

Abstract
Epigenetic modifiers are currently in clinical use for various tumor types. Recently, numerous studies supporting the combination of histone deacetylase inhibitors (HDACi) and DNA methyltransferase inhibitors have emerged, encouraging early clinical trials of these agents together. Here we show that MS-275, an HDACi, and 5-azacytidine, a methyltransferase inhibitor, display synergistic cytotoxicity and apoptosis in AML and ALL cells. Intracellular production of reactive oxygen species (ROS), such as superoxide and hydrogen peroxide, is a novel marker for this synergism in ALL cells. These data suggest that assessment of oxidative stress can serve as a marker of the concerted action of MS-275 and 5-azacytidine.
Keywords: MS-275, 5-azacytidine, histone deacetylase inhibitor, DNA methyltransferase, reactive oxygen species
1. Introduction
Epigenetic therapy, the reversal of aberrant epigenetic changes in tumor cells such as the silencing of tumor suppressor genes through means of DNA hypomethylation and histone deacetylation, has become a promising new tool in cancer treatment strategies (1, 2). Classes of drugs such as histone deacetylase inhibitors (HDACi) and DNA methyltransferase inhibitors are currently being used to target these epigenetic changes.
HDACi have been shown to promote differentiation, cell cycle arrest, apoptosis, and the expression of tumor-suppressor genes such as p21WAF1 (3, 4). MS-275 is a novel and highly active benzamide derivative HDACi that has exhibited in vivo and in vitro anti-proliferative activity toward pancreatic, gastric, lung, and ovarian cancer cells (5). As the first member of a new structural class of HDACi, the 2-aminophenyl-benzamides, MS-275 is unique in its profile of inhibition of HDAC family members with high selectivity for HDAC1 and HDAC3 but does not target HDAC8. Thus, it can be used as a tool to discern between relative contributions of these HDACs to cancer types. In addition, MS-275 has been evaluated in Phase I/II clinical trials to treat acute leukemias and solid tumors (6–8).
DNA methyltransferase inhibitors are another family of drugs that target epigenetic aberrations on cancer cells. 5-azacytidine (Vidaza) has been shown to reactivate tumor suppressor and DNA repair genes through the hypomethylation of cytosines (9). Currently, 5-azacytidine is a chemotherapeutic agent for acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) (10, 11). Its derivative, 5-aza-2-deoxyctyidine (decitabine), has already been FDA approved to treat MDS and is currently undergoing Phase III clinical trials (11).
The ability for both classes of drugs to reverse epigenetic alterations provided the rationale for combining the two into a single strategy to treat cancer, and the synergistic cytotoxic effects of the two classes have been confirmed in previous research (12–14). In fact, a recently published Phase I trial of combining phenylbutyrate and 5-azacytidine has demonstrated some complete or partial remissions in refractory solid and hematologic tumors (15). A separate clinical trial employed the same strategy but with different compounds, namely decitabine and valproic acid, and noted that transient DNA hypomethylation and global histone acetylation were induced with the combinatory treatment (16). However, other data, generated from a clinical trial combining MS-275 and 5-azacytidine, suggests that the hypomethylation of specific tumor suppressor genes, such as p15, CDH-1, DAPK-1 and SOCS-1, did not correlate with the clinical effect in myeloid malignancy patients (17). The lack of correlation between hypomethylation of these genes and response noted in that study suggests that other measures of the concerted action of these compounds should be considered. Therefore, this study sought to identify novel biological markers for activity of the combinatory effects of MS-275 and 5-azacytidine which could be quantified in future trials of these epigenetic strategies.
While previous research has shown that the combined treatment of HDACi and DNA methyltransferase inhibitors behave in a synergistic cytotoxic manner in vitro, this work is the first to demonstrate caspase activation, DNA fragmentation, and phosphatidylserine (PS) exposure in leukemia cells treated with MS-275, a novel HDACi, and 5-azacytidine. In addition, given data from several groups indicating that MS-275 as a single agent or combined with other anti-tumor agents generates reactive oxygen species (ROS) in certain tumor cell lines (18–32), the potentiation of ROS with the combined exposure was studied. Our findings show that the production of ROS in AML and ALL cells is an indicator for the synergistic, cytotoxic effects of MS-275 and 5-azacytidine. This ROS production is abrogated by cell permeable glutathione (GSH). Thus, assessment of oxidative stress induced by these compounds may be a novel biological marker for drug action which should be validated within the context of clinical trials.
2. Materials and methods
2.1 Cell Lines
Jurkat and ML-1 human leukemia cell lines were obtained from American Type Culture Collection (Rockville, MD). All cells were maintained in a humidified incubator with 5% CO2 at 37 °C. Cells were cultured in RPMI 1640 with 10% (v/v) heat-inactivation fetal bovine serum (Hyclone, Logan, UT), 2 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Sigma St. Louis, MO).
2.2 Reagents and antibodies
Trypsin-EDTA, propidium iodide (PI), Triton X-100, glutathione reduced ethyl ester, MS-275, and 5-azacytidine were purchased from Sigma (St Louis, MO). Dyes for the detection of intracellular peroxide (6-carboxy-2′,7′-dichlorofluorescein [H2DCF-DA]) and intracellular superoxide (dihydroethium [HEt]) were purchased from Molecular Probes (Eugene, OR). Caspase-3 substrate, DEVD-amc, was purchased from Biomol International, LP (Plymouth Meeting, PA). The pan caspase inhibitor, zVAD, was purchased from Calbiochem (San Diego, CA). Annexin-FITC was purchased from BD Pharmingen (San Jose, CA). Antibodies used were: polyclonal anti-acetyl-histone H3 (Cell Signaling, Beverly, MA), polyclonal anti-histone H3 (Abcam, Inc., Cambridge, MA), actin (Sigma, St. Louis, MO), and ECL anti-rabbit IgG (Amersham Bioscience, UK).
2.3 Western immunoblotting
After treatment, Jurkat cells (5 × 106) were incubated with indicated concentrations for 24 hr, washed in PBS, resuspended in lysis buffer (1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 20 mM Sodium Phosphate, pH 7.4) for 1 hr at 4 °C, and centrifuged at 13,000 × g for 15 min at 4 °C. Thirty μg of each lysate was loaded on a 12% SDS-PAGE acrylamide gels, transferred to nitrocellulose membrane, and blocked overnight at 4 °C with 5% non-fat dry milk in TBS-T (0.05% Tween-20). Membranes were probed with 1:1000 dilution of primary antibody in 5% milk in TBS-T, followed by 1:1000 dilution of secondary antibody in 5% milk in TBS-T. The bound antibodies were detected using the ECL plus Western Blotting detection system (Amersham Bioscience UK limited, Little Chalfont Buckinghamshire, England). Quantitative analysis of the blot was achieved using Gel Doc XR and Quantity One software (Bio-Rad Laboratories, Hercules, CA).
2.4 Assessment of DNA Fragmentation
The percentage of subdiploid of cells was determined by staining cells with propidium iodide (PI) followed by flow cytometric analysis as previously described (31). The cells were incubated for 24 hr, centrifuged at 15,000 × g for 5 min, and resuspended in 500 μL of PI solution (50 μg/mL PI, 0.1%Triton-X-100, and 0.1% sodium citrate in PBS). Samples were vortexed and assessed by flow cytometry on the FL-3 channel (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ). CellQuest software was used for the analysis of the data (BD Bioscence, Franklin Lakes NJ).
2.5 Quantitative analysis of intracellular peroxide and superoxide
The intracellular ROS levels were measured using cell-permeable dyes as previously described (32). After incubation for the designated time, cells were centrifuged and resuspended in 1 mL of RPMI medium containing 10 μM CM-H2DCF-DA to measure intracellular hydrogen peroxide levels or 10 μM HEt to measure intracellular superoxide levels. The samples were incubated for 30 min in the dark at 37 °C. Fluorescence intensity was assessed by the flow cytometer on the FL-1 (DCF) or FL-3 (HEt) channel and analyzed by CellQuest.
2.6 Caspase-3-Like Activity Assay
Cells were incubated for 12 hrs, centrifuged, resuspended in 100 μL PBS, and lysed by freezing and thawing. To each well, 50 μL of lysate and 150 μL of 50 μM DEVD-amc in DEVD buffer (10% sucrose, 0.001% IGEPAL, 0.1% CHAPS, 5 mM HEPES, pH 7.25) were added in duplicate on a 96-well plate. The release of fluorescence (amc) generated from the cleavage of DEVD-amc was measured using a spectrofluorometer (SpectraMax Gemini EM, Molecular Devices, Sunnyvale, CA) with an excitation of 355 nm and an emission of 460 nm.
2.7 Annexin V-FITC Staining
Cells were incubated for 12 hrs, centrifuged, washed twice with cold PBS, and resuspended in 100 μL of binding buffer (10mM Hepes/NaOH (pH 7.4) 140 mM NaCl, 2.5 mM CaCl2). Each sample was treated with 5 μL of annexin and 50 μg/mL of PI. Samples were incubated in the dark at room temperature for 15 min. Each sample was diluted in 1 mL of binding buffer. Fluorescence intensity was assed by flow cytometry on the FL-1 and F1–3 channel and analyzed on CellQuest.
2.8 Statistical analyses
For each condition, three experiments were performed and the results are presented as the mean ± S.D. The differences between two group conditions were analyzed using an independent, two-tailed t-test. Synergism was determined using the software program Calcusyn (Biosoft, Ferguson, MO), which incorporates the Chou and Talalay method (33). A combination index (CI) value < 1.0 indicates synergism.
3. Results
3.1 The combined exposure of MS-275 and 5-azacytidine increases cytotoxic effects of tumor cells
Previous in vitro data indicates the combination of HDACis, such as SAHA and valproic acid, and DNA methyltransferase inhibitors, such as decitabine, resulted in increased cytotoxic effects(4, 7, 10). To extend these studies to a benzamide derivative HDACi –MS-275, the potential, synergistic cytotoxicity of the combination of MS-275 and 5-azacytidine was investigated. ML-1 (AML) cells were treated with either a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or combined treatment in various ratios, incubated for 24 hr, and the percent viability was quantitated by trypan blue exclusion. There was a significant drop in ML-1 cell viability in the 1:1 ratio of the combined treatment compared to single doses (Figure 1a). At the 2:1 ratio of MS-275 to 5-azacytdine, the combined exposure had a combination index (CI) below 1, which indicates synergy compared to the single doses (Figure 1b). According to the CI values, synergistic cytotoxicity was observed at all ratios of combined treatment (Figure 1c).
Figure 1. The combined exposure of MS-275 and 5-azacytidine increases cytotoxic effects of tumor cells.


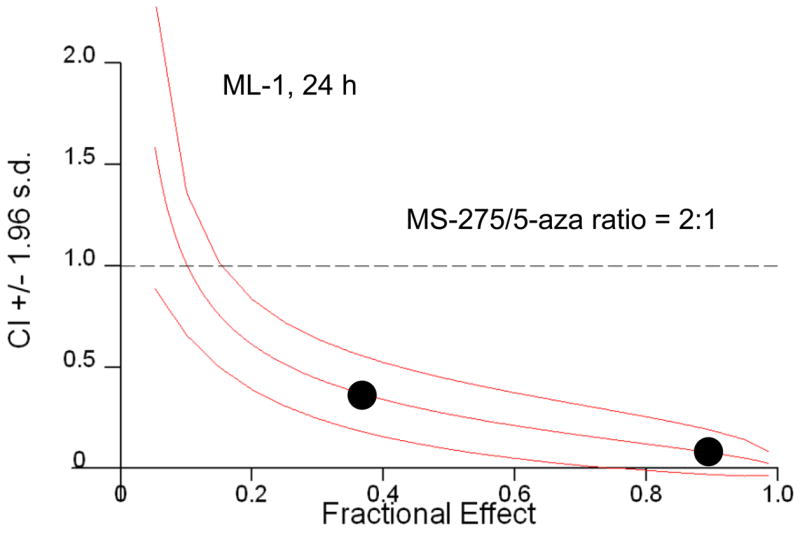
ML-1 cells were treated with either a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a combined treatment in various ratios, incubated for 24 hr, and the % viability was quantitated by trypan blue exclusion and counted on a Vi-Cell Coulter Counter. (a) Line graph representation of synergistic cytotoxicity using the combined treatments. The line graph represents either a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a 1:1 ratio of the combined treatment (2.5 to 10 μM of each drug). Each point represents 3 experiments. (b) Table representation of Combination Index (CI) values. A CI value less than 1 is considered synergistic. (c) Graph representation of the fractional effect values. ML-1 cells were treated with either a range of MS-275 (5 to 10 μM), a range of 5-azacytidine (2.5 to 5 μM), or a combined treatment at a fixed 2:1 ratio. The fractional effect values were determined using Calcusyn software.
3.2 The percentage of apoptotic cells synergistically increased with the combined exposure of MS-275 and 5-azacytidine
Since combinatory treatment resulted in a synergistic cytotoxic effect, the next step was to understand whether the induced cell death was a result of the activation of the apoptotic pathway. DNA fragmentation is a feature of apoptosis and can be quantitated by measuring cells with subdiploid amounts of DNA using PI. ML-1 and Jurkat (ALL) cells were treated with either a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a combined treatment in various ratios, incubated for 24 hr, incubated with PI reagent and assessed on the flow cytometer. For ML-1 cells, all ratios of combination resulted in CI values less than 1, which indicates a synergistic increase in DNA fragmentation with the combined exposure (Figure 2a and b). In the Jurkat cells, all of the dose combination was synergistic (Figure 2c and d). The results from both cell lines indicate that the synergistic cytotoxicity was due to the induction of the apoptotic pathway. In order to verify that the synergistic increase in DNA fragmentation of the cells was apoptotic rather than necrotic, Annexin V-FITC/PI staining was used to identify the percentage of cells that expressed PS on their cell surface, which is an obligate event for phagocytosis of apoptotic cells. This staining confirms that the percentage of Annexin V positive cells increases in the combination treatment relative to the individual doses (Figure 2e).
Figure 2. Apoptotic DNA fragmentation is synergistically increased by the combined exposure of MS-275 and 5-azacytidine.





ML-1 cells and Jurkat cells were treated either with a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a combined treatment in various ratios, incubated for 24 hr, incubated with the PI and assessed on the flow cytometer (FL-3). The bar graphs represent 3 experiments. Inset (marked by M1 region) shows the histograms for each treatment. (a) Bar graph and inset histograms of the synergistic increase in apoptotic DNA fragmentation via PI staining in ML1 cells using 5 μM MS-275 and 5 μM 5-azacytidine. (b) Table representation of the CI values of various ratios of the drugs in ML-1 cells. (c) Bar graph and inset histograms of the synergistic increase in apoptotic DNA fragmentation via PI staining in Jurkat cells using 5 μM MS-275 and 5 μM 5-azacytidine. (d) Table representation of the CI values of various ratios of the drugs in Jurkat cells. (e). Dot plot of the increase in population of apoptotic cells via Annexin-FITC staining in Jurkat cells using 5 μM MS-275 and 5 μM 5-azacytidine. Cells were treated with or without the combined treatment of 5 μM MS-275 and 5 μM 5-azacytidine, incubated with Annexin-FITC stain, and assessed on the flow cytometer (FL-1 and FL-3). The figure shows only one of the experiments, but all three experiments were comparable.
3.3 Caspase-3-like activity increases with the combined exposure of MS-275 and 5-azacytidine and is blocked by caspase inhibitor zVAD
In order to verify that the induction of the apoptotic pathway was caspase dependent, caspase-3-like activity was measured using a fluorogenic substrate DEVD-amc. Jurkat cells were treated with 5 μM MS-275, 5 μM 5-azacytidine, or a combination of the two, with or without a 30 minute pre-treatment with 10 μM zVAD, a pan-caspase inhibitor, and samples were incubated for a total of 12 hrs. Fluorescence, indicating liberation of the amc substrate from the caspase-3 substrate, was read on a spectrofluorometer. There was a statistically significant increase in caspase-3 activation with the combined treatment compared to either single dosage of MS-275 or 5-azacytidine (p-value = 0.006). In addition, the pan-caspase inhibitor blocked caspase activation in the combined exposure (p-value = 0.004). These results indicate the synergistic cytotoxicity was a result of caspase activation, further implicating an apoptotic pathway in the mechanism of cytotoxicity of these drugs (Figure 3).
Figure 3. Caspase-3-like activity is increased by the combined exposure of MS-275 and 5-azacytidine and is blocked by the caspase inhibitor zVAD.

Jurkat cells were treated with either 5 μM of MS-275, 5 μM of 5-azacytidine, or a combination of the two and incubated for 12 hrs. Samples were pre-treated with or without 10 μM of zVAD for 30 min and incubated for 12 hrs. The release of amc was measured using a spectrofluorometer.
3.4 The rate of histone- 3 acetylation dramatically increased with the combined exposure of MS-275 and 5-azacytidine
Previous studies have shown that HDACi prevent deacetylation of histones, which leads to an increase in expression of anti-tumor genes due to a higher rate of acetylated histones(1, 2). Analysis of specimens from clinical trials has shown histone acetylation is maintained with the combination of HDACi and DNA methyltransferase inhibitors in leukemia patients(16). To determine if histone acetylation was potentiated by MS-275 and 5-azacytidine, acetylated histone H-3 was measured by western blotting. Jurkat cells were treated with either 5 μM of MS-275, 5 μM of 5-azacytidine, or the combination of the two drugs, incubated for 24 hr, and lysed. The western blot showed a two to three fold increase in the acetylation of histone H-3 with the combined exposure compared to either single drug treatment (Figure 4). Therefore we find, in accordance with previous literature on different combinations of HDACi and hypomethylating agents, the synergistic cytotoxicity of using MS-275 and 5-azacytidine is also marked by an accumulation of histone acetylation(16).
Figure 4. Histone 3 acetylation is increased by the combined exposure of MS-275 and 5-azacytidine.
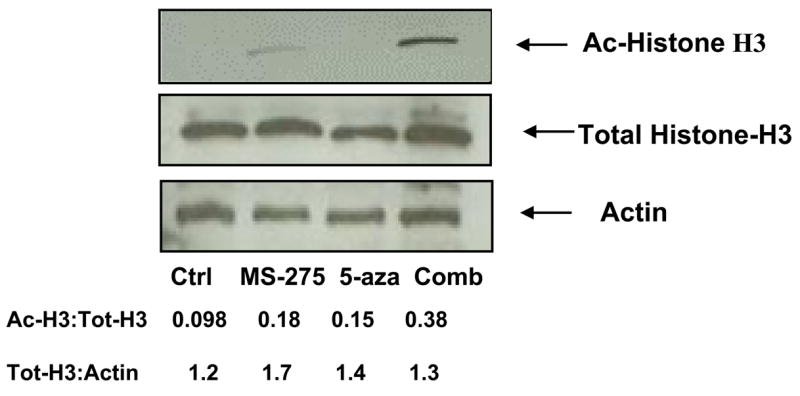
Jurkat cells were treated with either 5 μM of MS-275, 5 μM of 5-azacytidine, or the combination of the two drugs, incubated for 24 hr, and lysed. Western blotting for acetylated histone-H3, total histone-H3, and actin was performed (n = 3). Densitometry was used to quantitatively analyze the western blot.
3.5 Intracellular reactive oxygen species (ROS) levels synergistically increase by combined exposure of MS-275 and 5-azacytidine and are partially blocked with pretreatment of glutathione ester
Both MS-275 and the derivative of 5-azacytidine, decitabine, have been shown to increase intracellular ROS levels as single agents(3). To test if ROS production by the combination increases in a synergistic manner, intracellular peroxide and superoxide levels were measured. Jurkat cells were treated with either a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a combined treatment in various ratios, incubated for 16 hr, stained with dihydroethidium and assessed by flow cytometry. Cells treated with the combination showed increased levels of superoxide, and CI values for all the ratios of combination were less than 1 (Figure 5 a,b). Levels of intracellular peroxide were also synergistically increased in cells treated with both agents, with all but two of the CI values for the ratios of combination were less than 1 (Figure 5 c,d). Taken together, these CI values suggest that there was a definite synergistic increase in intracellular ROS levels using the combination of the two drugs. Therefore, ROS generation is an indicator for the synergistic effect of the combined exposure of the two drugs.
Figure 5. Intracellular ROS levels are synergistically increased by combined exposure to MS-275 and 5-azacytidine and ROS is partially blocked with glutathione ester pretreatment.






Intracellular superoxide synergistically increased with the combination of MS-275 and 5-azacytidine. Jurkat cells were treated either with a range of MS-275 (2.5 to 10 μM), a range of 5-azacytidine (2.5 to 10 μM), or a combined treatment in various ratios for 16 hr. Cells were then assessed for superoxide levels by dihydroethidium staining. (a) Bar graph represents 3 experiments using either 5 μM of MS-275, 5 μM of 5-azacytidine, or the combination of the two drugs. Inset: representative histogram showing M1 marker shows the increase in superoxide levels that is depicted by the bar graph. (b) Table represents the CI values for the synergistic increase in superoxide levels at various dosages using MS-275 and 5-azacytidine. Intracellular hydrogen peroxide synergistically increased with the combination of MS-275 and 5-azacytidine. Jurkat cells were treated with either 5 μM of MS-275, 5 μM of 5-azacytidine, or the combination of the two drugs, incubated for 16 hr. The CM-H2DCF-DA dye was used to detect intracellular peroxide, and fluorescence was assessed on a flow cytometer (FL-1). (c) Bar graph represents 3 experiments using either 5 μM MS-275, 5 μM 5-azacytidine, or the combination of the two drugs. Inset: Representative histogram showing M1 marker illustrates the increase in hydrogen peroxide levels that is depicted in the bar graph. (d) Table represents the CI values for the synergistic increase in hydrogen peroxide levels using MS-275 and 5-azacytidine. (e and f). The increased levels of ROS in Jurkat cells were partially blocked with glutathione ester pretratement. Jurkat cells were pretreated with 2 mM glutathione ester for 1 hr, then treated with or without the combined treatment of 5 μM MS-275 and 5 μM 5-azacytidine, incubated with for 16 hr, dyed with dihydroethidium staining, and assessed on the flow cytometer (FL-3). Bar graph represents 3 experiments.
In order to further implicate ROS in the mechanism of cytotoxicity of the combination, cells were pretreated with the cell permeable form of GSH, glutathione ethyl ester, before the combined treatment. Jurkat cells were pretreated with 2 mM of glutathione reduced ethyl ester for 1 hr and then treated with 5 μM MS-275 and 5 μM 5-azacytidine, incubated for 16 hr, stained with dihydroethidium or CM-H2DCF-DA dye, and assessed by flow cytometry. In figure 5e–f, experimental results showed that the pretreatment with glutathione ester significantly decreased the percentage of cells with high superoxide levels increase from an average of 52% to 43% (p-value = 0.042). This result indicates that the increase in ROS with the combined treatment can be partially blocked with the addition of glutathione.
4. Discussion
Since MS-275 and 5-azacytidine have undergone early clinical testing as single agents, and a new trial combining the two agents is underway, we set out to elucidate the mechanism of synergy between the two compounds in leukemia model systems. In this study, the combined exposure of MS-275 and 5-azacytidine resulted in a synergistic cytotoxic effect through the activation of the apoptotic pathway in AML and ALL cell lines. In addition, an increased rate of histone acetylation was observed in the AML cell line with the combinatory treatment. This result indicates that the epigenetic effects of MS-275 were maintained, if not enhanced, by the addition of 5-azacytidine. Furthermore, we find that an increase in intracellular ROS level parallels the cytotoxicity of the combined drug treatment. This data suggests that production of ROS is a novel marker for the synergistic nature of the two drugs.
Our study also shows for the first time that ROS generation by the combination of MS-275 and 5-azacytidine is inhibited by cell permeable GSH. Together with previous data demonstrating that N-acetyl-cysteine (NAC), a general oxidant, blocks DNA fragmentation by MS-275 in leukemia cells(20), this finding suggests that the intracellular oxidative environment may dictate sensitivity to HDACi containing regimens. NAC’s action as an antioxidant is thought to be due to heightened GSH levels, so our data is consistent with the notion that high intracellular GSH may be a determinant of HDACi sensitivity. Further studies measuring intracellular GSH levels within the context of clinical trials and in HDACi resistant cells will be required to test that hypothesis.
Given the quantitative nature of the ROS measurements conducted herein, the potential use of flow based assays for ROS production to measure in vivo efficacy of HDACi regimens is feasible. Our future plans include the evaluation of intracellular superoxide and peroxide levels in peripheral blood of patients prior to and after exposure to HDACi containing regimens in order to determine if this oxidative stress is measurable in vivo. This strategy may be applied to ongoing clinical trials of MS-275 and 5-azacytidine where clinical responses do not appear to correlate with hypomethylation of monitored tumor suppressor genes, underscoring the need to identify biological correlates. Future studies, such as those outlined above, are needed to explore the relationship between ROS induction and the synergistic cytotoxicity of the two agents and to determine a role for ROS measurements as a marker for drug action.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004 May 27;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 2.Hellebrekers DM, Griffioen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta. 2007 Jan;1775(1):76–91. doi: 10.1016/j.bbcan.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004 Feb 3;101(5):1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006 Jan;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 5.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999 Apr 13;96(8):4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005 Jun 10;23(17):3912–3922. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 7.Kouraklis G, Theocharis S. Histone deacetylase inhibitors: a novel target of anticancer therapy (review) Oncol Rep. 2006 Feb;15(2):489–494. [PubMed] [Google Scholar]
- 8.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, Karp JE. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007 Apr 1;109(7):2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, Wehrmann M, Gregor M, Lauer UM, Bitzer M. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer. 2007 May 15;109(10):2132–2141. doi: 10.1002/cncr.22652. [DOI] [PubMed] [Google Scholar]
- 10.Muller CI, Ruter B, Koeffler HP, Lubbert M. DNA hypermethylation of myeloid cells, a novel therapeutic target in MDS and AML. Curr Pharm Biotechnol. 2006 Oct;7(5):315–321. doi: 10.2174/138920106778521523. [DOI] [PubMed] [Google Scholar]
- 11.Atallah E, Kantarjian H, Garcia-Manero G. The role of decitabine in the treatment of myelodysplastic syndromes. Expert Opin Pharmacother. 2007 Jan;8(1):65–73. doi: 10.1517/14656566.8.1.65. [DOI] [PubMed] [Google Scholar]
- 12.Bovenzi V, Momparler RL. Antineoplastic action of 5-aza-2′-deoxycytidine and histone deacetylase inhibitor and their effect on the expression of retinoic acid receptor beta and estrogen receptor alpha genes in breast carcinoma cells. Cancer Chemother Pharmacol. 2001 Jul;48(1):71–76. doi: 10.1007/s002800100294. [DOI] [PubMed] [Google Scholar]
- 13.Hurtubise A, Momparler RL. Effect of histone deacetylase inhibitor LAQ824 on antineoplastic action of 5-Aza-2′-deoxycytidine (decitabine) on human breast carcinoma cells. Cancer Chemother Pharmacol. 2006 Nov;58(5):618–625. doi: 10.1007/s00280-006-0225-6. [DOI] [PubMed] [Google Scholar]
- 14.Zhu WG, Otterson GA. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents. 2003 May;3(3):187–199. doi: 10.2174/1568011033482440. [DOI] [PubMed] [Google Scholar]
- 15.Rudek MA, Zhao M, He P, Hartke C, Gilbert J, Gore SD, Carducci MA, Baker SD. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J Clin Oncol. 2005 Jun 10;23(17):3906–3911. doi: 10.1200/JCO.2005.07.450. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006 Nov 15;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamer E, Fandy HC, Licht Jonathan D, Jiemjit Anchalee, Baylin Stephen B, Herman James G, Kerns Patrick, Berkofsky-Fessler Windy, McConnell Melanie, Nasrallah Chris, Sun Yezhou, Zhang Weija, Gore Steven D. Reversal of methylation of candidate tumor suppressor genes is not required for clinical response in myeloid malignancy patients treated with sequential 5-azacitidine and the histone deacetylase inhibitor MS-275. American Association for Cancer Research Annual Meeting; 2007; Los Angeles, CA.: American Association for Cancer Research Annual Meeting: Proceedings; 2007. p. 2483. [Google Scholar]
- 18.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005 Jan 18;102(3):673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acharya MR, Figg WD. Histone deacetylase inhibitor enhances the anti-leukemic activity of an established nucleoside analogue. Cancer Biol Ther. 2004 Aug;3(8):719–720. doi: 10.4161/cbt.3.8.1065. [DOI] [PubMed] [Google Scholar]
- 20.Lin CT, Lin WH, Lee KD, Tzeng PY. DNA mismatch repair as an effector for promoting phorbol ester-induced apoptotic DNA damage and cell killing: implications in tumor promotion. Int J Cancer. 2006 Oct 15;119(8):1776–1784. doi: 10.1002/ijc.22068. [DOI] [PubMed] [Google Scholar]
- 21.Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, Flax EL, Wickham J, Reed JC, Byrd JC, Grever MR. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004 Jul;18(7):1207–1214. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- 22.Kawai Y, Arinze IJ. Valproic acid-induced gene expression through production of reactive oxygen species. Cancer Res. 2006 Jul 1;66(13):6563–6569. doi: 10.1158/0008-5472.CAN-06-0814. [DOI] [PubMed] [Google Scholar]
- 23.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003 Jul 1;63(13):3637–3645. [PubMed] [Google Scholar]
- 24.Martirosyan A, Leonard S, Shi X, Griffith B, Gannett P, Strobl J. Actions of a histone deacetylase inhibitor NSC3852 (5-nitroso-8-quinolinol) link reactive oxygen species to cell differentiation and apoptosis in MCF-7 human mammary tumor cells. J Pharmacol Exp Ther. 2006 May;317(2):546–552. doi: 10.1124/jpet.105.096891. [DOI] [PubMed] [Google Scholar]
- 25.Donadelli M, Costanzo C, Beghelli S, Scupoli MT, Dandrea M, Bonora A, Piacentini P, Budillon A, Caraglia M, Scarpa A, Palmieri M. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim Biophys Acta. 2007 Jul;1773(7):1095–1106. doi: 10.1016/j.bbamcr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Yu C, Friday BB, Lai JP, McCollum A, Atadja P, Roberts LR, Adjei AA. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res. 2007 Feb 15;13(4):1140–1148. doi: 10.1158/1078-0432.CCR-06-1751. [DOI] [PubMed] [Google Scholar]
- 27.Sato T, Suzuki M, Sato Y, Echigo S, Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int J Oncol. 2006 May;28(5):1233–1241. [PubMed] [Google Scholar]
- 28.Rosato RR, Almenara JA, Maggio SC, Atadja P, Craig R, Vrana J, Dent P, Grant S. Potentiation of the lethality of the histone deacetylase inhibitor LAQ824 by the cyclin-dependent kinase inhibitor roscovitine in human leukemia cells. Mol Cancer Ther. 2005 Nov;4(11):1772–1785. doi: 10.1158/1535-7163.MCT-05-0157. [DOI] [PubMed] [Google Scholar]
- 29.Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P, Spiegel S, Grant S. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res. 2005 Mar 15;65(6):2422–2432. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- 30.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004 Jun 1;10(11):3839–3852. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 31.Chandra J, Hackbarth J, Le S, Loegering D, Bone N, Bruzek LM, Narayanan VL, Adjei AA, Kay NE, Tefferi A, Karp JE, Sausville EA, Kaufmann SH. Involvement of reactive oxygen species in adaphostin-induced cytotoxicity in human leukemia cells. Blood. 2003 Dec 15;102(13):4512–4519. doi: 10.1182/blood-2003-02-0562. [DOI] [PubMed] [Google Scholar]
- 32.Chandra J, Tracy J, Loegering D, Flatten K, Verstovsek S, Beran M, Gorre M, Estrov Z, Donato N, Talpaz M, Sawyers C, Bhalla K, Karp J, Sausville E, Kaufmann SH. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood. 2006 Mar 15;107(6):2501–2506. doi: 10.1182/blood-2005-07-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]