

Abstract
Rapid changes in extracellular K+ concentration ([K+]o in the mammalian CNS are counteracted by simple passive diffusion as well as by cellular mechanisms of K+ clearance. Buffering of [K+]o can occur via glial or neuronal uptake of K+ ions through transporters or K+ -selective channels. The best studied mechanism for [K+]o buffering in the brain is called K+ spatial buffering, wherein the glial syncytium disperses local extracellular K+ increases by transferring K+ ions from sites of elevated [K+]o to those with lower [K+]o. In recent years, K+ spatial buffering has been implicated or directly demonstrated by a variety of experimental approaches including electrophysiological and optical methods. A specialized form of spatial buffering named K+ siphoning takes place in the vertebrate retina, where glial Müller cells express inwardly rectifying K+ channels (Kir channels) positioned in the membrane domains near to the vitreous humor and blood vessels. This highly compartmentalized distribution of Kir channels in retinal glia directs K+ ions from the synaptic layers to the vitreous humor and blood vessels. Here, we review the principal mechanisms of [K+]o buffering in the CNS and recent molecular studies on the structure and functions of glial Kir channels. We also discuss intriguing new data that suggest a close physical and functional relationship between Kir and water channels in glial cells.
Keywords: potassium channel, glia, Müller cells, astrocytes, retina, aquaporin
Neurons are bathed in an extracellular fluid that is rich in Na+ ions and relatively poor in K+ ions. The relative concentrations of these cations are reversed inside the cells, and the resulting chemical gradients across the cell membrane are crucial to many important processes, including fast electrical signaling involving Na+ influx and K+ efflux. Even modest neuronal K+ effluxes may elicit considerable changes in extracellular K+ concentration ([K+]o), due to the limited volume of the CNS extracellular space (Nicholson and Sykova, 1998; Kume-Kick et al., 2002) and the low baseline [K+]o. These [K+]o changes can impact a wide variety of neuronal processes, such as maintenance of membrane potential, activation and inactivation of voltage-gated channels, synaptic transmission, and electrogenic transport of neurotransmitters. Thus, it is not surprising that diverse cellular mechanisms for tight control of [K+]o are found in the CNS of vertebrates and invertebrates. When these mechanisms are disrupted in instances such as spreading depression, the extracellular [K+]o can reach values as high as 60 mM (Somjen, 2001, 2002) and CNS function is severely compromised.
In the brain, extracellular K+ is kept close to 3 mM independent of fluctuations in serum levels (Katzman, 1976; Somjen, 1979) although local variations in extracellular K+ do occur following neuronal activity changes. For example, light stimulation of cat retina promotes slow, transient [K+]o increases in the primary visual cortex (Fig. 1A; Singer and Lux, 1975; Connors et al., 1979). Likewise, light stimulation of frog or cat retinas induces [K+]o increases in the inner and outer plexiform layers and decreases in the outer retina and subretinal space (Fig. 1B; Karwoski et al., 1985; Frishman et al., 1992). In cat spinal cord, rhythmic flexion/extension of the knee joint causes [K+]o to increase by as much as 1.7 mM (Heinemann et al.,1990). Even higher elevations in [K+]o can be evoked by direct electrical stimulation of afferent pathways. In general, increases of neuronal activity induce local [K+]o increases by several millimolar until it reaches a plateau or ceiling level (Heinemann and Lux, 1977; Connors et al., 1982). This ceiling level is about 10–12 mM in the cat somatosensory cortex (Heinemann and Lux, 1977), in the rat optic nerve (Connors et al., 1982; Ransom et al., 1986), and in the cat thalamus (Gutnick et al., 1979). The ceiling level is only exceeded under pathophysiological conditions such as in anoxia (Vyskocil et al., 1972) or in spreading depression (Somjen, 2002).
Fig. 1.
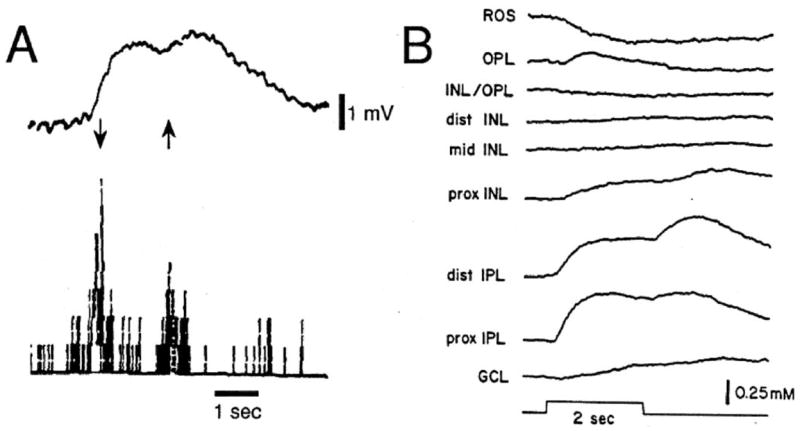
Changes in [K+]o in the cat striate cortex and frog retina with enhanced neuronal activity. (A) Upper trace. Dynamic [K+]o changes in the cat striate cortex upon stimulation of the receptive field of hypercomplex cells. [K+]o changes were measured with a double-barreled K+-sensitive microelectrode. Arrows represent bars of light moving down or up in a cell’s receptive field. The 1 mV scale bar corresponds to approximately 0.17 mM. Lower trace. Spike activity recorded in the reference barrel of the K+-sensitive microelectrode. [From Singer and Lux (1975) with permission.] (B) [K+]o changes within layers of the frog retina. A 2-s light stimulus evokes [K+]o increases in the inner plexiform layer (IPL) and outer plexiform layer (OPL) and decreases of [K+]o in the subretinal space (ROS). [From Karwoski et al. (1985), with permission.]
Overview of K+ regulatory mechanisms
Mechanisms of [K+]o buffering can be broadly categorized as either K+ uptake or K+ spatial buffering (Fig. 2; Newman, 1995; Amedee et al., 1997; Somjen, 2002). In the case of K+ uptake, excess K+ ions are temporarily sequestered into glial cells by transporters or K+ channels. To preserve electroneutrality, K+ influx into glial cells is accompanied by either influx of anions such as Cl− or by efflux of cations such as Na+ (Fig. 2A). Eventually, K+ ions accumulated in glia are released back into the extracellular space and the overall distribution of K+ across the cellular compartments is restored. It is expected that during the process of K+ uptake, ions will accumulate within the glia and the cells will experience water influx and swelling. This is in marked contrast to the K+ spatial buffering mechanism (Orkand et al., 1966), in which functionally coupled, highly K+ permeable glial cells transfer K+ ions from regions of elevated [K+]o to regions of lower [K+]o. The transfer of K+ is mediated by a K+ current flow through the glial network. The K+ current is driven by the difference between the glial syncytium membrane potential (Vm) and the local K+ equilibrium potential (EK). In regions of [K+]o increases, there is a net driving force causing K+ to flow into the glial cells (Fig. 2B). This K+ entry causes a local depolarization which propagates electrotonically through the glial cell network. As a result, there is a net driving force causing K+ to flow out of the glial cells. Such glial-mediated K+ movements in the K+ spatial buffering mechanism disperse local [K+]o increases with little net gain of K+ ions within the glial cells. The overall efficiency of the spatial buffer process will depend, in part, on the electrical space constant of the glial cell syncytium (Newman, 1995). In certain CNS regions close to fluid reservoirs, such as the retina, K+ spatial buffer currents flow within single glial cells rather than through a network of cells. In these cases, K+ influx occurs in one region of the glial cell and efflux occurs through another cell region (see “K+ siphoning in the retina,” below).
Fig. 2.
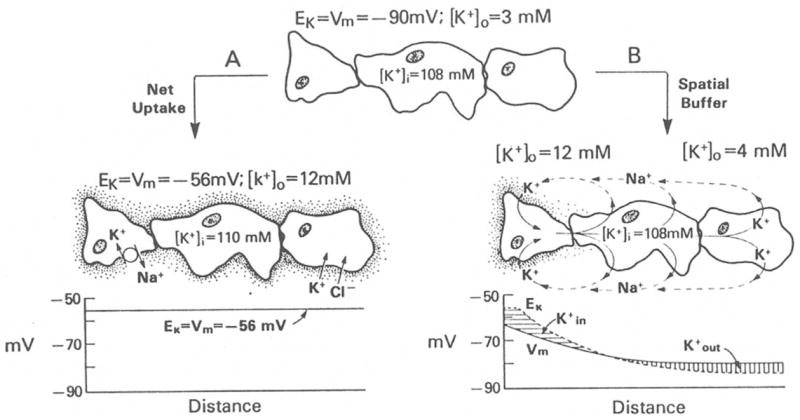
Diagram depicting the role of glial cells in [K+]o homeostasis. Top: Glial cells are electrically coupled via gap junctions forming a functional syncytium. With [K+]o equaling 3 mM, the glial syncytium has a membrane potential of approximately −90 mV. (A) Net K+ uptake mechanism. When [K+]o is increased, glial cells accumulate K+ either by the activity of a Na,K–ATPase or by a pathway in which K+ is cotranported with Cl−. In this mechanism of [K+]o regulation, the membrane potential in the glial syncytium is spatially uniform at −56 mV. (B) K+ spatial buffering mechanism. Local increases of [K+]o produce a glial depolarization that spreads through the glial syncytium. The local difference in Vm and EKdrives the K+ uptake in regions of elevated [K+]o and K+ outflow at distant regions. The intracellular currents are carried by K+ and extracellular currents are mediated by other ions such as Na+. [From Orkand (1986), with permission.]
Net K+ uptake is mediated mainly by Na,K–ATPase pumps (sodium pumps) and Na–K–Cl cotransporters. The sodium pump is a transmembrane enzyme that acts as an electrogenic ion transporter in the plasma membrane of all cells (Kaplan, 2002; Jorgensen et al., 2003). Each cycle of the sodium pump activity expels three Na+ ions and moves two K+ ions into the cell. The primary role of the sodium pump is therefore to maintain high intracellular K+ and low intracellular Na+ (Kaplan, 2002; Jorgensen et al., 2003). Upon local [K+]o increases, sodium pumps are expected to transport K+ ions into the cells in exchange for Na+ ions, assuming that the pump extracellular K+ site is not saturated and the intracellular Na+ concentration is not limiting (Sweadner, 1995). It has been suggested that the sodium pump expressed in glial cells is better suited for [K+]o buffering than the neuronal isoform given observations that the glial isoform has a lower affinity for extracellular K+ (Franck et al., 1983; Reichenbach et al., 1992). In retina, for example, glial cells (Müller cells) express a sodium pump isoform with maximal activity at about 10–15 mM [K+]o while the isoform found in rod photoreceptors saturate at [K+]o as low as 3 mM (Reichenbach et al., 1992).
There have been several reports in support to the notion that sodium pumps help to buffer [K+]o in the CNS. For instance, a transient accumulation of K+ ions and simultaneous depletion of Na+ ions in glial cells is detected in guinea-pig cortices following electrical stimulation (Ballanyi et al., 1987). A substantial fraction of this K+ accumulation is prevented by pharmacological blockade of sodium pumps (Ballanyi et al., 1987). Likewise, in hippocampal slices, blockade of sodium pumps increases baseline [K+]o and prevents the rapid clearance of extracellular K+ ions following neuronal stimulation (Fig. 3A) (D’Ambrosio et al., 2002). In the rat optic nerve, clearance of K+ accumulation following axonal stimulation is highly temperature dependent (Q10 = 2.6) as expected for a carrier-mediated process, and is largely blocked by Na,K–ATPase pump inhibitors (Ransom et al., 2000).
Fig. 3.
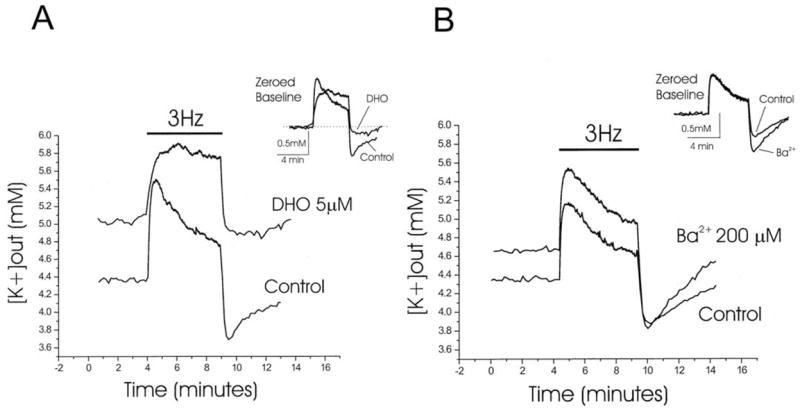
Differential roles of Na,K–ATPase pumps and Kir channels for the [K+]o regulation in rat hippocampus slice. (A) In control condition, 3-Hz antidromic stimulation induces a rise of [K+]o in area CA3 that peaks at approximately 5.5 mM followed by a decline to approximately 4.7 mM. With addition of the sodium pump inhibitor di-hydro-ouabain (DHO), the baseline [K+]o increases to approximately 5.1 mM. Antidromic stimulation (3-Hz) induces a rise of [K+]o to approximately 5.9 mM but there is no [K+]o recovery phase. Also absent is the undershoot of [K+]o following the stimulation period. In the inset the two traces are shown superimposed, and the baseline is zeroed. (B) In control condition, 3-Hz antidromic stimulation induced a rise of [K+]o that peaked at approximately 5.2 mM followed by an undershoot in [K+]o. With Ba2+ (200 μM) in the bath, baseline [K+]o increased in relation to control levels and the undershoot in [K+]o following antidromic stimulation is more pronounced. In the inset the two traces are shown superimposed, and the baseline is zeroed. [From D’Ambrosio (2002), with permission.]
Transient accumulation of K+ ions in cultured astrocytes has also been linked to the activity of Na–K–Cl co-transporters, which are integral membrane proteins that transport Na, K, and Cl ions into and out of cells in an electrically neutral manner, often with a stoichiometry of 1Na:1K:2Cl (Haas and Forbush, 1998). To date, two Na–K–Cl cotransporter isoforms have been identified: NKCC1, which is present in a wide variety of secretory epithelia and non-epithelial cells; and NKCC2, which is present exclusively in the kidney (Haas and Forbush, 1998). Both NKCC isoforms are members of a diverse family of cation-chloride cotransport proteins that share a common predicted membrane topology and are sensitive to loop diuretics such as bumetanide and furosemide (Haas and Forbush, 1998). In cultured astrocytes, intracellular K+ accumulation following increased [K+]o can be partially prevented by furosemide or bumetanide or by removal of external Na+ and Cl− (Kimelberg and Frangakis, 1985; Walz, 1992; Rose and Ransom, 1996). More recently, the role of Na–K–Cl co-transporters in [K+]o homeostasis has also been demonstrated by optical methods (MacVicar et al., 2002). In the rat optic nerve, intrinsic optical signals (see below) revealed that increased [K+]o induces astrocyte swelling, and this swelling is reversibly depressed by furosemide and bumetanide (MacVicar et al., 2002). A monoclonal antibody to the NKCC1 form of the Na–K–2Cl cotransporter showed that NKCC1 is expressed in astrocytes from optic nerves (MacVicar et al., 2002), suggesting the involvement of this particular Na–K–Cl cotransporter isoform in K+ buffering.
However, the exact role of glial K+ uptake or spatial buffering in the various parts of the CNS has remained uncertain. This question is under considerable debate, and research has shown that the relative contributions may vary across species and between CNS regions (Newman, 1995). In the rat optic nerve, for example, [K+]o buffering seems more dependent on the active uptake mechanism than on K+ spatial buffering as post-stimulus recovery of [K+]o is highly sensitive to sodium pump inhibition but not to glial K+ channel blockers (Ransom et al., 2000). By contrast, in the CA3 region of rat hippocampus, both the sodium pumps and glial K+ channels seem critical for setting the baseline [K+]o and the post-stimulus recovery of [K+]o (D’Ambrosio et al., 2002; Fig. 3). In this preparation, sodium pumps are necessary for the clearance of excess K+ during afferent stimulation, whereas glial K+ channels are necessary to prevent large [K+]o undershoots following post-tetanus (D’Ambrosio et al., 2002). In retina, K+ spatial buffering has a major role in regulating the [K+]o (Newman, 1995; see below).
K+ spatial buffering
Two conditions are necessary for efficient K+ spatial buffering as originally proposed by Orkand et al. (1966): 1) the glial cells should form a syncytium in which K+ currents can traverse relatively long distances; and 2) these cells should be highly and selectively permeable to K+, which both enters and exits through the glial cell membranes. As described in the section on “K+ siphoning in the retina,” below, spatial buffer currents can efficiently dissipate [K+]o increases by flowing through single cells as well as through a syncytium of coupled glial cells. Several lines of evidence demonstrate that astrocytes do indeed form a functional syncytium that allows intercellular diffusion of ions and other signaling molecules (Nagy and Rash, 2000; Rouach et al., 2002). Such extensive cellular coupling is due to the high density of gap junctional proteins (connexins) in glial cells (Dermietzel, 1998; Rouach et al., 2002). Immunocytochemical and in situ hybridization methods reveal that astrocytes express multiple connexins, including Cx30, Cx40, Cx43 and Cx45 (Dermietzel, 1998; Dermietzel et al., 2000; Zahs et al., 2003). Among these, Cx43 seems to be the most important for coupling, which is significantly reduced in cultured astrocytes derived from Cx43 knockout mice (Dermietzel et al., 2000).
Additionally, numerous studies have also shown that glial cell membranes are highly and almost exclusively permeable to K+ ions (Sontheimer, 1994). The principal K+ channels found in glial cells are the inwardly rectifying K+ (Kir) channels, which allow K+ ions to flow much more readily in the inward than outward direction (Doupnik et al., 1995; Stanfield et al., 2002). These channels have high open probability at the normal resting membrane potential and thus allow both glial K+ influx and efflux. Kir channels in glia have been described in many CNS regions, including mammalian astrocytes from the optic nerve (Barres et al., 1990), spinal cord (Ransom and Sontheimer, 1995) and other brain regions (Sontheimer, 1994). Although glia may express additional types of K+ channels, such as calcium and voltage-dependent K+ channels (Sontheimer, 1994), these other channel types are mostly inactive at the hyperpolarized glial resting membrane potential (−60 to −90 mV; Kuffler et al., 1966; Dennis and Gerschenfeld, 1969). Another important biophysical property of Kir channels is that their slope conductance increases with elevations in [K+]o by a square root relation (Stanfield et al., 2002). This unique property of Kir channels allows K+ conductance increases in glial cells, and therefore enhanced K+ clearance rates, when [K+]o is raised (Newman, 1993).
Finally, an important implicit assumption in the K+ spatial buffering mechanism is the low permeability of glial cells to anions such as Cl− as the low anion conductance insures that net uptake of KCl will not occur upon rises of [K+]o. Unfortunately, there is no consensus concerning values for Cl− permeability of glial cells in the native tissue (Walz, 2002). While a relatively high basal Cl− conductance has been reported in glial cells of guinea-pig olfactory cortex (Ballanyi et al., 1987) other studies have failed to demonstrate a significant Cl− conductance for glial cells in situ (Walz, 2002).
Evidence for K+ spatial buffering
The first support for K+ spatial buffering was provided in the classic work by Orkand et al. (1966), who reported that in amphibians, stimulation of the optic nerve leads to slow depolarization and repolarization of the glial cells surrounding the nonmyelinated axons. The slow depolarization and repolarization of these glial cells was thought to reflect K+ transfer by glial cells via the K+ spatial buffering mechanism. Subsequently, extensive support for the K+ spatial buffering hypothesis came from measurements of extracellular field potentials. In the case of K+ spatial buffering, the transcellular transfer of K+ ions from areas of elevated to lower [K+]o generates return current loops in the extracellular space giving rise to extracellular field potentials. These activity-induced slow extracellular field potentials are generated in various CNS regions including the cortex and retina (Gardner-Medwin et al., 1981; Dietzel et al., 1989). In the retina, slow extracellular field potentials (slow PIII and M waves) are generated upon light stimulation of the retinas (Xu and Karwoski, 1997; Karwoski and Xu, 1999). Current source density analysis indicates that these waves originate from K+ spatial buffering by retinal glial cells (Xu and Karwoski, 1997; Karwoski and Xu, 1999). The transfer of K+ ions by retinal glial cells, which generates the slow PIII wave, buffers the photoreceptor-based light-evoked decrease in [K+]o in the outer retina. As expected, these K+ spatial buffering fluxes are abolished by blocking retinal glial cell K+ conductance with Ba2+ (Oakley et al., 1992; Kofuji et al., 2000).
Measurements of activity-dependent [K+]o changes in the cortex and cerebellum show that [K+]o varies with depth and time in a manner consistent with transcellular transfer of K+ ions (Gardner-Medwin and Nicholson, 1983). These [K+]o changes were not abolished by sodium pump inhibitors, indicating that they are likely due to a passive K+ transport mechanism such as K+ spatial buffering (Gardner-Medwin and Nicholson, 1983). Similar results were obtained in the drone retina, where it was estimated that about 10 times more K+ ions move as a result of spatial buffering than by simple diffusion through the extracellular space (Coles et al., 1986).
More direct evidence for K+ spatial buffering has been provided by optical imaging of brain slices (Holthoff and Witte, 2000). When K+ ions are transferred via glial cells, there should be shrinkage of the extracellular space in areas of K+ influx and swelling in areas of K+ efflux (Dietzel et al., 1980). Changes in extracellular volume following neuronal stimulation could be demonstrated by monitoring intrinsic optic signals (IOS) in brain slices. As predicted for K+ spatial buffering, stimulation of cortical areas promoted shrinkage of the extracellular space followed by swelling in the layers above and below the stimulated area (Fig. 4; Holthoff and Witte, 2000). This swelling in the extracellular space was associated with local increases in [K+]o and was dependent on gap junctional coupling (Holthoff and Witte, 2000). Such a pattern of changes in extracellular space volume and cellular coupling dependency is consistent with K+ spatial buffering in the mammalian cortex.
Fig. 4.
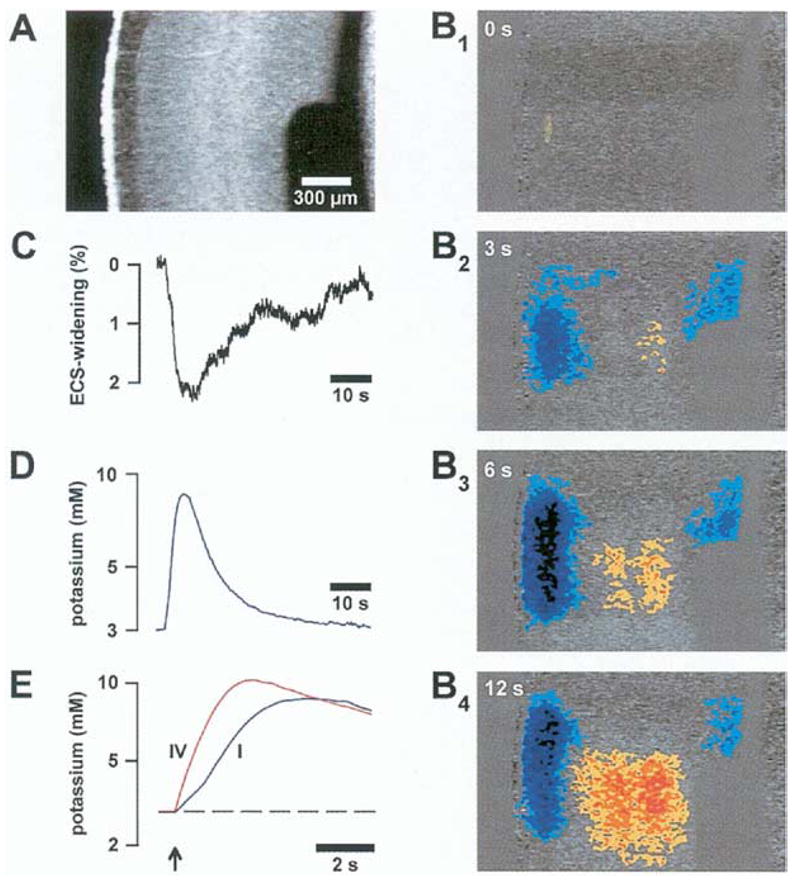
Evidences for K+ spatial buffering in the rat cortex. (A) Image of the brain slice viewed with darkfield optics. (B1–4) Time course of IOS changes upon neuronal stimulation in middle cortical layers. Red colors represent IOS increases while blue colors represent IOS decreases. These correspond, respectively, to shrinkage and widening of the extracellular space. Notice the extracellular space shrinkage in the middle cortical layers and widening in the most superficial and deep cortical layers as predicted for a K+ spatial buffering mechanism. (C) Time course of extracellular space widening in the cortical layer I measured independently validates the interpretation of IOS. (D) Time course of [K+]o increase in layer I. (E) Time course of [K ]o increase in layer I (blue) and in layer IV (red). [From Holthoff and Witte (2000), with permission.]
K+ siphoning in the retina
In addition to the brain, K+ spatial buffering has been investigated in the retina, where the Müller cell is the principal glial cell type (Newman and Reichenbach, 1996). Müller cells display morphological polarization with an endfoot process in close apposition to the vitreous and apical microvilli projecting into the subretinal space (Newman and Reichenbach, 1996). Like many other glial cells, Müller cells can be distinguished from their neuronal counterparts by a resting membrane potential that closely follows the EK following changes in [K+]o (Newman, 1985). This high selectivity of Müller cell membranes to K+ ions is due to the high density and localized expression of Kir channels (Newman, 1993; Kofuji et al., 2000).
Physiological studies in Müller cells provided the first indication that K+ conductances are unevenly distributed along their plasma membrane. By focally increasing the [K+]o along amphibian Müller cells and monitoring the resulting depolarizations, Newman (1984) was able to map the subcellular distribution of K+ conductance in this glial cell type. He found that K+ conductance was highly concentrated in the endfoot process, with 94% of the total K+ conductance localized to this relatively small subcellular domain (Newman, 1984). Single channel patch clamp recordings later confirmed these findings (Brew et al., 1986).
The observation of a highly non-uniform distribution of Kir channels in Müller cells led to the hypothesis that excess K+ from the inner plexiform layer of retina, where most retinal synapses are located, are “siphoned” by Müller cells to the vitreous humor (Newman et al., 1984; Newman, 1987a). This hypothesis, called K+ siphoning, is a specialized form of the spatial buffering mechanism in which the subcellular distribution of K+ channels in glial cells directs clearance of K+ into large reservoirs such as the vitreous humor (Fig. 5). In mammalian retinas, the pattern of Kir channel distribution is similar to that found in amphibians (Newman, 1987b). The major difference is that additional regions of high K+ conductance are found in more distal processes in cells of species with vascularized retina such as mouse, cat and monkey. This suggests that Müller cell’s perivascular processes may also possess high K+ conductance (Newman, 1993) and direct K+ to spaces near blood vessels (Newman, 1987b).
Fig. 5.
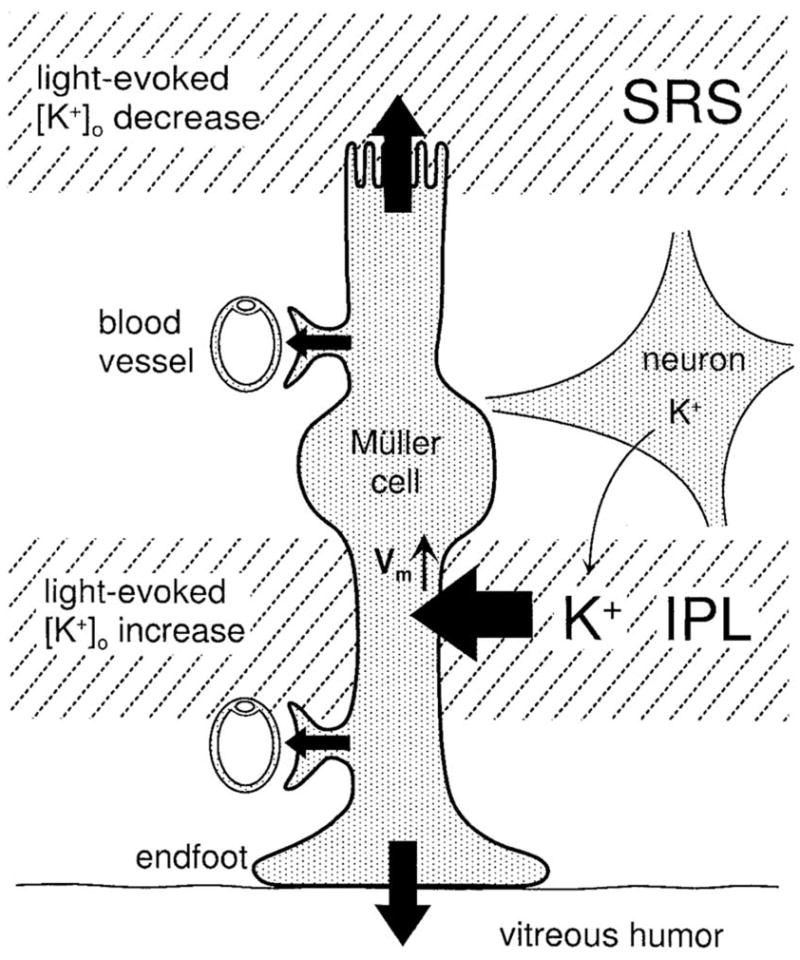
K+ siphoning in the retina. K+ released in the inner plexiform layer (IPL) upon neuronal stimulation enters the principal glial cell of retina (Müller cell). K+ leaves the glial cell preferentially from the endfoot processes enveloping blood vessels and the vitreal endfoot, as K+ conductance is maximal in these membrane regions. Light-evoked [K+ efflux from apical processes. [From Newman ]o decrease in the subretinal space (SRS) lead to K (1996b), with permission.]
The K+ siphoning hypothesis was supported by the observation that in the amphibian retina, light flashes promote increased [K+]o in both the inner plexiform layer and the vitreous humor (Karwoski et al., 1989). These light-evoked increases of [K+]o in the vitreous humor were mediated by Kir channels expressed in Müller cells, as shown by the observation that vitreal increases were blocked by superfusion of retinas with barium, a Kir channel blocker. Similarly, in cat retinas, light-evoked [K+]o increases in the inner plexiform layer are three-fold larger following blockade of Kir channels (Frishman et al., 1992), confirming that glial-mediated K+ spatial currents play an important role in curtailing large changes of [K+]o.
Glial cells and K+ channels
Because Kir channels most likely underlie K+ spatial buffering in the CNS, there is considerable interest in determining their macromolecular structure, mechanisms of targeting and modulation by intracellular and extracellular factors. The Kir channels have been recently cloned, and over 20 genes are currently known to encode various Kir channel subunits (Nichols and Lopatin, 1997; Stanfield et al., 2002). Site-directed mutagenesis and heterologous channel expression have been used to identify structural elements involved in specific Kir channel functions. These studies have revealed the basic Kir channel design of two transmembrane domains and a re-entry loop (P-loop), with intracellular amino and carboxyl termini (Nichols and Lopatin, 1997; Stanfield et al., 2002). The Kir channel subunits are usually categorized into seven major subfamilies (Kir1 to Kir7) that are diversely regulated by intracellular and extracellular factors (Stanfield et al., 2002).
Of these family members, immunocytochemical and in situ hybridization studies demonstrate that the Kir4.1 channel is broadly expressed in brain (Poopalasundaram et al., 2000; Higashi et al., 2001), and different reports have suggested that it is expressed only in glial cells (Higashi et al., 2001) or in both neurons and glia (Li et al., 2001). Kir4.1 immunoreactivity can be demonstrated in cultured (Li et al., 2001) and in situ astrocytes (Poopalasundaram et al., 2000; Higashi et al., 2001). In the olfactory bulb, Kir4.1 immunoreactivity is detected in about half of the glial fibrillary acidic protein-positive astrocytes, but not in neurons (Higashi et al., 2001). Immunogold microscopic examination reveals that Kir4.1 channels are enriched in the processes of astrocytes enveloping synapses and blood vessels (Higashi et al., 2001). In addition to Kir4.1 channels, other Kir channel subunits may also be expressed in various glial cell types. Kir2.2 channels are expressed in Bergman glial cells and astrocytes in the cerebellum (Leonoudakis et al., 2001), and Kir2.1 channels are found in astrocytes and oligodendrocytes in the forebrain (Stonehouse et al., 1999). Finally, single cell in situ PCR experiments in astrocytes from mouse hippocampal slices identified transcripts for Kir2.1, Kir2.2, Kir2.3 and Kir4.1 channels (Schroder et al., 2002). Thus, a large variety of Kir channel subtypes may be expressed in glial cells and this may explain the wide range of single channel Kir conductances reported in glial cells (Sontheimer, 1994).
The properties of specific Kir channel subunits have been assessed in heterologous expression systems such as Xenopus oocytes or transfected cells. Kir2.1 currents show steep inwardly rectifying current-voltage relationships with minimal outward currents at membrane potentials positive to EK (Kubo et al., 1993). In contrast, Kir4.1 channels are weakly rectifying, allowing substantial outward currents (Takumi et al., 1995). A further complexity is provided by the fact that Kir channels are tetrameric proteins, and in heterologous expression systems Kir subunits can form either homomeric or heteromeric channels (Stanfield et al., 2002). The expression of Kir5.1 subunits in Xenopus oocytes or mammalian cell lines does not result in functional channels, but co-expression with Kir4.1 channels leads to formation of heteromeric channels that are highly sensitive to intracellular changes in pH (Tucker et al., 2000).
As in retinal Müller cells (see below), the distribution of K+ conductance in astrocytes seems to be non-homogenous. Freshly dissociated salamander astrocytes have an approximately 10-fold higher conductance in their endfeet than other cell regions (Newman, 1986). So far, only Kir4.1 channels have been mapped to astrocytic end-feet (Higashi et al., 2001). In conclusion, although it is clear that K+ conductance in astrocytes is likely mediated by Kir channels, further investigations are required to determine the precise molecular composition of such channels.
Kir channel subtypes expressed in Müller cells
In contrast to astrocytes in the brain, retinal Müller cells are relatively homogenous and are thus an attractive model for studying Kir channel composition. Recently, several groups have examined the distribution of Kir channels in Müller cells using immunocytochemical and molecular biological techniques (Ishii et al., 1997; Kofuji et al., 2000). These studies demonstrate highly concentrated expression of the weakly rectifying Kir4.1 channels in Müller cell endfeet and on the processes enveloping blood vessels (Fig. 6A, C, E), a distribution that correlates well with the previously mentioned electrophysiological studies (Newman, 1987b, 1993). Several additional lines of evidence argue that Kir4.1 channels are the principal Kir channel subtype in Müller cells: 1) patch clamp recordings in rabbit Müller cells and in transfected 293 cells expressing Kir4.1 channels show similar single channel conductances and open probabilities (Tada et al., 1998); 2) genetic ablation of Kir4.1 channels in mice decreases the membrane conductance of Müller cells by 10-fold (Kofuji et al., 2000); and 3) the slow PIII wave of the electroretinogram, which is generated by K+ fluxes through Müller cells, is absent in Kir4.1 knockout animals. This effect was not caused by overall impairment of neuronal function as indicated by the fact that the a- and b-waves, associated with neuronal activity, were not decreased in the knockout animals (Kofuji et al., 2000).
Fig. 6.
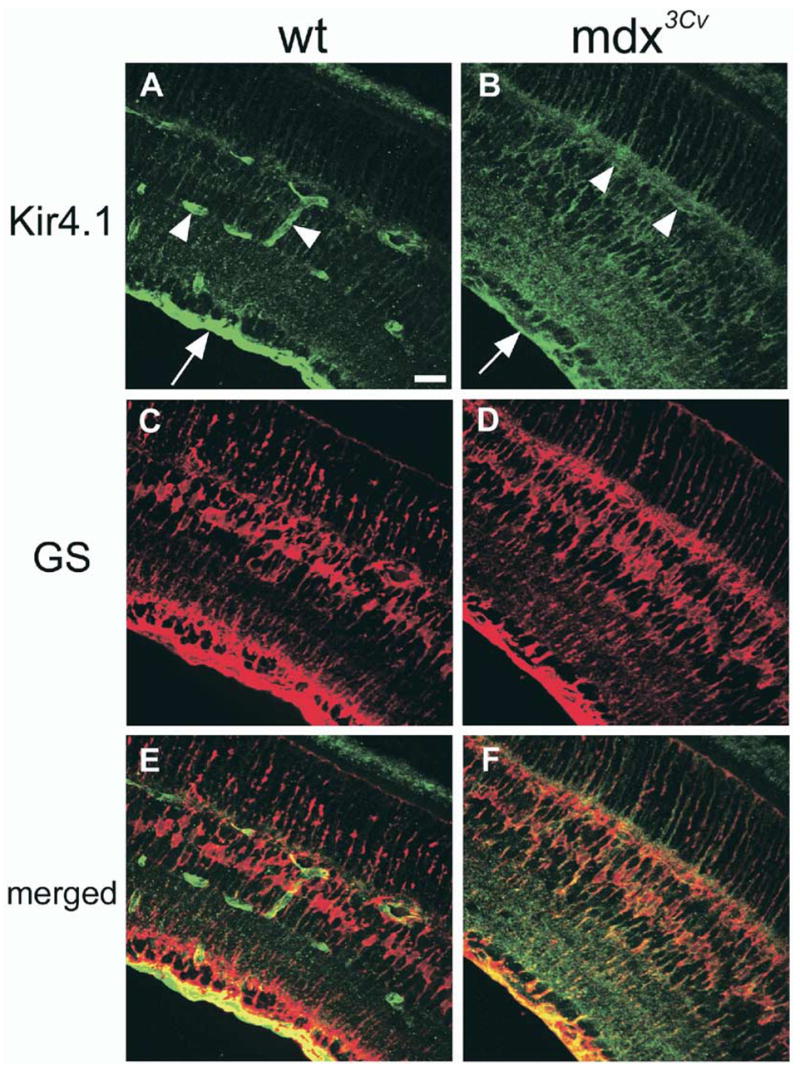
Kir4.1 channel localization in wild type and mdx3Cv (dystrophin knockout) mouse retinal sections. (A) Kir4.1 is concentrated at the inner limiting membrane (arrow) and to processes around blood vessels (arrowheads) in wild-type retina. (B) In the mdx3Cv mouse, Kir4.1 is more evenly distributed throughout the retina with a reduction in staining at the inner limiting membrane (arrow) and no apparent enrichment of Kir4.1 around blood vessels (arrowheads). The Müller-specific marker glutamine synthetase (C, D), and merged images (E, F) suggest the localization of Kir4.1 to Müller cells. Scale bar = 25 μm (A). [From Connors and Kofuji (2002), with permission.]
Other Kir channels, such as the strongly rectifying Kir2.1 channels, may also be expressed in Müller cells. In mouse Müller cells, Kir2.1 channels are expressed along the plasma membrane in a uniform manner that does not resemble the clustered distribution seen for Kir4.1 channels (Kofuji et al., 2002). Such differential expression of Kir2.1 and Kir4.1 channels may enhance the efficiency of K+ siphoning in the retina (Kofuji et al., 2002). Expression of weakly rectifying Kir4.1 channels in selective membrane domains would allow K+ ions to leave Müller cells and be stored in extracellular sinks such as the vitreous humor whereas expression of strongly rectifying Kir2.1 channels would allow greater influx of K+ in the synaptic layers (Kofuji et al., 2002).
A recent additional study suggests the expression of Kir5.1 channels in the soma and stalks of Müller cells; immunoprecipitation assays show that a fraction of the Kir4.1 subunits in retina are coassembled with Kir5.1 subunits (Ishii et al., 2003). Because heterologously expressed Kir4.1 and Kir5.1 form heteromeric Kir channels that are highly sensitive to physiological changes in intracellular pH, it has been suggested that expression of Kir5.1 subunits in Müller cells promotes the coordinated coupling between acid–base regulation and K+ buffering in the retina (Ishii et al., 2003). In this hypothesis, increases in [K+]o and the resulting glial depolarization would increase the activity of the electrogenic Na+ -HCO3− co-transporter (Newman, 1996a). The increased influx of HCO3− would then cause intracellular alkalinization and subsequent increases in the activity of heteromeric Kir4.1/Kir5.1 channels, ultimately enhancing K+ uptake into Müller cells (Ishii et al., 2003).
In summary, investigations in Müller cells have provided compelling evidence demonstrating a major role for Kir4.1 channels in retinal K+ buffering. Kir4.1 channels appear to have the functional and anatomical distributions that best match the physiological studies performed in Müller cells over the past decade. Biochemical and immunocytochemical work also suggests that other Kir channels, including Kir2.1 and Kir5.1, may be involved in differential modification of K+ entry pathways into these cells.
Kir channel accessory proteins in Müller cells: localization and function
The focal aggregation of Kir4.1 channels in Müller cells raises the intriguing question of how these channels are targeted to such precise glial subcellular domains. This is an important question, as the efficiency of the retinal K+ siphoning process is highly dependent on the clustered and non-homogenous distribution of Kir channels in Müller cells (Newman, 1995). It has recently been shown that the water channel aquaporin 4 (AQP4) is also highly enriched in the endfeet and perivascular processes of Müller cells (Nagelhus et al., 1999). At this time, the physiological significance of such channel co-localization is not precisely known. Transfer of K + ions by K+ siphoning in the retina is expected to generate osmotic imbalances and parallel water fluxes may be needed to dissipate such imbalances. It is possible that the highly asymmetric distribution of AQP4 channels allows funneling of water to the appropriate compartments in the retina, from Müller cells into the vitreous humor and regions near blood vessels (Nagelhus et al., 1999).
This spatial overlap of Kir and aquaporins, two highly non-homologous channel types, suggests that there may be a common molecular mechanism for their subcellular distribution and targeting. Although the Kir4.1 and AQP4 channels are highly divergent in their primary sequences, they share a key -S-X-V-COOH motif in their C-termini. This sequence is able to bind to PDZ domains, which are modular amino acid motifs implicated in many protein-protein interactions (Hung and Sheng, 2002). Proteins possessing these domains are abundantly expressed in the nervous system and include post-synaptic density protein-95, Chapsyn-110/PSD-93, SAP-102 and hDlg/SAP97 (Hung and Sheng, 2002). Positioning of several proteins in the postsynaptic density in excitatory synapses is critically dependent on their interactions with PDZ-domain containing proteins (Sheng and Sala, 2001).
Although specific PDZ domain-containing protein(s) have yet to be unequivocally identified in Müller cells, the SAP97 protein, which is present in Müller cells, has been shown to increase Kir4.1 currents in heterologous systems (Horio et al., 1997). Another candidate is the PDZ domain-containing adapter protein, α-syntrophin. In tissues where α-syntrophin is present, it is localized to the cell membrane by its association with the multi-protein dystrophin glycoprotein complex (DGC; Ahn and Kunkel, 1995). The DGC spans the cell membrane, forming a molecular bridge between basal lamina proteins in the extracellular space and an array of signaling molecules in the intracellular domain. Immunolocalization studies in retina revealed that the DGC components, α-dystroglycan and the short dystrophin isoform, Dp71, appear to be localized in a fashion very similar to that of Kir4.1 in Müller cells (Claudepierre et al., 2000b). The role of Dp71 in the localization of Kir4.1 has recently been investigated in the dystrophin null mutant mouse, mdx3Cv (Connors and Kofuji, 2002). Immunohistochemistry experiments revealed that the polarized subcellular distribution of Kir4.1 is altered in Müller glial cells from mdx3Cv mice, displaying a more homogeneous distribution pattern (Fig. 6B, D, F), though immunoblotting and whole cell patch clamp experiments revealed that the channel is expressed at normal levels at the plasma membrane and its electrophysiological properties are unchanged (Connors and Kofuji, 2002). Similar findings have also been reported for a null mouse line for the dystrophin isoform Dp71 (Dalloz et al., 2003).
These results strongly suggest that the DGC is important to the localization of Kir4.1 in Müller cells. It is possible that the DGC targets the Kir channels to the membrane domains facing the vitreous or blood vessels by binding of extracellular portions of the DGC to the basal lamina of these regions (Fig. 7). This assumes the existence of an intermediate protein containing a PDZ domain. As mentioned previously, the best candidate for such an adaptor protein is α-syntrophin. α-Syntrophin is expressed in Müller cells and is putatively part of the Müller cell-specific DGC (Claudepierre et al., 2000a). In addition, α-syntrophin has been shown to interact with AQP4 in astrocytes in a PDZ-dependent manner, and is required for the membrane expression and localization of AQP4 (Neely et al., 2001). Therefore, α-syntrophin could underlie the co-localization of Kir4.1 and AQP4 seen in Müller cells.
Fig. 7.
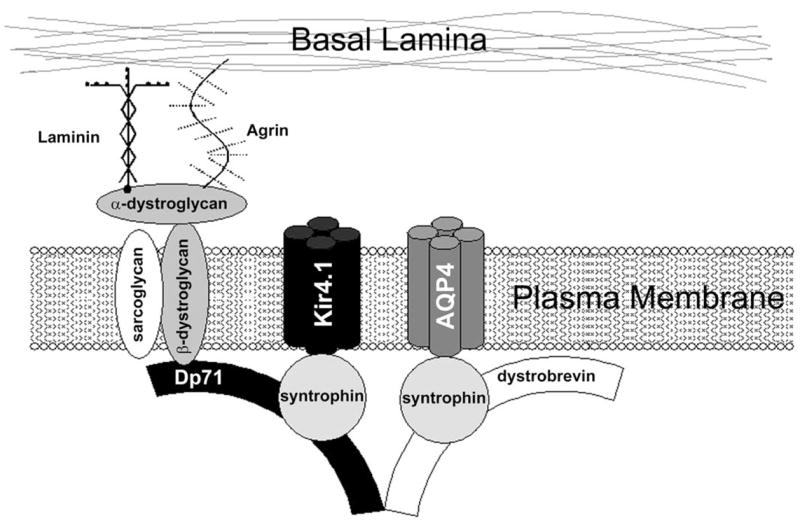
Schematic representation of the glial cell DGC and associated Kir and water channels. The complex is shown with the putative interactions between a syntrophin isoform, Kir4.1 and AQP4. [From Kofuji and Connors (2003), with permission.]
In astrocytes, the direct participation of DGC proteins in the targeting of Kir4.1 and AQP4 channels has been demonstrated in an α-syntrophin knockout mouse line. In astrocytes, as in Müller cells, AQP4 and Kir4.1 are strongly expressed in glial membranes (endfeet) that are in direct contact with capillaries and the pia (Nielsen et al., 1997; Higashi et al., 2001). Quantitative immunoelectromicroscopy has shown that in the hippocampus of α-syntrophin knockout mice, the expression of AQP4 in astrocyte end-feet is greatly diminished, while expression of Kir4.1 channels is less affected (Amiry-Moghaddam et al., 2003). These results indicate that α-syntrophin is critical for the targeting and clustering of AQP4 channels to astrocytic endfeet, but is perhaps not as vital for targeting of Kir4.1 channels. Further work will be needed to establish whether Kir4.1 channels are indeed linked to the DGC in astrocytes via syntrophin isoforms.
Despite the apparent lack of major rearrangements in Kir4.1 channel localization and expression in hippocampus astrocytes, the clearance of extracellular K+ following neuronal stimulation was significantly impaired in hippocampus slices from α-syntrophin knockout mice (Amiry-Moghaddam et al., 2003). Such studies provide the first evidence for a direct functional coupling between AQP4 and Kir4.1 channels. Perhaps, as suggested by Amiry-Moghaddam et al. (2003), the efficiency of K+ spatial buffering is decreased by impairment of the accompanying water flux. This tantalizing observation suggests that impaired targeting or function of AQP4 or/and Kir4.1 channels may have clinical relevance in conditions such as epilepsy or brain edema. It remains to be seen, however, whether Kir4.1 channels play an important role in K+ spatial buffering in the brain and in the overall buffering of [K+ ]o. Future studies in mice with genetic inactivation of Kir4.1 channels should provide an answer to this important question.
CONCLUSIONS
It has been almost 40 years since the initial proposal of K+ spatial buffering as a mechanism for [K+]o buffering in the CNS (Orkand et al., 1966). Since then, it has become clear that [K+ ions ]o regulation involves both net uptake of K and K+ spatial buffering. The relative importance of these K+ homeostatic mechanisms may vary from site to site in the CNS. K+ spatial buffering has been best investigated in the retina, where it is called K+ siphoning to reflect the directed K+ transport from the plexiform layers to the vitreous and to blood vessels (Newman et al., 1984). Molecular studies indicate that Kir4.1 channel localization is critical to the highly asymmetric K+ conductance found in Müller cells (Kofuji et al., 2000). Furthermore, Kir2.1 and possibly Kir5.1 are also expressed in Müller cells (Kofuji et al., 2002). K+ buffering in the retina may be facilitated by concerted action of the strongly rectifying Kir2.1 channels, which allow K+ entry into Müller cells, and the weakly rectifying Kir4.1 channels, which allow K+ exit to large sinks such as the vitreous humor (Kofuji et al., 2002). Moreover, recent reports indicate that dystrophin and dystrophin-associated proteins may promote the clustering and subcellular distribution of Kir4.1 channels and the water channel AQP4 in astrocytes (Amiry-Moghaddam et al., 2003). The co-localization of water and K+ channels in glial cell membranes suggests that K+ buffering and water flux are tightly coupled in the brain.
Although significant progress has been made in the past decade, we still do not have a coherent picture of the relative importance of the various mechanisms for [K+]o buffering in the CNS. Given recent advances in optical, electrophysiological and genetic methods, it is plausible that this picture will gain greater clarity in the near future.
Abbreviations
- DGC
dystrophin glycoprotein complex
- EK
equilibrium potential
- IOS
intrinsic optic signal
- [K+]o
extracellular K+ concentration
- Kir
inwardly rectifying K+
- Vm
glial syncytium membrane potential
References
- Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amedee T, Robert A, Coles JA. Potassium homeostasis and glial energy metabolism. Glia. 1997;21:46–55. doi: 10.1002/(sici)1098-1136(199709)21:1<46::aid-glia5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci USA. 2003;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, ten Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol. 1987;382:159–174. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Koroshetz WJ, Chun LL, Corey DP. Ion channel expression by white matter glia: the type-1 astrocyte. Neuron. 1990;5:527–544. doi: 10.1016/0896-6273(90)90091-s. [DOI] [PubMed] [Google Scholar]
- Brew H, Gray P, Mobbs P, Attwell D. Endfeet of retinal glial cells have higher densities of ion channels that mediate K+ buffering. Nature. 1986;324:466–468. doi: 10.1038/324466a0. [DOI] [PubMed] [Google Scholar]
- Claudepierre T, Dalloz C, Mornet D, Matsumura K, Sahel J, Rendon A. Characterization of the intermolecular associations of the dystrophin-associated glycoprotein complex in retinal Müller glial cells. J Cell Sci. 2000a;113(Pt 19):3409–3417. doi: 10.1242/jcs.113.19.3409. [DOI] [PubMed] [Google Scholar]
- Claudepierre T, Mornet D, Pannicke T, Forster V, Dalloz C, Bolanos F, Sahel J, Reichenbach A, Rendon A. Expression of Dp71 in Müller glial cells: a comparison with utrophin- and dystrophin-associated proteins. Invest Ophthalmol Vis Sci. 2000b;41:294–304. [PubMed] [Google Scholar]
- Coles JA, Orkand RK, Yamate CL, Tsacopoulos M. Free concentrations of Na, K, and Cl in the retina of the honeybee drone: stimulus-induced redistribution and homeostasis. Ann NY Acad Sci. 1986;481:303–317. doi: 10.1111/j.1749-6632.1986.tb27160.x. [DOI] [PubMed] [Google Scholar]
- Connors B, Dray A, Fox P, Hilmy M, Somjen G. LSD’s effect on neuron populations in visual cortex gauged by transient responses of extracellular potassium evoked by optical stimuli. Neurosci Lett. 1979;13:147–150. doi: 10.1016/0304-3940(79)90032-6. [DOI] [PubMed] [Google Scholar]
- Connors BW, Ransom BR, Kunis DM, Gutnick MJ. Activity-dependent K+ accumulation in the developing rat optic nerve. Science. 1982;216:1341–1343. doi: 10.1126/science.7079771. [DOI] [PubMed] [Google Scholar]
- Connors NC, Kofuji P. Dystrophin Dp71 is critical for the clustered localization of potassium channels in retinal glial cells. J Neurosci. 2002;22:4321–4327. doi: 10.1523/JNEUROSCI.22-11-04321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalloz C, Sarig R, Fort P, Yaffe D, Bordais A, Pannicke T, Grosche J, Mornet D, Reichenbach A, Sahel J, Nudel U, Rendon A. Targeted inactivation of dystrophin gene product Dp71: phenotypic impact in mouse retina. Hum Mol Genet. 2003;12:1543–1554. doi: 10.1093/hmg/ddg170. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na+/K+ -pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Dennis MJ, Gerschenfeld HM. Some physiological properties of identified mammalian neuroglial cells. J Physiol. 1969;203:211–222. doi: 10.1113/jphysiol.1969.sp008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermietzel R. Diversification of gap junction proteins (connexins) in the central nervous system and the concept of functional compartments. Cell Biol Int. 1998;22:719–730. doi: 10.1006/cbir.1999.0393. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Lux HD. Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia. 1989;2:25–44. doi: 10.1002/glia.440020104. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res. 1980;40:432–439. doi: 10.1007/BF00236151. [DOI] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Franck G, Grisar T, Moonen G. Glial and neuronal Na+, K+ pump. In: Fedoroff S, Hertz L, editors. Advances in neurobiology. New York: Academic Press; 1983. pp. 139–159. [Google Scholar]
- Frishman LJ, Yamamoto F, Bogucka J, Steinberg RH. Light-evoked changes in [K+]o in proximal portion of light-adapted cat retina. J Neurophysiol. 1992;67:1201–1212. doi: 10.1152/jn.1992.67.5.1201. [DOI] [PubMed] [Google Scholar]
- Gardner-Medwin AR, Nicholson C. Changes of extracellular potassium activity induced by electric current through brain tissue in the rat. J Physiol. 1983;335:375–392. doi: 10.1113/jphysiol.1983.sp014540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner-Medwin AR, Coles JA, Tsacopoulos M. Clearance of extracellular potassium: evidence for spatial buffering by glial cells in the retina of the drone. Brain Res. 1981;209:452–457. doi: 10.1016/0006-8993(81)90169-4. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Heinemann U, Lux HD. Stimulus induced and seizure related changes in extracellular potassium concentration in cat thalamus (VPL) Electroencephalogr Clin Neurophysiol. 1979;47:329–344. doi: 10.1016/0013-4694(79)90284-0. [DOI] [PubMed] [Google Scholar]
- Haas M, Forbush B., 3rd The Na-K-Cl cotransporters. J Bioenerg Biomembr. 1998;30:161–172. doi: 10.1023/a:1020521308985. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 1977;120:231–249. doi: 10.1016/0006-8993(77)90903-9. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Schaible HG, Schmidt RF. Changes in extracellular potassium concentration in cat spinal cord in response to innocuous and noxious stimulation of legs with healthy and inflamed knee joints. Exp Brain Res. 1990;79:283–292. doi: 10.1007/BF00608237. [DOI] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Witte OW. Directed spatial potassium redistribution in rat neocortex. Glia. 2000;29:288–292. doi: 10.1002/(sici)1098-1136(20000201)29:3<288::aid-glia10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Horio Y, Hibino H, Inanobe A, Yamada M, Ishii M, Tada Y, Satoh E, Hata Y, Takai Y, Kurachi Y. Clustering and enhanced activity of an inwardly rectifying potassium channel, Kir4.1, by an anchoring protein, PSD-95/SAP90. J Biol Chem. 1997;272:12885–12888. doi: 10.1074/jbc.272.20.12885. [DOI] [PubMed] [Google Scholar]
- Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol. 2003;285:C260–267. doi: 10.1152/ajpcell.00560.2002. [DOI] [PubMed] [Google Scholar]
- Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T, Uchiyama Y, Kurachi Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;17:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu Rev Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Xu X. Current source-density analysis of light-evoked field potentials in rabbit retina. Vis Neurosci. 1999;16:369–377. doi: 10.1017/s0952523899162163. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Müller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwoski CJ, Newman EA, Shimazaki H, Proenza LM. Light-evoked increases in extracellular K+ in the plexiform layers of amphibian retinas. J Gen Physiol. 1985;86:189–213. doi: 10.1085/jgp.86.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman R. Maintenance of a constant brain extracellular potassium. Fed Proc. 1976;35:1244–1247. [PubMed] [Google Scholar]
- Kimelberg HK, Frangakis MV. Furosemide- and bumetanide-sensitive ion transport and volume control in primary astrocyte cultures from rat brain. Brain Res. 1985;361:125–134. doi: 10.1016/0006-8993(85)91282-x. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Connors NC. Molecular substrates of potassium spatial buffering in glial cells. Mol Neurobiol. 2003;28:195–208. doi: 10.1385/MN:28:2:195. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia. 2002;39:292–303. doi: 10.1002/glia.10112. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of Amphibia. J Neurophysiol. 1966;29:768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- Kume-Kick J, Mazel T, Vorisek I, Hrabetova S, Tao L, Nicholson C. Independence of extracellular tortuosity and volume fraction during osmotic challenge in rat neocortex. J Physiol. 2002;542:515–527. doi: 10.1113/jphysiol.2002.017541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J Cell Sci. 2001;114:987–998. doi: 10.1242/jcs.114.5.987. [DOI] [PubMed] [Google Scholar]
- Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- Nagelhus E, Horio Y, Inanobe A, Fujita A, Haug F, Nielsen S, Kurachi Y, Ottersen O. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci USA. 2001;98:14108–14113. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Regional specialization of retinal glial cell membrane. Nature. 1984;309:155–157. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Membrane physiology of retinal glial (Müller) cells. J Neurosci. 1985;5:2225–2239. doi: 10.1523/JNEUROSCI.05-08-02225.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–454. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Regulation of potassium levels by Müller cells in the vertebrate retina. Can J Phys Pharm. 1987a;65:1028–1034. doi: 10.1139/y87-162. [DOI] [PubMed] [Google Scholar]
- Newman EA. Distribution of potassium conductance in mammalian Müller (glial) cells: a comparative study. J Neurosci. 1987b;7:2423–2432. [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom B, editors. Neuroglia. New York: Oxford University Press; 1995. pp. 717–731. [Google Scholar]
- Newman EA. Acid efflux from retinal glial cells generated by sodium bicarbonate cotransport. J Neurosci. 1996a;16:159–168. doi: 10.1523/JNEUROSCI.16-01-00159.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Regulation of extracellular K+ and pH by polarized ion fluxes in glial cells: the retinal Muller cell. Neuroscientist. 1996b;2:109–117. [Google Scholar]
- Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Reichenbach A. The Müller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–312. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley BI, Katz B, Xu Z, Zheng J. Spatial buffering of extracellular potassium by Müller (glial) cells in the toad retina. Exp Eye Res. 1992;55:539–550. doi: 10.1016/s0014-4835(05)80166-6. [DOI] [PubMed] [Google Scholar]
- Orkand RK. Glial-interstitial fluid exchange. Ann NY Acad Sci. 1986;481:269–272. doi: 10.1111/j.1749-6632.1986.tb27157.x. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of Amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–372. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Carlini WG, Connors BW. Brain extracellular space: developmental studies in rat optic nerve. Ann NY Acad Sci. 1986;481:87–105. doi: 10.1111/j.1749-6632.1986.tb27141.x. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol. 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach A, Henke A, Eberhardt W, Reichelt W, Dettmer D. K+ ion regulation in retina. Can J Physiol Pharmacol. 1992;70(Suppl):S239–247. doi: 10.1139/y92-267. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Mechanisms of H+ and Na+ changes induced by glutamate, kainate, and D-aspartate in rat hippocampal astrocytes. J Neurosci. 1996;16:5393–5404. doi: 10.1523/JNEUROSCI.16-17-05393.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. 2002;19:447–458. doi: 10.1006/mcne.2001.1080. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Singer W, Lux HD. Extracellular potassium gradients and visual receptive fields in the cat striate cortex. Brain Res. 1975;96:378–383. doi: 10.1016/0006-8993(75)90751-9. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Extracellular potassium in the mammalian central nervous system. Annu Rev Physiol. 1979;41:159–177. doi: 10.1146/annurev.ph.41.030179.001111. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- Sontheimer Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–172. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- Stanfield PR, Nakajima S, Nakajima Y. Constitutively active and G-protein coupled inward rectifier K+ channels: Kir2.0 and Kir3.0. Rev Physiol Biochem Pharmacol. 2002;145:47–179. doi: 10.1007/BFb0116431. [DOI] [PubMed] [Google Scholar]
- Stonehouse AH, Pringle JH, Norman RI, Stanfield PR, Conley EC, Brammar WJ. Characterisation of Kir2.0 proteins in the rat cerebellum and hippocampus by polyclonal antibodies. Histochem Cell Biol. 1999;112:457–465. doi: 10.1007/s004180050429. [DOI] [PubMed] [Google Scholar]
- Sweadner KJ. Na,K-ATPase and its isoforms. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford University Press; 1995. pp. 259–272. [Google Scholar]
- Tada Y, Horio Y, Kurachi Y. Inwardly rectifying K+ channel in retinal Müller cells: comparison with the KAB-2/Kir4.1 channel expressed in HEK293T cells. Jpn J Physiol. 1998;48:71–80. doi: 10.2170/jjphysiol.48.71. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyami H, Sohmiya K, Nakanishi S, Kurachi Y. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–16346. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem. 2000;275:16404–16407. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- Vyskocil F, Kritz N, Bures J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res. 1972;39:255–259. doi: 10.1016/0006-8993(72)90802-5. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of Na/K/Cl cotransport in astrocytes. Can J Physiol Pharmacol. 1992;70(Suppl):S260–262. doi: 10.1139/y92-270. [DOI] [PubMed] [Google Scholar]
- Walz W. Chloride/anion channels in glial cell membranes. Glia. 2002;40:1–10. doi: 10.1002/glia.10125. [DOI] [PubMed] [Google Scholar]
- Xu X, Karwoski C. The origin of slow PIII in frog retina: current source density analysis in the eyecup and isolated retina. Vis Neurosci. 1997;14:827–833. doi: 10.1017/s0952523800011561. [DOI] [PubMed] [Google Scholar]
- Zahs KR, Kofuji P, Meier C, Dermietzel R. Connexin immunoreactivity in glial cells of the rat retina. J Comp Neurol. 2003;455:531–546. doi: 10.1002/cne.10524. [DOI] [PubMed] [Google Scholar]