

Abstract
The lanthanide cation, gadolinium (Gd) attenuates post-ischemic myocardial stunning. This study tests the hypothesis that Gd also preconditions the myocardium against infarction following ischemia-reperfusion (IR), and explores potential mechanisms underlying Gd-induced cardioprotection. Regional myocardial infarction was induced in rats by occluding the left anterior descending artery for 30 minutes and reperfusing for 120 minutes. Rats (n = 6/group) were administered intravenous Gd (1 to 100 μmol/kg) 15 minutes prior to ischemia. Hearts were excised after reperfusion to determine infarct size (IS) and area at risk (AAR). The ratio IS/AAR (%) was reduced by Gd in a “U”-shaped, dose-dependent manner. The minimum dose that reduced IS/AAR was 5 μmol/kg (52 ± 5% vs. 64 ± 4%), while the dose that reduced IS/AAR maximally was 20 μmol/kg (44 ± 4%). Gd also reduced IS/AAR when given 1 minute before reperfusion (47 ± 3%) but not when given 10 seconds after reperfusion (60 ± 3%). Cardioprotection was maintained if I-R was delayed 24–72 hours after Gd administration. Cardioprotection by Gd was abolished by inhibition of JAK-2 with AG-490, of p42/44 MAPK with PD98059 or of KATP channels with glibenclamide. None of these agents given alone altered IS/AAR compared with controls. Inhibition of JAK-2 also blocked Gd-induced delayed cardioprotection. Gd may have broad potential roles in IR, as it confered immediate cardioprotection when given prior to ischemia or prior to reperfusion, and delayed cardioprotection for up to 72 hours after administration. The mechanism underlying Gd-induced preconditioning appears to be multi-factorial, involving JAK-2, STAT-3 and p44 MAPK pathways, as well as KATP channels.
INTRODUCTION
The trivalent lanthanide cation, gadolinium (Gd), modulates cardiac contractile dysfunction in a variety of settings. Gd prevents contractile dysfunction resulting from abnormal, axial stretch of isolated papillary muscles (1), attenuates post-ischemic regional contractile dysfunction (stunning) in vivo (2, 3), and restores normal contractile function in dilated cardiomyopathy (4). Importantly, Gd attenuates myocardial stunning if given prior to onset of ischemia or if given during ischemia (prior to reperfusion). Gd does not enhance contractile function of normal myocardium (1–4), suggesting that its effects result from interruption of pathophysiologic events that cause contractile dysfunction. The precise mechanism(s) underlying its effects remain unknown.
Based on the cardioprotective effects of Gd on stunned myocardium, we hypothesized that Gd might also precondition the myocardium to reduce infarct size following ischemia-reperfusion (IR). While previous reports have suggested that Gd abolishes the preconditioning effects of certain mechanical interventions (5, 6), we hypothesized that Gd would modulate the effects of specific mediators of IR-induced injury similar to its apparent mechanism of effect in stunned myocardium. Potential targets for Gd-induced preconditioning include the JAK-STAT pathway (7), p42/44 MAPK (7), KATP channels (7) and free radicals (8). Accordingly, the objectives of the present study were to determine (1) the effect of Gd on infarct size in a rodent model of regional myocardial ischemia/reperfusion; (2) to demonstrate possible temporal characteristics of Gd-mediated cardioprotection; and (3) to determine the potential roles of JAK, STAT, p42/44 MAP kinase, KATP chann els and free radicals in Gd-associated cardioprotection.
MATERIALS AND METHODS
Male Sprague-Dawley (220–250 g) rats were used for all studies. All animal work was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin. The studies were conducted in a blinded manner.
Instrumentation, Ischemia-Reperfusion Protocol and Gd treatment
Rats were anesthetized with 20–40 mg/kg intraperitoneal sodium pentobarbital. The right jugular vein was cannulated for delivery of saline solution. A catheter was inserted in the left femoral artery to measure both blood pressure and heart rate, and to withdraw blood. Pressure and rate measurements were monitored using a Gould PE50 or PE23 pressure transducer connected to a Grass model 7 polygraph. The rats were intubated with a 14 gauge catheter and ventilated at 38–45 breaths/min (Harvard Apparatus, model 683; South Natick, MA) with supplemental oxygen. Atelectasis was prevented by maintaining a positive end-expiratory pressure of 5–10 mmH2O. Arterial pH, pCO2, and pO2 were monitored with an AVL 995 pH/blood gas analyzer, and normal values were maintained by adjusting respiratory rate, tidal volume and/or oxygen flow. Body temperature was maintained between 35–37°C using a heating pad.
A left thoracotomy was performed, the pericardium was opened and the left coronary artery was identified. A ligature (6-0 Prolene) was passed around the proximal segment of the left coronary artery and the ends of the suture were threaded through a propylene tube to form a snare. Regional left ventricular ischemia was induced by tightening the snare for 30 minutes. Coronary artery occlusion was confirmed by epicardial cyanosis and a decrease in blood pressure. Reperfusion was achieved by releasing the snare and was confirmed by a marked hyperemic response of the myocardium. The heart was reperfused for 120 minutes then excised and assessed for extent of tissue injury.
Gadolinium chloride hexahydrate (1 to 100 μmol/kg dissolved in 0.9% NaCl solution; Sigma, Milwaukee, WI) was given as an intravenous (i.v.) bolus 15 minutes before inducing myocardial ischemia. To determine potential mechanisms of action of Gd, certain pharmacological blocking agents were administered i.v. to some animals 5 minutes prior to Gd treatment. To determine important temporal relations of Gd-mediated effects with respect to ischemia-reperfusion, Gd was given to some animals during ischemia (1 or 15 minutes prior to reperfusion) and to others 10 sec following reperfusion. In another series of rats, ischemia-reperfusion was delayed 24 – 120 hours after Gd treatment through the tail vein. In these animals, an isolated heart preparation was employed as described below to exclude Gd from the coronary perfusate.
Isolated heart perfusion
Isolated rat hearts were perfused in a retrograde manner at constant perfusion pressure (98 mmHg) with a balloon inflated in the left ventricle. End-diastolic pressure was set to 5 mmHg and developed pressure was recorded during steady-state conditions (9). The standard perfusate was modified Krebs-Henseleit bicarbonate buffer, containing (in mM): 118.5 NaCl, 25.0 NaHCO3, 4.8 KCl, 0.6 MgSO4, 1.2 H2O, and 1.2 KH2PO4 (pH 7.4 when gassed with 95% O2-5% CO2), in which the calcium content was reduced to 1.8 mM. Glucose (11.1 mM) was added to the perfusate. Before use, all perfusion fluids were filtered through cellulose acetate membranes with a pore size of 5.0 μm to remove particulate matter. Hearts were allowed to beat spontaneously and were kept in temperature-controlled chambers to maintain myocardial temperature at 37 °C during periods of perfusion and ischemia. Regional ischemia was induced by snaring the left coronary artery for 30 minutes, after which the snare was released and the heart reperfused for 120 minutes. Recovery of left ventricular developed pressure was determined after 60 minutes reperfusion and was expressed as a percentage of its pre-ischemic value. Hearts were removed from the apparatus and assessed for extent of tissue injury.
Determination of tissue injury
Infarct size (IS), left ventricular (LV) size and area at risk (AAR) were determined as described previously (9, 10). For the in vivo studies following 2 hours of reperfusion, the heart was excised, cross sectioned into four to five 1-mm-thick transverse sections and separated into normal zone and AAR. The pieces were incubated in 1% (w/v) 2,3,5-triphenyltetrazolium chloride (TTC) to determine infarct size. The heart was then incubated overnight at 4°C in 10% formaldehyde and the infarcted tissue was dissected from the AAR. Infarct size was expressed as a percent of the AAR (IS/AAR). For the in vitro studies following 2 hours of reperfusion, the ligature was again occluded and the AAR was determined by patent-blue-negative staining. TTC was delivered to the isolated heart via a syringe attached to the three-way stopcock. The latex balloon was removed from the left ventricle before the hearts were stained. TTC was administered at a rate of 1 ml/min for 10 min. Hearts were then rapidly removed from the perfusion apparatus and sliced across the long axis of the left ventricle from apex to base into four to five 1-mm-thick transverse sections, transferred into a vial containing 10% formaldehyde, and stored at 4°C overnight. The infarcted tissue was then separated from the noninfarcted tissue and measured as a percent (%infarct = infarct area/area at risk × 100) using a digital color camera with a 55-mm lens (Nikon). Imaging analysis software (Imaging Research; St. Catharines, Ontario, Canada) was programmed to recognize infarcted tissue in proportion to the entire left ventricle. IS was expressed as a percentage of the AAR (IS/AAR).
Western analysis
In some animals, the free wall of the left ventricle from in vivo treated hearts was excised and snap-frozen for analysis. This was done either at 15 minutes following Gd administration or at 5 minutes following reperfusion. Frozen hearts were powdered in a pre-chilled mortar and pestle with liquid nitrogen. Ice-cold lysis buffer pH 7.0 (20mM MOPS, pH 7.0, 2mM EGTA, 5mM EDTA, 30mM sodium fluoride, 40mM β-glycerophosphate, pH 7.2, 10mM sodium pyrophosphate, 2mM sodium orthovanadate, 1mM PMSF, 0.5% NP-40) was added to powdered tissue and homogenized for 20 strokes. The homogenate was centrifuged at 13,000×g at 4 °C for 30 min. Protein concentration was measured (BCA method, Interchim, Montluçon, France). Aliquots of supernatant containing equal amounts of protein were boiled in sample loading buffer for 5 min before loading on 10% SDS–polyacrylamide gel and transferred onto Nitrocellulose membrane (Bio-rad). The non-specific binding sites on the membrane were blocked with 5% non-fat milk in tris-buffered saline (Bio-Rad) plus 0.1% Tween-20 (TBST). The membrane was incubated in TBST buffer containing 3% Bovine Albumin and 0.1% Sodium Azide with specific antibodies against phosphorylated (Tyr 705) and non phosphorylated STAT-3, phosphorylated STAT-5 (Tyr 694), phosphorylated (Thr 202/Tyr 204) and non-phosphorylated p42/44 MAPK (antibodies all from Cell Signaling) diluted 1:1000 overnight at 4 °C. All membranes were incubated in horseradish peroxidase-conjugated anti-rabbit IgG antibody (Bio-rad) in 5% non-fat milk in TBST for 1 h at room temperature. Finally, the blots were developed by ECL.
Statistical analysis
All groups consisted of 6 animals. All values were expressed as the mean ± SD. For the infarct size, densitometry, hemodynamic data, left ventricular mass, and AAR, statistical significance was determined by performing a one-way ANOVA with Bonferroni’s multiple comparison test as the post hoc test. Significance was attributed to those groups that had a P value < 0.05.
RESULTS
A total of 283 rats were used to obtain 276 successful experiments. Seven rats were excluded due to ventricular fibrillation during reperfusion (5), respirator malfunction (1), and the suture breaking during negative staining (1).
Dose-Response Studies
Rats treated with Gd 15 minutes before inducing myocardial ischemia exhibited increased resistance to ischemia as determined by a reduction in the ratio of infarct size-to-area at risk (IS/AAR; %). Gadolinium decreased infarct size in a U-shaped dose-dependent manner (Figure 1A). The minimum dose of Gd that reduced myocardial infarction compared with controls was 5 μmol/kg (52 ± 5% vs. 64 ± 4%), while the dose that afforded maximum reduction in infarct size was 20 μmol/kg (44 ± 4%). Results for heart rate and mean arterial pressure during the dose-response experiments are summarized in the Table. Administration of Gd at a dose of 20 μmol/kg or more resulted in a decrease in blood pressure. Animals treated with any dose of Gd exhibited decreased blood pressure at 15 minutes of ischemia and all animals exhibited decreased blood pressure after 120 minutes reperfusion. Based on the dose-response data, the dose of Gd used in all further experiments was 20 μmol/kg, as it resulted in maximal cardioprotection with minimal hemodynamic effects.
Figure 1.
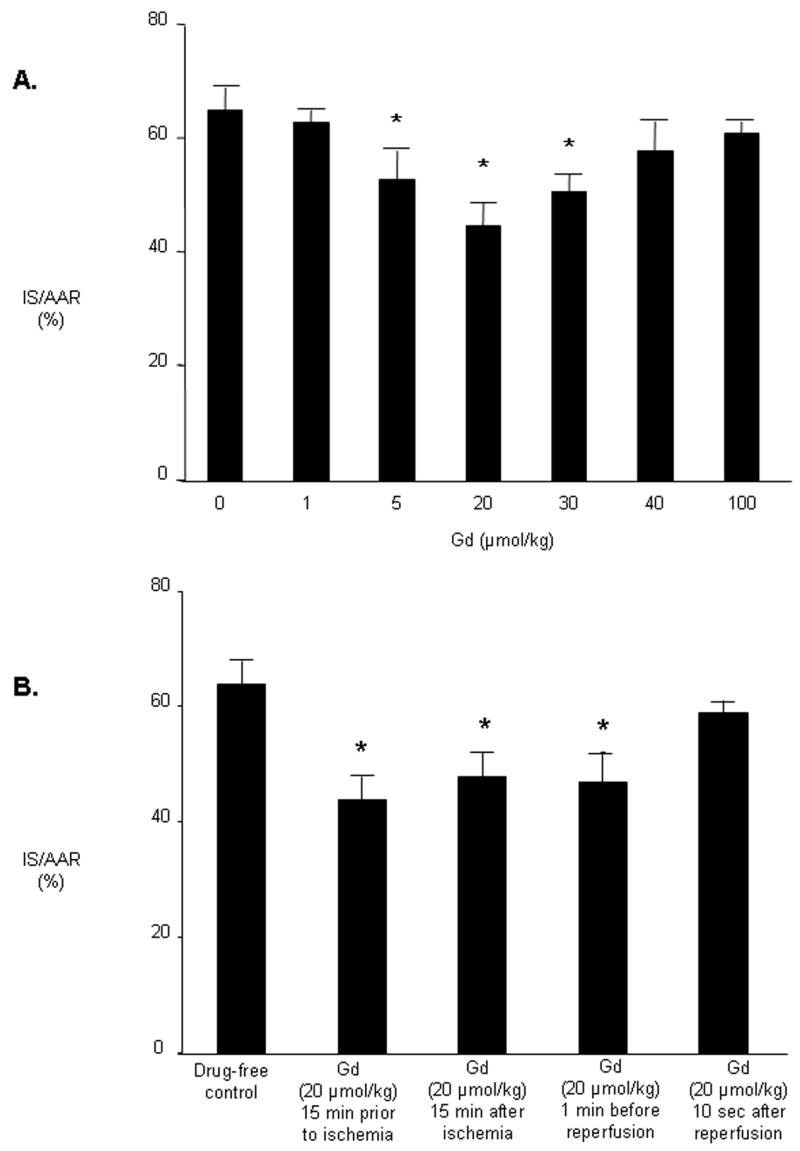
A. Dose-response studies of Gd immediate ischemic cardio-protection. Rats received Gd (1 to 100 μmol/kg IV) 15 minutes before ischemia. Infarct size was determined after 30 minutes of regional ischemia and 120 minutes of reperfusion. B. Phase of action of Gd. AAR, area at risk; IS, infarct size. Data are mean ± SD, n = 6/group. * = P < 0.05, Gd vs drug-free control.
Temporal Relations
Administration of Gd during ischemia (1 or 15 minutes prior to reperfusion) reduced infarct size to the same extent as when Gd was given before onset of ischemia, but administration of Gd after reperfusion was ineffective in reducing IS (Figure 1B).
Delayed cardioprotection was observed in isolated hearts 24–72 hours following administration of Gd (Figure 2). Reduction in infarct size compared with controls (42 ± 5 to 14 ± 5%) and increased recovery of post-ischemic developed pressure (61 ± 6 to 77 ± 5%) was maximal 72 hours after Gd administration.
Figure 2.
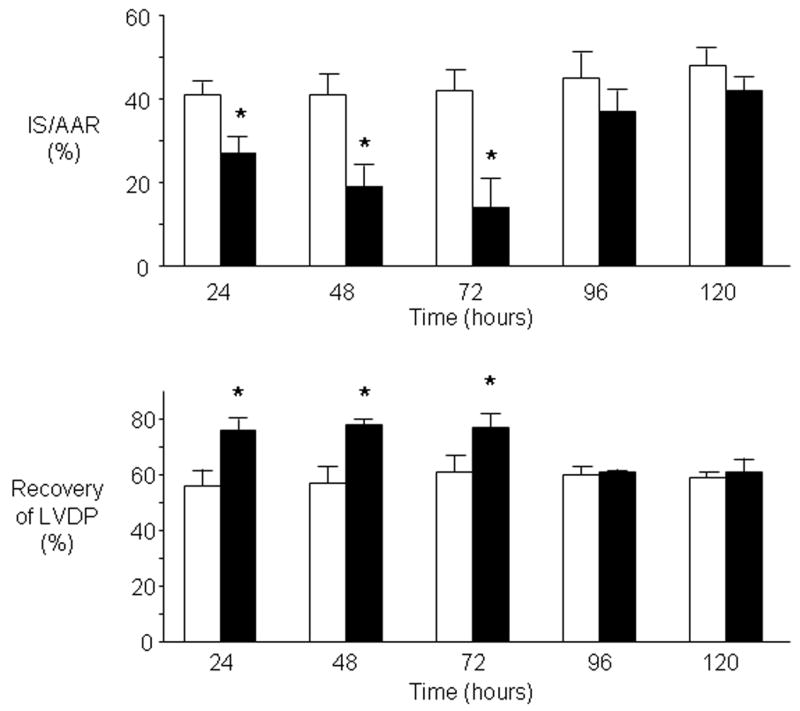
Gd confers delayed ischemic cardio-protection. Rats (n = 6 per group) were treated with a single dose of Gd (20 μmol/kg) via the tail vein and allowed to recover 24–120 hours. Hearts were then isolated and subjected to 30 minutes regional myocardial ischemia and 180 minutes reperfusion with Gd excluded from the coronary perfusate. Open bars represent data from saline treated rats. Solid bars represent data from Gd treated rats. LVDP, left ventricular developed pressure. Bar graphs represent the mean ± SD. * P < 0.05, Gd vs saline-treated.
Mechanism of Action
Administration of the Janus kinase (JAK-2) inhibitor AG-490 (3 mg/kg) in vivo 5 minutes before Gd treatment abolished Gd-induced cardioprotection but had no effect on infarct size when given alone prior to ischemia (Figure 3). Western analysis with anti-signal transducers and activators of transcription (STAT) antibodies demonstrated that reperfusion initiates increased phosphorylation of STAT-3 (Tyr 705) (Figure 4). Administration of Gd in the absence of ischemia also increased STAT-3 phosphorylation, but administration of Gd did not further increase reperfusion-induced phosphorylation of STAT-3 compared with reperfusion in the absence of Gd. Stripping and reprobing at the same blots with non phosphorylated antibodies confirmed that equal amounts of protein were loaded. Gd had no effect on phosphorylation of STAT-5 (Tyr 694) either before or after ischemia (data not shown). Pretreatment with the JAK-2 inhibitor AG-490 15 minutes before Gd treatment also blocked the delayed cardioprotective effect of Gd 24 hours later (Figure 5). AG-490 alone had no effect on delayed cardioprotection.
Figure 3.
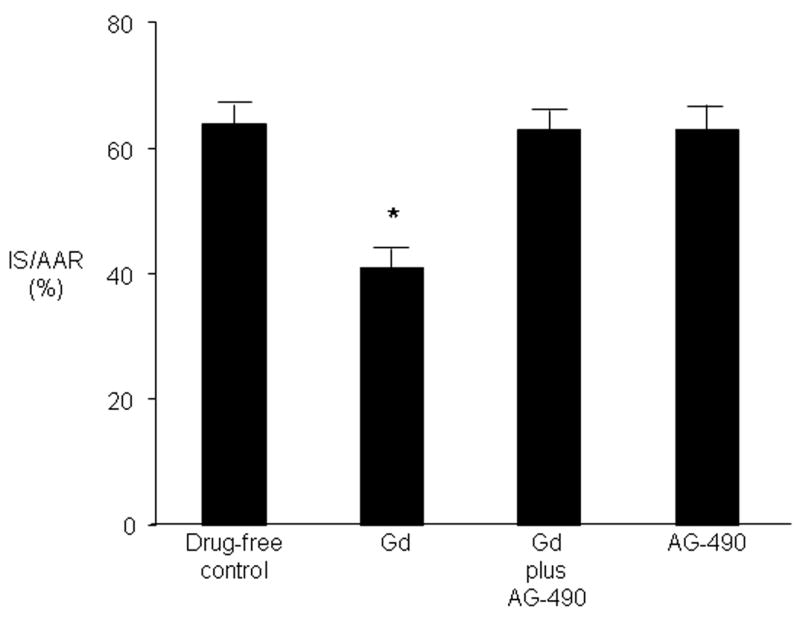
JAK-2 is required for immediate Gd ischemic cardio-protection. Rats were untreated or were administered Gd (20 μmol/kg) alone or in the presence of AG-490 (3 mg/kg) or received the inhibitor alone. Infarct size was determined after 30 minutes of regional ischemia and 120 minutes of reperfusion. Values are mean ± SD. * = P < 0.05 vs drug-free control.
Figure 4.
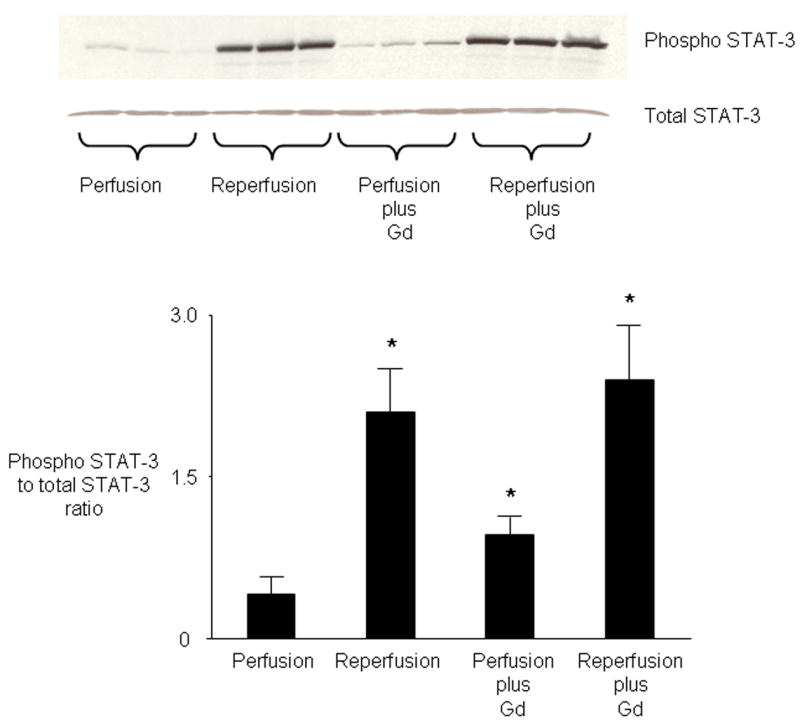
Role of STAT in mediating protection by Gd. Western analysis of cell lysates from hearts demonstrates that Gd (20 μmol/kg) given before ischemia (Perfusion plus Gd) causes phosphorylation of STAT-3. Reperfusion in the absence of Gd (Reperfusion) also induces phosphorylation of STAT-3 (Tyr-705). Pre-ischemic treatment with Gd does not further increase reperfusion-induced phosphorylation of STAT-3 (Reperfusion plus Gd). Total STAT-3 results shown in lower panel. Bar graphs represent the mean ± SD. * P < 0.05, vs perfusion.
Figure 5.

Involvement of JAK-2 in triggering delayed cardioprotection by Gd. Rats were pretreated with the JAK inhibitor AG-490 (3 mg/kg) or saline 15 min before Gd treatment (20 μmol/kg) and resistance to injury from IR determined 24–120 hours later. Infarct size and recovery of post-ischemic left ventricular developed pressure was determined after 30 minutes of regional ischemia and 120 minutes of reperfusion. Values are mean ± SD. * = P < 0.05 vs drug-free control.
Administration of the selective p42/44 MAPK inhibitor PD98059 (1 mg/kg) 5 minutes before Gd treatment abolished Gd-induced cardioprotection but had no effect on infarct size when given alone prior to ischemia (Figure 6). Reperfusion after ischemia was associated with increased phosphorylation of both p42 and p44 MAPK (Figure 6). Administration of Gd in the absence of ischemia had no effect on phosphorylation of p42/44 MAPK but further increased phosphorylation of p42 and p44 MAPK during reperfusion. Stripping and reprobing of the same blots with non phosphorylated antibodies confirmed that equal amount of proteins were loaded.
Figure 6.

Involvement of p42/44 MAPK in mediating protection by Gd. Rats were untreated or were administered Gd (20 μmol/kg) alone or in the presence of PD90859 (1mg/kg) or received the inhibitor alone. Infarct size was determined after 30 minutes of regional ischemia and 120 minutes of reperfusion. Values are mean ± SD. * = P < 0.05 vs untreated. Western analysis of cell lysates from hearts demonstrates that Gd (20 μmol/kg) does not increase phosphorylation of p42/44 MAPK before ischemia (Perfusion plus Gd). Reperfusion in the absence of Gd induces phosphorylation of p42/44 MAPK (Reperfusion). Gd further increases phosphorylation of p42/p44 MAPK during reperfusion (Reperfusion plus Gd). Total p42/44 MAPK results shown in lower panel. Bar graphs represent the mean ± SD. * P < 0.05, perfusion plus Gd vs perfusion. + P < 0.05, reperfusion plus Gd vs reperfusion.
Administration of the general KATP channel inhibitor glibenclamide (3 mg/kg) 5 minutes before Gd treatment abolished Gd-induced cardioprotection but had no effect on infarct size when given alone prior to ischemia (Figure 7).
Figure 7.

Involvement of KATP channels in mediating protection by Gd. Rats were untreated or were administered Gd (20 μmol/kg) alone or in the presence of the KATP channel blocker glibenclamide (3 mg/kg) or received inhibitor alone. Infarct size was determined following 30 minutes of regional ischemia and 120 minutes of reperfusion. Values are mean ± SD. * P < 0.05 vs drug-free control.
Administration of the free radical scavengers 2-mercaptoproprionyl glycine (20 mg/kg) or N-acetyl cysteine (150 mg/kg) 5 minutes before Gd treatment had no effect on Gd-induced cardioprotection and had no effect on infarct size when given alone (results not shown).
DISCUSSION
The present study is the first to demonstrate myocardial preconditioning against infarction by the lanthanide cation, gadolinium (Gd). We have previously demonstrated that Gd attenuates contractile dysfunction associated with various pathophysiologic conditions, including abnormal stretch, ischemia-reperfusion (stunning) and cardiomyopathy (1–4). In the current study, administration of Gd in the setting of ischemia-reperfusion (IR) reduced infarct size in a dose-dependent manner by a mechanism involving activation of both JAK/STAT and p44 MAPK pathways, as well as KATP channels. The infarct reducing effect of Gd is dose-dependent, with maximum reduction in infarct size manifest at a dose of 20 μmol/kg. We did not observe a reduction in infarct size with Gd at 40 μmol/kg, the dose used by Gysembergh et al to block volume overload-induced cardioprotection (5), however we did observe protection with Gd at doses of 5–30 μmol/kg. These findings illustrate the dose-dependent nature of protection against infarction with Gd and highlight the need to perform dose-response studies when determining the potential cardioprotective properties of novel agents.
The temporal effects of Gd with respect to IR observed in the present study are important, as they suggest potential broad applications for the use of Gd in different clinical settings. Similar to previous observations on stunned myocardium (3), Gd was effective in the present study at protecting the heart from infarction when given prior to onset of ischemia or when given during ischemia (in particular, when given only one minute prior to reperfusion). Thus Gd may offer cardioprotection when administered prior to ischemia in planned settings where IR might occur, such as elective coronary bypass surgery (CABG) or elective percutaneous intervention (PCI). It may also be useful however, in acute coronary syndromes, where administering Gd immediately prior to reperfusion (by either emergency CABG or PCI) could help reduce infarct size. The magnitude of immediate protection afforded by Gd in the current study is less than the protection reported with ischemic preconditioning (31% vs. 86% reduction in infarct size) (11), but the fact that Gd can be given one minute before reperfusion and still provide significant infarct reduction makes it perhaps more clinically important than ischemic preconditioning. In addition to these temporal relationships, we observed that a single dose of Gd confers delayed cardioprotection for up to 72 hours to an extent that is comparable to delayed cardioprotection observed with ischemic preconditioning (66% vs. 67% reduction in infarct size) (12). This effect may also have important clinical applications in revascularization procedures, where recurrent ischemia may occur early after the procedure from graft occlusion or PCI failure.
The cardioprotective effects of Gd observed in the present study appear to be mediated through multiple mechanisms, including both the JAK/STAT pathway and the p42/p44 MAPK component of the Reperfusion Injury Salvage Kinase pathway. The JAK/STAT pathway plays an integral role in the response of the myocardium to various insults, including myocardial infarction. Furthermore it has a prominent role in cardioprotective therapies such as ischemic preconditioning. Further studies are needed to determine if other components of preconditioning such as PKC, adenosine receptors, and Akt/PI3 kinase play a role in Gd-induced cardioprotection. The JAK/STAT pathway also plays an essential role in the development of delayed preconditioning (13). Our study demonstrates that activation of JAK/STAT is necessary for both the immediate and delayed cardioprotective effects of Gd to be manifest. We propose that Gd stimulates the heart to conduct a signal via the JAK/STAT pathway to the nucleus to activate nuclear transcription factors, resulting in the synthesis of genes such as inducible nitric oxide synthase, hemoxygenase-1, aldose reductase, and Mn superoxide dismutase to confer delayed protection against injury from myocardial infarction. As JAK resides in the vicinity of the sarcolemma, we speculate that Gd may be acting through a cell surface receptor to activate JAK/STAT. The notion that Gd can act as a ligand to a receptor protein is supported by recent studies showing the metabotropic glutamate receptor 1α binds Gd resulting in activation of two distinct pathways involving calcium (mediated through the Gq family of heterotrimeric guanine nucleotide-binding proteins) and adenosine 3′,5′-monophosphate (cAMP, mediated through Gs proteins) (14). Furthermore it is known that the JAK/STAT and p42/44 MAPK pathways can be simultaneously activated in the heart (15, 16) to confer protection against injury from ischemia/reperfusion.
The present study suggests that the cardioprotective effects of Gd are also mediated in part through ATP-dependent potassium (KATP) channels. These channels, highly expressed in myocardial sarcolemmal and thought to be expressed in myocardial mitochondria, have been found to serve as mediators of cardioprotection (17–19). Gadolinium is known to exert effects on a broad range of ion channels in the heart. Recently Babich et al (20) proposed that Gd binding stabilizes ion channels in an inactivated state similar to the actions of other calcium channel blockers such as the dihydropyridines. In addition to L-type calcium channels and the sodium-calcium exchanger, another pathway exists for entry of calcium into the ventricular myocyte that is gadolinium-sensitive and distinct from the B-type channel (21). Furthermore Gd is commonly used to interrupt stretch-mediated changes in intracellular ion flux in the heart through a presumed blockade of specific stretch-activated ion channels (22–24). Since mechanical stretch causes pathophysiologic changes in the myocardium (1, 22, 25, 26), and regional myocardial ischemia results in abnormal stretch (2, 27, 28), the effects of Gd observed herein may also be related to modulation of ion fluxes that are caused by ischemia-induced stretch. Effects on KATP and perhaps other ion channels may be of particular importance when Gd is administered one minute before reperfusion, as cell signaling to induce gene expression (as postulated above) would be a less likely factor in such a short time frame.
The effects of Gd on infarct size were not altered by treatment with free radical scavengers, suggesting that the mechanism underlying the immediate cardioprotective effects of Gd is not related to oxidative stress. The selection of the in vivo doses for the radical scavengers 2-mercaptoproprionyl glycine (20mg/kg) and N-acetyl cysteine (150 mg/kg) were based on previous studies from our lab (29) and from another laboratory, respectively (30).
In summary, a single treatment with Gd conferred immediate protection against injury caused by regional ischemia and reperfusion in the rat heart, with protection mediated partially by JAK-2, STAT3, p42/p44 MAPK and KATP channels. Gd also conferred delayed cardioprotection by a mechanism associated with the activation of JAK-2. Further studies are warranted to identify additional pathways by which Gd confers cardioprotection. Our results suggest that Gd directly protects the heart and may represent a novel approach for the treatment of myocardial IR injury.
Table 1.
Hemodynamic values for Gd dose-response studies. Values are mean ± SD, n = 6/group. Hearts subjected to 30 minutes regional ischemia followed by 2 hours of reperfusion.
| BASELINE
|
15 min POST DRUG
|
15min ISCHEMIA
|
2hr REPERFUSION
|
|||||
|---|---|---|---|---|---|---|---|---|
| Heart rate, beats/min | MAP, mmHg | Heart rate, beats/min | MAP, mmHg | Heart rate, beats/min | MAP, mmHg | Heart rate, beats/min | MAP, mmHg | |
| Drug-free control | 353 ± 3 | 128 ± 6 | - | - | 370 ± 17 | 127 ± 6 | 347 ± 9 | 96 ± 6* |
| Gd (1 μmol/kg) | 354 ± 5 | 131 ± 4 | 360 ± 7 | 127 ± 5 | 366 ± 7 | 120 ± 3* | 362 ± 6 | 80 ± 9* |
| Gd (5 μmol/kg) | 377 ± 12 | 134 ± 5 | 378 ± 14 | 126 ± 7 | 377 ± 12 | 119 ± 8* | 353 ± 11 | 81 ± 6* |
| Gd (20 μmol/kg) | 387 ± 11 | 117 ± 5 | 382 ± 9 | 100 ± 4* | 382 ± 8 | 90 ± 5* | 372 ± 8 | 66 ± 4* |
| Gd (30 μmol/kg) | 373 ± 11 | 126 ± 3 | 377 ± 8 | 102 ± 4* | 380 ± 7 | 89 ± 5* | 363 ± 8 | 68 ± 5* |
| Gd (40 μmol/kg) | 380 ± 4 | 134 ± 2 | 376 ± 9 | 104 ± 5* | 380 ± 5 | 88 ± 2* | 358 ± 3* | 69 ± 3* |
| Gd (100 μmol/kg) | 364 ± 13 | 130 ± 3 | 360 ± 13 | 98 ± 2* | 358 ± 14 | 94 ± 4* | 352 ± 13 | 51 ± 7* |
= P < 0.05, vs baseline group. MAP = mean arterial pressure.
Acknowledgments
The clerical assistance of Mary Lynne Koenig is gratefully acknowledged.
The authors claim no financial interest in the products mentioned within this article. This research was supported in part by National Institutes of Health grants HL 54075 to John E. Baker and HL 08311 to Garrett J. Gross.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nicolosi AC, Kwok CS, Contney SJ, Olinger GN, Bosnjak ZJ. Gadolinium prevents stretch-mediated contractile dysfunction in isolated papillary muscles. Am J Physiol Heart Circ Physiol. 2001;280:H1122–8. doi: 10.1152/ajpheart.2001.280.3.H1122. [DOI] [PubMed] [Google Scholar]
- 2.Nicolosi AC, West G, Markley JG, Logan B, Olinger GN. Gadolinium attenuates regional stunning in the canine heart in vivo. J Thorac Cardiovasc Surg. 2002;124:57–62. doi: 10.1067/mtc.2002.122524. [DOI] [PubMed] [Google Scholar]
- 3.Nicolosi AC, Kwok CS, Logan B. Effects of gadolinium on regionally stunned myocardium: temporal considerations. J Surg Res. 2007;139:286–91. doi: 10.1016/j.jss.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Nicolosi AC, Kwok CS, Bosnjak ZJ. Antagonists of stretch-activated ion channels restore contractile function in hamster dilated cardiomyopathy. J Heart Lung Transplant. 2004;23:1003–7. doi: 10.1016/j.healun.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Gysembergh A, Margonari H, Loufoua J, Ovize A, Andre-Fouet X, Minaire Y, et al. Stretch-induced protection shares a common mechanism with ischemic preconditioning in rabbit heart. Am J Physiol. 1998;274:H955–64. doi: 10.1152/ajpheart.1998.274.3.H955. [DOI] [PubMed] [Google Scholar]
- 6.Ovize M, Kloner RA, Przyklenk K. Stretch preconditions canine myocardium. Am J Physiol. 1994;266:H137–46. doi: 10.1152/ajpheart.1994.266.1.H137. [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–53. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 8.Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol. 2007;292:H19–27. doi: 10.1152/ajpheart.00712.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y, Hutchins W, Su J, Siker D, Hogg N, Pritchard K, Jr, et al. Delayed cardioprotection with isoflurane: role of reactive oxygen and nitrogen. Am J Physiol. 2005;288:H175–H84. doi: 10.1152/ajpheart.00494.2004. [DOI] [PubMed] [Google Scholar]
- 10.Baker J, Hsu A, Fu X, Kozik D, Tweddell J, Gross G. Darbepoetin alfa protects the rat heart against infarction: dose-response, phase of action and mechanisms. Journal of Cardiovascular Pharmacology. 2007;49:337–45. doi: 10.1097/FJC.0b013e318040cf81. [DOI] [PubMed] [Google Scholar]
- 11.Fryer R, Pratt P, Hsu A, Gross G. Differential activation of extracellular signal regulated kinase isoforms in preconditioning and opioid-induced cardioprotection. J Pharmacol Exp Ther. 2001;296:642–9. [PubMed] [Google Scholar]
- 12.Rajesh KG, Sasaguri S, Zhitian Z, Suzuki R, Asakai R, Maeda H. Second window of ischemic preconditioning regulates mitochondrial permeability transition pore by enhancing Bcl-2 expression. Cardiovasc Res. 2003;59:297–307. doi: 10.1016/s0008-6363(03)00358-4. [DOI] [PubMed] [Google Scholar]
- 13.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–5. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tateyama M, Kubo Y. Dual signaling is differentially activated by different active states of the metabotropic glutamate receptor 1alpha. Proc Natl Acad Sci U S A. 2006;103:1124–8. doi: 10.1073/pnas.0505925103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–53. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 16.Rafiee P, Shi Y, Su J, Pritchard K, Jr, Tweddell JS, Baker JE. Erythropoietin protects the infant heart against ischemia-reperfusion injury by triggering multiple signaling pathways. Basic Res Cardiol. 2005;100:187–97. doi: 10.1007/s00395-004-0508-1. [DOI] [PubMed] [Google Scholar]
- 17.Cole WC, McPherson CD, Sontag D. ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ Res. 1991;69:571–81. doi: 10.1161/01.res.69.3.571. [DOI] [PubMed] [Google Scholar]
- 18.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, DiAlonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Potential mechanism of cardioprotection. Circ Res. 1997;81:1072–82. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 19.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–8. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 20.Babich O, Matveev V, Harris AL, Shirokov R. Ca2+-dependent inactivation of CaV1.2 channels prevents Gd3+ block: does Ca2+ block the pore of inactivated channels? J Gen Physiol. 2007;129:477–83. doi: 10.1085/jgp.200709734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kupittayanant P, Trafford AW, Diaz ME, Eisner DA. A mechanism distinct from the L-type Ca current or Na-Ca exchange contributes to Ca entry in rat ventricular myocytes. Cell Calcium. 2006;39:417–23. doi: 10.1016/j.ceca.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Bode F, Katchman A, Woosley RL, Franz MR. Gadolinium decreases stretch-induced vulnerability to atrial fibrillation. Circulation. 2000;101:2200–5. doi: 10.1161/01.cir.101.18.2200. [DOI] [PubMed] [Google Scholar]
- 23.Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol. 1998;275:C619–21. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- 24.Li GR, Baumgarten CM. Modulation of cardiac Na(+) current by gadolinium, a blocker of stretch-induced arrhythmias. Am J Physiol Heart Circ Physiol. 2001;280:H272–9. doi: 10.1152/ajpheart.2001.280.1.H272. [DOI] [PubMed] [Google Scholar]
- 25.Downing SW, Savage EB, Streicher JS, Bogen DK, Tyson GS, Edmunds LH., Jr The stretched ventricle. Myocardial creep and contractile dysfunction after acute nonischemic ventricular distention. J Thorac Cardiovasc Surg. 1992;104:996–1005. [PubMed] [Google Scholar]
- 26.Hansen DE, Craig CS, Hondeghem LM. Stretch-induced arrhythmias in the isolated canine ventricle. Evidence for the importance of mechanoelectrical feedback. Circulation. 1990;81:1094–105. doi: 10.1161/01.cir.81.3.1094. [DOI] [PubMed] [Google Scholar]
- 27.Edwards CH, 2nd, Rankin JS, McHale PA, Ling D, Anderson RW. Effects of ischemia on left ventricular regional function in the conscious dog. Am J Physiol. 1981;240:H413–20. doi: 10.1152/ajpheart.1981.240.3.H413. [DOI] [PubMed] [Google Scholar]
- 28.Tennant R, Wiggers C. The effect of coronary occlusion on myocardial contraction. Am J Physiol. 1935;112:351–61. [Google Scholar]
- 29.Peart JN, Gross GJ. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res Cardiol. 2005;100:328–36. doi: 10.1007/s00395-005-0526-7. [DOI] [PubMed] [Google Scholar]
- 30.Choi H, Kim SH, Chun YS, Cho YS, Park JW, Kim MS. In vivo hyperoxic preconditioning prevents myocardial infarction by expressing bcl-2. Exp Biol Med (Maywood) 2006;231:463–72. doi: 10.1177/153537020623100412. [DOI] [PubMed] [Google Scholar]