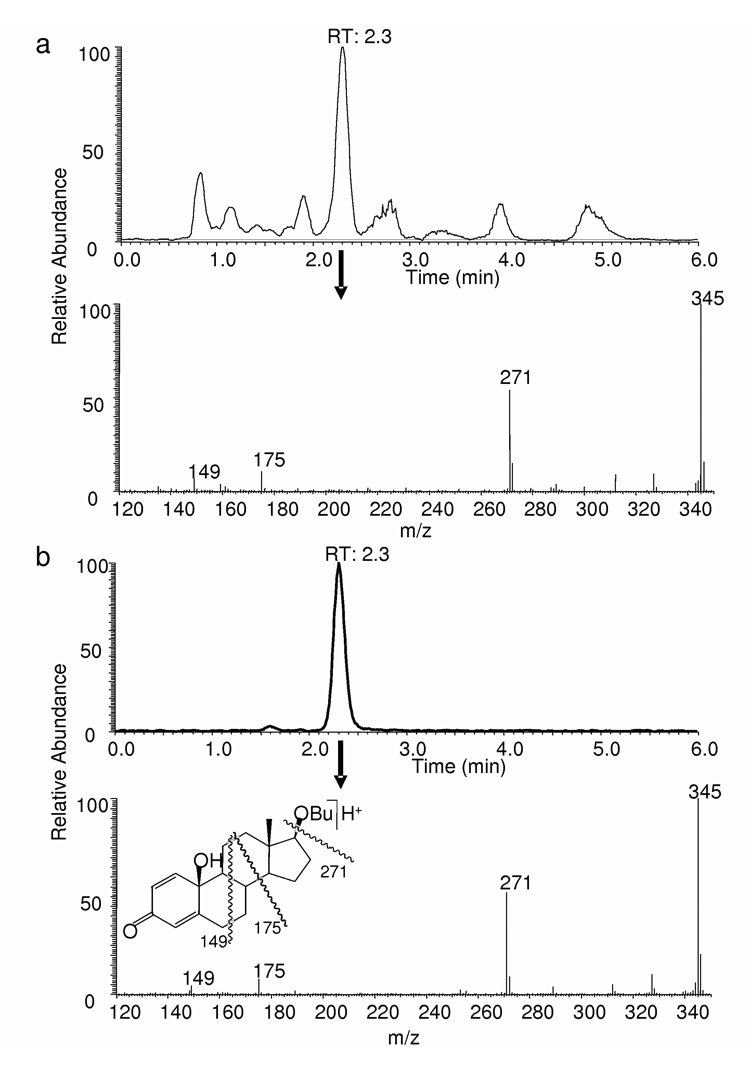
LC-MS analysis of the product formed from 17β-butoxy-1,3,5(10)-estratriene-3-ol (17OBu-E2) upon exposure to •OH generated by the Fenton reaction. (a) Extracted ion-current chromatogram (m/z 345) and APCI mass spectrum from the major LC peak of the reaction product at RT: 2.3 min; (b) Extracted ion-current chromatogram (m/z 345) and APCI mass spectra are from the major LC peak at RT: 2.3 min for synthetic 10β-hydroxy,17βbutoxyestra-1,4-dien-3,17-dione (17OBu-E2-quinol).
We have also unambiguously confirmed the in vitro formation of 2 in a biological medium (in OVX rat brain homogenate) that exposed the substrate (1) to the radical product of the Fenton reaction. It is important to recognize that this “chemical shield” erected by 1 and leading to the conversion to para-quinol strips the A-ring of the steroid from the quintessential functional group (the phenolic hydroxyl group) required for antioxidant neuroprotection. However, we have shown that, similarly to cyclohexadienones [43], para-quinols can be converted back to their phenolic precursors by reductive aromatization. Specifically, the neuroprotective 1 is recovered by a NAD(P)H-depended enzyme-catalyzed reaction without causing oxidative stress (Fig. 4). As such, a true antioxidant cycle exists between the phenolic A-ring synthetic estrogen (1) and its corresponding para-quinol (2) during •OH detoxification (Fig. 6).
Fig. 6.
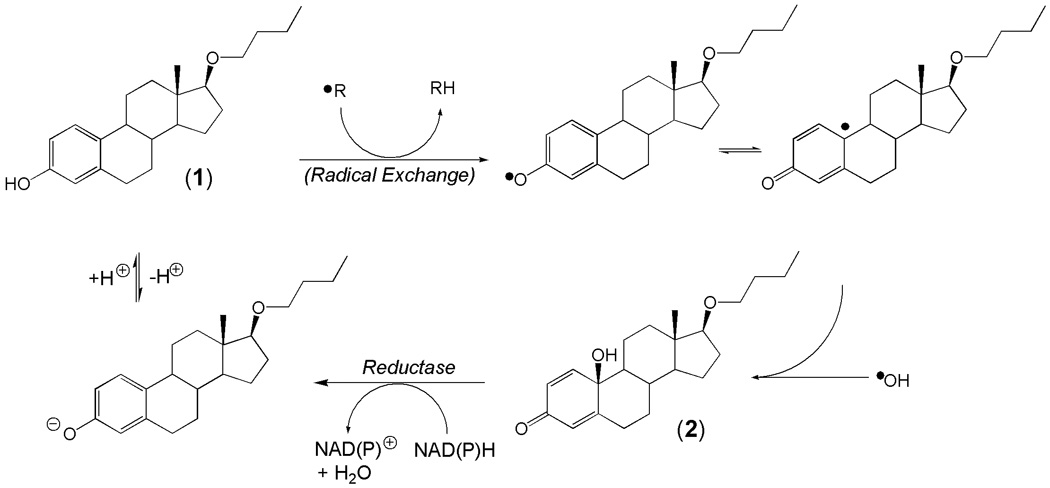
Hydroxyl-radical scavenging antioxidant cycle for 17OBu-E2 (1).
The regeneration of 1, which is a more potent inhibitor of lipid peroxidation (Table 1) based on its higher lipophilicity (with ClogP of 5.46) than the parent estrogen (E2, ClogP = 4.01), from 2 also implicates that the latter may be used as a biochemical precursor (prodrug) of 1. In addition to the lack of intrinsic estrogenicity (Table 1), pharmaceutically relevant physicochemical properties of 2 such as decreased lipophilicity (hydrophobicity) [52] may also be considered more favorable that those of 1.
The existence of the antioxidant cycle and the prodrug mechanism [conversion of para-quinol (2) to phenolic estrogen derivative (1)] were also confirmed by the protection of cultured HT-22 cells against glutamate-induced oxidative stress in the presence of 2 and using 1 as positive control (Fig. 5). The observation that dosing with a non-phenolic para-quinol protected hippocampus-derived HT-22 neurons against glutamate-induced oxidative stress despite its lack of activity as an antioxidant supported our hypothesis for its reductive conversion to a neuroprotective phenolic A-ring steroid (Fig. 3). The shift in the dose–response curve between the parent estrogen derivative (1) and its quinol (2) in our studies was due to the treatment schedule. When compound 2 was added simultaneously with the start of the oxidative insult, the neuroprotective compound 1 evolved in time through activation from 2 as its precursor. Until an adequate concentration of 1 is reached during this activation period, oxidative stress eventually kills a fraction of the cells. On the other hand, a 5-h pretreatment of the cultured HT-22 cells with 2 before the oxidative stressing improved cell survival, and the shift in the dose–response curve compared to the parent phenolic compound practically disappeared (Fig. 5).
In conclusion, we presented the first (bio)chemical evidence of a “chemical shield” provided by a neuroprotective estrogen derivative. In this process an antioxidant cycle exists between the synthetic phenolic A-ring estrogen 1 and its corresponding p-quinol 2 (Fig. 6). The latter may also be used as precursors (prodrugs) for the highly lipophilic parent steroids. These findings may lay the foundation for a rational design of novel neuroprotective agents sought to improve therapeutic safety compared to estrogens.
Acknowledgements
The authors wish to thank Dr. Evelyn J. Perez for completing the cell survival and receptor binding studies. This work was supported by the National Institute of Health Grants: NS44765 and RR12028. Laszlo Prokai is the Robert A. Welch Professor of the University of North Texas Health Science Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chrichton R, Ward R, editors. Metal-based neurodegeneration: from molecular mechanisms to therapeutic strategies. Chichester: John Wiley & Sons; 2006. [Google Scholar]
- 2.Schipper HM, Bernier L, Mehindate K, Frankel D. Mitochondrial iron sequestration in dopamine-challenged astroglia: Role of heme oxygenase-1 and the permeability transition pore. J Neurochem. 1999;2:1802–1811. doi: 10.1046/j.1471-4159.1999.0721802.x. [DOI] [PubMed] [Google Scholar]
- 3.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 4.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Rad Biol Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 6.Cuzzocrea S, Riley DP, Caputi AP, Salvemini M. Antioxidant therapy: A new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Reviews. 2001;53:135–215. [PubMed] [Google Scholar]
- 7.Leranth C, Roth RH, Elsworth JD, Naftolin F, Horvath TL, Redmond DE. Estrogen is essential for maintaining nigrostriatal dopamine neurons in primates: implications for Parkinson's disease and memory. J Neurosci. 2000;20:8604–8609. doi: 10.1523/JNEUROSCI.20-23-08604.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bimonte-Nelson HA, Francis KR, Umphlet CD, Granholm A-C. Progesterone reverses the spatial memory enhancements initiated by tonic and cyclic oestrogen therapy in middle-aged ovariectomized female rats. Eur J Neurosci. 2006;24:229–242. doi: 10.1111/j.1460-9568.2006.04867.x. [DOI] [PubMed] [Google Scholar]
- 9.Simpkins JW, Yang SH, Liu R, Perez E, Cai ZY, Covey DF, Green PS. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004;35:2648–2651. doi: 10.1161/01.STR.0000143734.59507.88. [DOI] [PubMed] [Google Scholar]
- 10.Simpkins JW, Wang J, Wang X, Perez E, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. CNS & Neurol. Disorders - Drug Targets. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- 11.Lacort M, Leal AM, Liza M, Martin C, Martinez R, Ruiz-Larrea MB. Protective effect of estrogens and catecholestrogens against peroxidative membrane damage in vitro. Lipids. 1995;30:141–146. doi: 10.1007/BF02538267. [DOI] [PubMed] [Google Scholar]
- 12.Shwaery GT, Vita JA, Keaney FR., Jr. Antioxidant protection of LDL by physiological concentratons of 17β-estradiol. Requirement for estradiol modification. Circulation. 1997;95:1378–1385. doi: 10.1161/01.cir.95.6.1378. [DOI] [PubMed] [Google Scholar]
- 13.Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- 14.Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Prog Neurobiol. 2001;63:29–60. doi: 10.1016/s0301-0082(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 15.Keller JN, Germeyer A, Begley JG, Mattson MP. 17Beta-estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J Neurosci Res. 1997;50:522–530. doi: 10.1002/(SICI)1097-4547(19971115)50:4<522::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 16.Branna DW, Dhandapania K, Wakadea C, Mahesha VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: Basic mechanisms and clinical implications. Steroids. 2007;72:382–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prokai L, Simpkins JW. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol Therapeut. 2007;114:1–12. doi: 10.1016/j.pharmthera.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niki E, Nakano M. Estrogens as antioxidants. Methods in Enzymology. 1990;186:330–333. doi: 10.1016/0076-6879(90)86126-g. [DOI] [PubMed] [Google Scholar]
- 19.Ayres S, Tang M, Subbiah MT. Estradiol-17β as an antioxidant: some distinct features when compared with common fat-soluble antioxidants. J Lab Clin Med. 1996:367–375. doi: 10.1016/s0022-2143(96)80008-4. [DOI] [PubMed] [Google Scholar]
- 20.Moorsmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green PS, Gridley KE, Simpkins JW. Nuclear estrogen receptor-independent neuroprotection by estratrienes: A novel interaction with glutathione. Neuroscience. 1998;84:7–10. doi: 10.1016/s0306-4522(97)00595-2. [DOI] [PubMed] [Google Scholar]
- 22.Miller CP, Jirkovsky I, Hayhurst DA, Adelman SJ. In vitro antioxidant effects of estrogens with a hindered 3-OH function on the copper-induced oxidation of low density lipoprotein. Steroids. 1996;61:305–308. doi: 10.1016/0039-128x(95)00234-h. [DOI] [PubMed] [Google Scholar]
- 23.Badeau M, Adlercreutz H, Kaihovaara P, Tikkanen MJ. Estrogen A-ring structure and antioxidative effect on lipoproteins. J Steroid Biochem Mol Biol. 2005;96:271–278. doi: 10.1016/j.jsbmb.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 24.Green PS, Gordon K, Simpkins JW. Phenolic A ring requirements for the neuroprotective effects of steroids. J Steroid Biochem Mol Biol. 1997;63:229–235. doi: 10.1016/s0960-0760(97)00124-6. [DOI] [PubMed] [Google Scholar]
- 25.Prokai L, Oon SM, Prokai-Tatrai K, Abboud KA, Simpkins JW. Synthesis and biological evaluation of 17-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J Med Chem. 2001;44:110–114. doi: 10.1021/jm000280t. [DOI] [PubMed] [Google Scholar]
- 26.Kagan VE, Tyurina YY. Recycling and redox cycling of phenolic antioxidants. Ann N Y Acad Sci. 1998;854:425–434. doi: 10.1111/j.1749-6632.1998.tb09921.x. [DOI] [PubMed] [Google Scholar]
- 27.Packer JE, Slater TF, Wilson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1079;278:737–738. doi: 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- 28.Prokai L, Prokai-Tatrai K, Perjesi P, Simpkins JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev Res. 2005;66:118–125. [Google Scholar]
- 29.Walling C. Fenton’s reagent revisited. Acc Chem Res. 1075;28:125–131. [Google Scholar]
- 30.Prokai-Tatrai K, Rivera-Portalatin NM, Rauniyar N, Prokai L. A facile microwave-assisted synthesis of p-quinols by lead(IV) acetate oxidation. Lett Org Chem. 2007;4:265–267. [Google Scholar]
- 31.Gold AM, Schwenk E. Synthesis and reaction of steroidal quinols. J Am Chem Soc. 1958;80:5683–5687. [Google Scholar]
- 32.Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez J, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci USA. 2003;100:11741–11746. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nutter LM, Wu YY, Ngo EO, Sierra EE, Gutierrez PL, Abul- Hajj YL. An oquinone form of estrogen produces free radicals in human breast cancer cells: correlation with DNA damage. Chem Res Toxicol. 1994;7:23–28. doi: 10.1021/tx00037a004. [DOI] [PubMed] [Google Scholar]
- 34.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 35.Koch RB, Stern B, Ferrari CG. Linoleic acid and trilinolein as substrates for soybean lipoxidase(s) Arch Biochem Biophys. 1958;78:165–167. doi: 10.1016/0003-9861(58)90325-4. [DOI] [PubMed] [Google Scholar]
- 36.Ramarathnam N, Osawa T, Namiki M, Kawakishi S. Chemical studies on novel rice hull antioxidants. 1. Isolation, fractionation, and partial characterization. J Agric Food Chem. 1988;36:732–737. [Google Scholar]
- 37.Osawa T, Namiki M. A novel type of antioxidant isolated from leaf wax of Eucalyptus leaves. Agric Biol Chem. 1981:735–739. [Google Scholar]
- 38.Du Z, Bramlage WJ. Modified thiobarbituric acid assay for measuring lipid oxidation in sugar-rich plant tissue extracts. J Agric Food Chem. 1992;40:1566–1570. [Google Scholar]
- 39.Green PS, Perez EJ, Callway T, Simpkins JW. Estradiol attenuation of beta-amyloid-induced toxicity: A comparison of MTT and calcein AM assays. J Neurocytol. 2000;29:419–423. doi: 10.1023/a:1007173509470. [DOI] [PubMed] [Google Scholar]
- 40.Markovic Z, Solaja B, Milic D, Juranic I, Gasic MJ. Molecular orbital study of the oxidation of steroidal phenols into quinols and epoxyquinols. J Serb Chem Soc. 2000;65:491–496. [Google Scholar]
- 41.Ruscic B, Wagner AF, Harding LB, Asher RL, Feller D, Dixon DA, Peterson KA, Song Y, Qian X, Ng CY, Liu J, Chen W, Schwenke DW. On the enthalpy of formation of hydroxyl radical and gas-phase bond dissociation energies of water and hydroxyl. J Phys Chem A. 2002;106:2727–2747. [Google Scholar]
- 42.Logan SR. Physical chemistry for the biomedical sciences. London: Taylor & Francis; 1998. pp. 98–99. [Google Scholar]
- 43.Magdziak D, Meek SJ, Pettus TRR. Cyclohexadienone ketals and quinols: four building blocks potentially useful for enantioselective synthesis. Chem Rev. 2004;104:1383–1430. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]
- 44.Samuni AM, Chuang EY, Krishna MC, Stein W, DeGraff W, Russo A, Mitchell JB. Semiquinone radical intermediate in catecholic estrogen-mediated cytotoxicity and mutagenesis: Chemoprevention strategies with antioxidants. PNAS USA. 2003;100:5390–5395. doi: 10.1073/pnas.0930078100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prokai-Tatrai K, Prokai L. The impact of metabolism on the safety of estrogen therapy. Ann. N. Y. Acad. Sci. 2005;1052:243–247. doi: 10.1196/annals.1347.018. [DOI] [PubMed] [Google Scholar]
- 46.Rivera-Portalatin NM, Vera-Serrano JL, Prokai-Tatrai K, Prokai L. Comparison of estrogen-derived ortho-quinone and para-quinol concerning induction of oxidative stress. J Steroid Biochem Mol Biol. 2007;105:71–75. doi: 10.1016/j.jsbmb.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buxton JV, Greenstock CL, Helman WP, Ross AB. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms, and hydroxyl radicals in aqueous solution. J Phys Chem Ref Data. 1988;17:513–886. [Google Scholar]
- 48.Levin ER. Cell localization, physiology and nongenomic actions of estrogen receptors. J Applied Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 49.Tikkanen MJ, Vihma V, Jauhiainen M, Hockerstedt A, Helisten H, Kaamanen M. Lipoprotein-associated estrogens. Cardiovasc Res. 2002;56:184–188. doi: 10.1016/s0008-6363(02)00535-7. [DOI] [PubMed] [Google Scholar]
- 50.Liang Y, Belford S, Tang F, Prokai L, Simpkins JW, Hughes JA. Membrane fluidity effects of estratrienes. Brain Res Bull. 2001;54:661–668. doi: 10.1016/s0361-9230(01)00483-x. [DOI] [PubMed] [Google Scholar]
- 51.Kappe CO. Controlled microwave heating in modern organic synthesis. Angew Chem Int Eng Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 52.Hansch C, Björkroth JP, Leo A. Hydrophobicity and central nervous system agents: On the principle of minimal hydrophobicity in drug design. J Pharm Sci. 1987;76:663–687. doi: 10.1002/jps.2600760902. [DOI] [PubMed] [Google Scholar]