

Abstract
Overexpression of the c-Myc oncoprotein is observed in a large number of hematopoietic malignancies, and transgenic animal models have revealed a potent role for c-Myc in the generation of leukemias and lymphomas. However, the reason for high c-Myc protein levels in most cases is unknown. We examined whether aberrant protein stabilization could be a mechanism of c-Myc overexpression in leukemia cell lines and in primary bone marrow samples from pediatric acute lymphoblastic leukemia (ALL) patients. We found that c-Myc protein half-life was prolonged in the majority of leukemia cell lines and bone marrow samples tested. There were no mutations in the c-myc gene in any of the leukemia cell lines that could account for increased c-Myc stability. However, abnormal phosphorylation at two conserved sites, Threonine 58 and Serine 62, was observed in leukemia cell lines with stabilized c-Myc. Moreover, stabilized c-Myc from the ALL cell lines showed decreased affinity for glycogen synthase kinase3β, the kinase that phosphorylates c-Myc at Threonine 58 and facilitates its degradation. These findings reveal that deregulation of the c-Myc degradation pathway controlled by Serine 62 and Threonine 58 phosphorylation is a novel mechanism for increased expression of a potent oncoprotein known to be involved in hematopoietic malignancies.
Keywords: oncogene, ubiquitylation, phosphorylation, GSK3β, PI(3)K
Introduction
The c-myc proto-oncogene encodes a transcription factor that is critically involved in cell-fate decisions, including proliferation, differentiation and apoptosis.1-5 Myc proteins are widely expressed during embryogenesis, and homozygous deletion of c-myc results in embryonic lethality.6 In general, c-Myc promotes cell growth and proliferation, and reduced c-Myc expression appears to be necessary for terminal differentiation.1-5 Therefore, control of c-Myc expression is vital to maintaining regulated cell proliferation in normal cells.
Deregulated c-Myc expression has been shown to play a significant role in cancer. Overexpression of c-Myc is observed in most human cancers, and transgenic animal models demonstrate c-Myc-induced tumorigenesis in multiple tissues, including leukemia and lymphoma.7-10 In addition, high c-Myc expression in many human hematopoietic malignancies has been correlated with a poor prognosis.11 In Burkitt’s lymphoma, increased c-myc transcription is driven by the t(8;14) translocation, which juxtaposes the immunoglobulin heavy-chain enhancer to the c-myc gene.12 Also, c-myc gene amplification has been reported in some leukemia cell lines.13 However, in most leukemias, the reason for c-Myc overexpression has not been elucidated.
c-Myc expression is tightly regulated in normal cells in response to growth stimulatory signals. Control of c-Myc expression occurs at transcriptional, post-transcriptional, translational and post-translational levels.14-16 c-Myc protein rapidly accumulates when a quiescent cell is stimulated into the cell cycle. This transient increase in c-Myc protein levels, which potentiates S-phase entry, is enhanced by post-translational stabilization of the protein.17 c-Myc levels subsequently diminish to low basal levels with progression through the cell cycle and continued proliferation. The clearance of c-Myc in continuously cycling cells and the maintenance of low levels in quiescent cells are largely owing to rapid ubiquitin-mediated proteolysis.18,19
The regulated stabilization and degradation of c-Myc protein is controlled by ordered phosphorylation at two specific sites near the N terminus of the protein: Serine 62 and Threonine 58.20 Phosphorylation at these two sites occurs sequentially.21 After a growth stimulatory signal, Serine 62 phosphorylation stabilizes c-Myc. Subsequent Threonine 58 phosphorylation promotes ubiquitylation and degradation by the 26S proteasome.22,23 Furthermore, removal of the stabilizing Serine 62 phosphate by protein phosphatase 2A (PP2A) facilitates the polyubiquitylation and degradation of c-Myc.24
Phosphorylation at Serine 62 and Threonine 58 is regulated by two Ras-dependent pathways.17,20 Activation of the first pathway, the Raf/MEK/ERK (extracellular signal-regulated protein kinase) kinase cascade, can enhance c-Myc protein stability by ERK-mediated phosphorylation of Serine 62. The second pathway, involving phosphatidylinositol 3-kinase (PI(3)K) signaling, can also contribute to c-Myc stability by leading to the inhibition of glycogen synthase kinase (GSK)3β, which phosphorylates at Threonine 58. As growth stimulatory signals diminish, GSK3β becomes active and c-Myc is phosphorylated at Threonine 58.25 Threonine 58 phosphorylation enhances PP2A-mediated removal of the Serine 62 phosphate and also allows recognition by the E3 ubiquitin ligase, SCF,Fbw7 which polyubiquitinates c-Myc and targets it for proteasome-mediated degradation.22-24 This complex mechanism of post-translational control is important for regulated proliferation of normal cells.
Some evidence exists that disruption of the c-Myc degradation pathway controlled by Serine 62 and Threonine 58 phosphorylation may occur in cancer. For example, c-Myc that is mutated from Threonine to Alanine at position 58 shows impaired degradation and can facilitate the transformation of primary human cells.20,24 Moreover, this stable c-MycT58A mutant can accelerate lymphomagenesis in animal models.26 Interestingly, point mutations at or adjacent to the c-Myc Threonine 58 phosphorylation site are frequently seen in human Burkitt’s lymphoma.27,28 These mutations prevent phosphorylation at Threonine 58 and result in stabilized c-Myc protein in certain Burkitt’s lymphoma cells.19,29,30 Whether stabilization of c-Myc occurs in other human hematopoietic malignancies has not previously been reported.
In this study, we describe the presence of aberrant stabilization of c-Myc protein in human leukemia cells. We present evidence that overexpression of c-Myc in certain leukemia cell lines and primary patient samples correlates with prolonged c-Myc half-life. The increase in c-Myc half-life occurs in the absence of genetic alterations such as gene amplification or mutations in the c-myc coding sequence. Furthermore, we demonstrate that specific abnormalities in the phosphorylation/dephosphorylation pathway that regulates c-Myc stabilization and degradation occur in some acute lymphoblastic leukemia (ALL) cell lines. These studies have therapeutic implications, as understanding the mechanism leading to aberrant expression of c-Myc could help direct identification of new targeted drugs.
Materials and methods
Cell lines
REH, SupB15, K562, HL-60 and CA46 cells were purchased from the American Type Culture Collection (Rockville, MD, USA). JY cells were a generous gift from Grover Bagby (OHSU). All leukemia cell lines and JY cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin and 2mm l-glutamine at 37°C in 5% CO2. Cells were kept between concentrations of 5 × 105/ml and 2 × 106/ml and were passed at least three times per week.
Primary cells
Consent was obtained from the Institutional Review Board at Oregon Health & Science University to obtain cryopreserved ficolled cells that were originally obtained by bone marrow aspiration at diagnosis from pediatric ALL patients. After thawing, cells were maintained in RPMI 1640 with 20% FBS and 2mm l-glutamine for 16–20 h before performing assays.
Peripheral blood mononuclear cells (PBMCs) were prepared from human blood. For each preparation, 30 ml whole blood was drawn from a healthy volunteer donor into sterile ethylenediaminetetraacetic acid (EDTA) tubes, diluted 1:1 with Hank’s balanced salt solution (BSS), and separated using Histopaque ficoll reagent. After centrifugation at 1400 r.p.m. for 30 min, the white cell layer was extracted. Cells were washed two times with Hank’s BSS. Cells were maintained in RPMI 1640 medium with 20% FBS and 2mm l-glutamine.
Fluorescent in situ hybridization
The leukemia cell lines were grown following standard protocols. Primary patient samples were cultured as above, except phytohemaglutinin was included in the culture medium. At the time of harvest, Colcemid (Sigma, St Louis, MO, USA) was added at a final concentration of 150 ng/ml. After 4–6 h, cells were trypsinized, placed in hypotonic media consisting of 5% FBS and 75mm KCl, and fixed to slides. The slides were baked for 6 min at 90°C in PBS, washed with 2 × SSC at 37°C for 30 min, dehydrated through graded alcohols and then air-dried. Probes for c-myc (fluorescein isothiocyanate) and the centromeric region of chromosome 8 (CEP8) (Aqua) (Abbott Laboratories, Abbott Park, IL, USA) were mixed, denatured at 75°C for 10 min and preannealed at 37°C for 30 min. The probes were then added onto the slides, and hybridization was carried out using HYBrite (Vysis, Downers Grove, IL, USA; 72°C denaturation temperature, 2 min denaturation time and reannealing at 37°C overnight). The next day, the slides were washed with 0.4 × SSC/0.1% NP-40 at 73°C for 2 min, with 2 × SSC/ 0.1% NP-40 (Sigma, St Louis, MO, USA) at room temperature for 1 min, and then counterstained with 125 μg/μl 4’,6’-diamidino-2-phenylindole II (Vysis). Cells were observed using a Nikon E800 fluorescence microscope, and captured using CytoVision software from Applied Imaging (San Jose, CA, USA).
Antibodies and reagents
The c-Myc antibodies, N262 and C33, were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The actin and tubulin antibodies are from Sigma (St Louis, MO, USA). The c-Myc Serine 62 phospho-specific antibody was prepared as described previously.20 The Threonine 58 phospho-specific and GSK3β antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Lactacystin was obtained from EJ Corey Lab, Harvard University, and LY294002 was from purchased from EMD Biosciences (San Diego, CA, USA).
Western blotting and data quantitation
Cells were harvested using Ab lysis buffer (20mm Tris (pH 7.5), 50mm NaCl, 0.5% Nonidet P40, 0.5% sodium deoxycholate, 0.5% SDS, 1mm EDTA, 10mm sodium fluoride, 100mm sodium vanadate, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 0.5 μg/ml leupeptin, 0.2 mg/ml 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF) and 1.6 mg/ml iodoacetamide). Protein concentration for each sample prepared with Ab lysis buffer was determined using BCA solutions kit (Sigma, St Louis, MO, USA). Alternatively, equal cell numbers were lysed in SDS sample buffer and adjusted for equal actin levels. Equal protein for each sample was separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilon-P or Immobilon-FL membrane (Millipore, Billerica, MA, USA). Membranes were blocked using Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) or 5% non-fat milk in PBS with 0.05% Tween-20. Primary antibodies were in Odyssey blocking buffer, except for anti-phospho-Threonine 58, which was in 2.5% non-fat milk. Primary antibodies were detected using secondary antibodies labeled with the near-infrared fluorescent dyes IRDye800 (Rockland, Philadelphia, PA, USA) and Alexa Fluor 680 (Molecular Probes, Eugene, OR, USA) to allow two-color imaging and band overlay. Secondary antibodies were diluted 1:10 000 in Odyssey blocking buffer, and blots were scanned with a LI-COR Odyssey Infrared Imager (Lincoln, NE, USA) to visualize proteins and allow simultaneous anti-rabbit and anti-mouse dual wavelength detection. Band intensities for Western blots were quantitated after background subtraction using LI-COR Odyssey Infrared Imager software version 1.2, which allows for linear protein concentration analysis over four orders of magnitude. Error bars were calculated using Excel (Microsoft, Redmond, WA, USA).
Sequencing the c-myc coding region
Total RNA was isolated from each cell line and subjected to reverse transcription to generate cDNA followed by PCR to amplify the entire c-myc coding region, which was then sequenced.
Pulse-chase experiments
Cells were placed in Dulbecco’s modified Eagle’s medium (DMEM) lacking l-methionine and l-cysteine with 10% dialyzed FBS for 15 min. Cells were then labeled in vivo with 35S-methionine/cysteine (Perkin-Elmer, Boston, MA, USA) in DMEM medium (-l-met/l-cys) with 10% dialyzed FBS for 20–30 min for leukemia cell lines and JY cells, and 2–4 h for primary leukemia bone marrow cells, ficolled normal bone marrow cells and PBMCs owing to their slow metabolic rate. Cells were then washed one time with ‘chase’ medium consisting of RPMI with 10% FBS, 5mm l-methionine and 3mm l-cysteine. Equal numbers of cells were incubated in the chase media for the indicated chase times and then harvested in Ab lysis buffer as described previously.17 Labeled c-Myc was immunoprecipitated from an equal number of cells for each time point using the c-Myc C33 monoclonal antibody-conjugated beads (Santa Cruz Biotechnology). Immunoprecipitated c-Myc was separated by SDS-PAGE, and labeled c-Myc was quantitated using a phosphorimager as described previously.17 After background subtraction, band intensity was plotted on a log scale for each time point using Microsoft Excel graphing function, and c-Myc half-life was determined from the exponential line equation.
Co-immunoprecipitation
Equal cell numbers were lysed in 10 × cell volumes of Co-immunoprecipitation (Co-IP) buffer (20mm Tris, pH 7.5, 12.5% glycerol, 0.2% NP-40, 200mm NaCl, 1mm EDTA, 1mm ethyleneglycol tetraacetate, 1mm dithiothreitol, 10mm sodium fluoride, 100mm sodium vanadate, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 0.5 μg/ml leupeptin, 0.2 mg/ml AEBSF and 1.6 mg/ml iodoacetamide) and pre-cleared with protein A + G agarose (Calbiochem, San Diego, CA, USA). Samples were then incubated with either 1:50 dilution of anti-c-Myc (C33)-conjugated beads (Santa Cruz Biotechnology) or control protein A + G agarose. Precipitated proteins were washed three times in 10 × cell volumes of Co-IP buffer, released by boiling in SDS sample buffer and separated by SDS-PAGE. Gels were transferred and Western blot analysis was performed.
Results
c-Myc is overexpressed in leukemia cell lines without c-myc gene amplification or coding sequence mutations
Overexpression of c-Myc has been reported in most cancers, including various types of leukemia.2,11,31,32 In some cases, c-myc gene amplification has been described. We assessed c-myc gene status and c-Myc protein expression in two precursor B-ALL cell lines (REH and SupB15), one acute myelogenous leukemia (AML) cell line (HL-60) and one chronic myeloid leukemia (CML) cell line (K562). To evaluate the presence of c-myc gene amplification in the four leukemia cell lines, the OHSU Cytogenetics laboratory performed fluorescence in situ hybridization (FISH) using a c-myc probe (Figure 1a). Consistent with previous reports, we found amplification of the c-myc gene, in the form of a large block of c-myc signal-positive material, on a chromosome other than chromosome 8 in the HL-60 cell line (red arrows).13 Conversely, none of the three remaining cell lines had either large blocks of amplified sequence or double minutes detected by FISH with the c-myc probe. Two normal c-myc signals were seen on chromosome 8 in the SupB15 cell line. One extra c-myc signal was seen in the K562 cell line owing to trisomy 8. Two extra copies of c-myc were located on chromosomes other than chromosome 8 without gene amplification in the REH cell line. Therefore, only in the HL-60 cell line did we find evidence of c-myc gene amplification.
Figure 1.
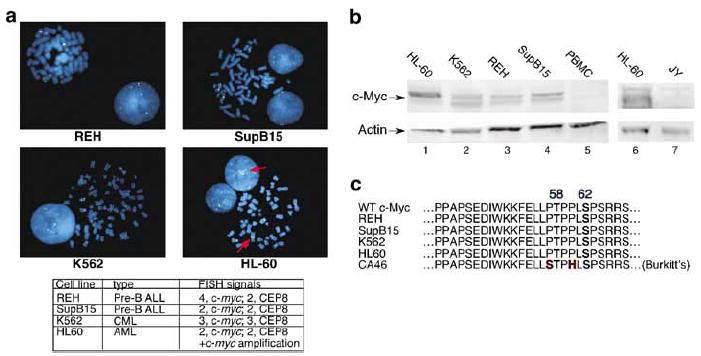
c-Myc overexpression occurs in leukemia cell lines that lack c-myc gene amplification or coding sequence mutations. (a) c-myc gene amplification is seen in HL-60 cells, but not REH, SupB15 or K562 cells. One hundred cells (20 metaphase and 80 interphase) from each cell line were analyzed by FISH using a c-myc probe (blue) and a CEP8 centromere probe as an identifier of chromosome 8 (green). Two c-myc signals were seen in SupB15 cells on chromosome 8. Four c-myc signals were seen in REH cells, two on chromosome 8 and two additional signals on other chromosomes. Three signals for c-myc and three for CEP8 were seen in K562 cells, indicating trisomy 8. In addition to two normal c-myc signals on chromosome 8, c-myc gene amplification, in the form of a block of c-myc-positive signal on another metaphase chromosome and multiple signals in interphase cells (red arrows), was detected in the HL-60 cell line. (b) c-Myc protein levels are elevated in leukemia cell lines. Whole-cell lysates were prepared from HL-60 (AML), K562 (CML), REH (ALL) and Sup-B15 (ALL) leukemia cell lines as well as normal PBMCs and the EBV-transformed B-lymphocyte cell line, JY. Western blot analysis was performed with 25 μg protein from each sample. The blots were probed with the C33 anti-c-Myc (lanes 1–5) and N262 anti-c-Myc (lanes 6 and 7) along with anti-actin antibodies. (c) No c-Myc coding sequence mutations are present in HL-60, K562, REH and SupB15 cells. Total RNA from each cell line was used for reverse transcription with PCR to amplify the entire c-Myc coding region, which was sequenced. The amino acids for the Box I region of c-Myc, a hot spot for mutations in Brukitt’s lymphomas, for each of the four leukemia cell lines is shown along with the CA46 Burkitt’s lymphoma line with known mutations, for comparison.
We next assessed c-Myc protein levels in the four leukemia cell lines by Western blotting. We found elevated levels of c-Myc protein in all four of the leukemia cell lines, with the highest levels in the HL-60 cells (Figure 1b). In contrast, both normal PBMCs from a healthy donor and the Epstein–Barr Virus (EBV)-transformed human B-lymphocyte cell line, JY, showed very low c-Myc protein levels. Our observation of high c-Myc levels in all four leukemia cell lines corroborates previous reports of c-Myc overexpression in leukemia cells.11,31 However, with the exception of HL-60 cells that have amplified c-myc, the reason for elevated c-Myc levels in these leukemia cell lines is unclear.
To examine the mechanism of increased c-Myc expression in the leukemia cell lines without c-myc gene amplification, we initially looked for mutations that could affect c-Myc protein turnover. Mutations in the coding region of c-myc at or adjacent to Threonine 58 result in impaired proteasome-mediated degradation and are frequently found in Burkitt’s lymphoma and AIDS-associated lymphomas.27,33-35 To discover whether mutations exist in the leukemia cell lines under study, we sequenced the entire c-myc coding region in each of the four leukemia lines. No coding sequence mutations were found (Figure 1c). Taken together, these results suggest that the c-Myc overexpression observed in some leukemia cell lines can occur in the absence of c-myc genetic alterations.
c-Myc protein is stabilized in leukemia cell lines without c-myc mutations or gene amplification
Post-translational control of c-Myc protein stability is important for preventing overexpression of the oncoprotein in normal cells.36 To assess whether aberrant stabilization of c-Myc protein could explain high c-Myc expression in the leukemia cell lines under study, we performed 35S-methionine/cysteine pulse-chase analyses to measure directly the half-life of c-Myc in the REH, SupB15, K562 and HL-60 cell lines. For controls, pulse-chase analysis of c-Myc was performed in PBMCs from a healthy donor and in the EBV-transformed B-lymphocyte cell line, JY. As shown in Figure 2a, three of the leukemia cell lines (REH, SupB15 and K562) showed slower turnover of c-Myc than what is seen in the PBMCs or JY cells. Interestingly, HL60 cells showed a similar half-life to the JY cells. Based on multiple independent experiments, the mean half-life (±s.e.) of c-Myc was 62.0±7.4 min in REH cells, 69.7±7.8 min in SupB15 cells and 41.5±4.1 min in the K562 cell line (Figure 2b). In contrast, the half-life of c-Myc was 20.5±0.3 min in the HL-60 cell line, compared to 20.9±3.3 min in JY cells and 12.1±1.9 min in PBMCs. PBMCs do not proliferate in culture without specific cytokine stimulation, and their short half-life of c-Myc is consistent with the reported half-life of c-Myc in quiescent cells.17,20 However, the EBV-transformed human B-lymphocyte cell line, JY, is highly proliferative and its average half-life for c-Myc of 20 min is consistent with previous reports of the half-life of c-Myc in proliferating cells.37,38 In comparison to the other leukemia cell lines, c-Myc turnover in the HL-60 cell line is relatively rapid. We therefore conclude that c-Myc is not significantly stabilized in HL-60 cells. In conclusion, high c-Myc levels can correlate with either gene amplification (HL-60 cell line) or protein stabilization (REH, SupB15 and K562 cell lines).
Figure 2.
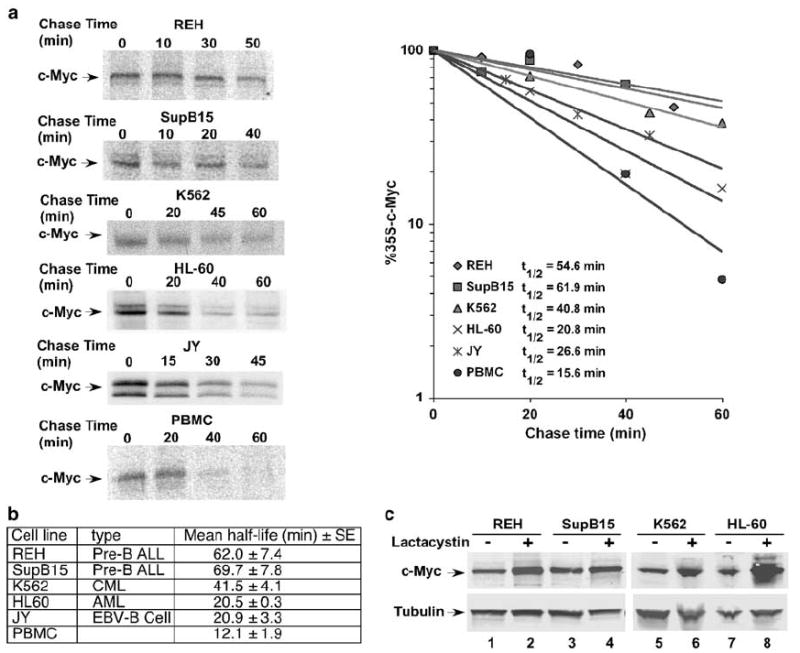
Leukemia cell lines without c-myc mutations or gene amplification have stabilized c-Myc protein. (a) The half-life of c-Myc is prolonged in REH, SupB15 and K562 leukemia cell lines. Leukemia cells, JY cells and PBMCs were pulse-labeled in vivo with 35S-methionine/cysteine and chased in medium with excess unlabeled methionine and cysteine for the indicated times as described in Materials and methods. Endogenous c-Myc was immunoprecipitated from an equal number of cells for each time point and analyzed by gel electrophoresis. 35S-labeled c-Myc from each sample was quantitated by phosphoimager. The rate of degradation of endogenous c-Myc for each leukemia cell line as well as JY and normal PBMCs is represented in the graph by best-fit exponential lines. Half-lives of c-Myc were calculated from exponential line equations and are shown for each cell type. Pulse-chase results shown here are representative of 2–3 independent experiments for each cell line. (b) Table showing mean half-life±s.e. for c-Myc in four leukemia cell lines, EBV-transformed JY cells and PBMCs calculated from 2–3 independent experiments for each cell type. (c) Proteasome inhibition increases c-Myc levels to a greater degree in HL-60 cells than in REH, SupB15 and K562 cells. Equal numbers of cells from each cell line were treated with 10 μm lactacystin for 4 h to inhibit proteasome-mediated degradation. Cells were then harvested and gel electrophoresis and Western blot analysis were performed using equal protein from each sample. Blots were probed with a c-Myc (N262) antibody and anti-tubulin antibody.
Leukemia cell lines with stabilized c-Myc have impaired 26S proteasome-mediated degradation
Normal control of c-Myc protein levels requires ubiquitylation and 26S proteasome-mediated degradation.18,19 Blocking the 26S proteasome in cells with normal turnover of c-Myc results in the accumulation of the protein. In contrast, proteasome inhibition should have significantly less of an effect on c-Myc levels in cells that already have impaired c-Myc degradation. We therefore assessed whether chemical inhibition of the 26S proteasome would have disparate effects on cells with varying levels of c-Myc protein stability.
REH, SupB15, K562 and HL-60 cells were treated for 4 h with lactacystin, a specific inhibitor of the 26S proteasome. As shown in Figure 2c, proteasome inhibition had only a modest effect on c-Myc levels in the three leukemia cell lines (REH, SupB15 and K562) where c-Myc is more stable (lanes 1–6). In contrast, a substantial increase in c-Myc protein was seen with lactacystin treatment of HL-60 cells (Figure 2c, lanes 7 and 8). Thus, while HL-60 cells have increased expression of c-Myc protein owing to c-myc gene amplification, the marked increase in c-Myc upon proteasome inhibition reflects the instability and normal rapid turnover of c-Myc in these cells. These findings suggest that in the three cell lines that show less of an effect with lactacystin, a perturbation exists upstream of 26S proteasome-mediated degradation, which results in prolonged c-Myc half-life.
The levels of Serine 62 and Threonine 58 phosphorylated c-Myc are altered in leukemia cell lines with stable c-Myc protein
Previous experiments have shown that Serine 62 phosphorylation precedes Threonine 58 phosphorylation in the normal c-Myc degradation pathway.20,21 However, polyubiquitinated c-Myc is primarily phosphorylated at Threonine 58, with little Serine 62 phosphorylation.20,24 Therefore, both Threonine 58 phosphorylation and subsequent dephosphorylation at Serine 62 are important for polyubiquitylation and timely degradation of c-Myc.24 We assessed the phosphorylation status of c-Myc at Serine 62 and Threonine 58 in the leukemia cell lines under investigation using phospho-specific antibodies. Double labeling for total and phosphorylated c-Myc allowed us to quantitate the ratio of phospho-Serine 62 c-Myc to total c-Myc and phospho-Threonine 58 c-Myc to total c-Myc (Figure 3, graph). As shown in Figure 3, in HL-60 cells, we detected very little Serine 62-phosphorylated c-Myc, while Threonine 58-phosphorylated c-Myc was clearly evident (lanes 2 and 9, and graph). This finding is consistent with previous experiments examining the phosphorylation status of unstable c-Myc in other cell types, and suggests that c-Myc in HL-60 cells is rapidly processed through the Serine 62 phosphorylation/Threonine 58 phosphorylation/Serine 62 dephosphorylation degradation pathway.20 In contrast, in the leukemia cell lines in which we observe more stable c-Myc protein, we consistently see a substantially higher level of Serine 62-phosphorylated c-Myc, whereas Threonine 58-phosphorylated c-Myc appears on average somewhat lower than that observed in the HL-60 cells (Figure 3, lanes 1,3,4, 6–8, and graph). This increased level of Serine 62-phosphorylated c-Myc and somewhat lower level of Threonine 58-phosphorylated c-Myc, as compared to unstable c-Myc from HL-60 cells, is most evident in the REH and SupB15 ALL cell lines, which had the longest c-Myc half-life (Figure 2b). These results suggest that the c-Myc degradation pathway, controlled by Serine 62 and Threonine 58 phosphorylation, is perturbed in leukemia cell lines with prolonged c-Myc half-life.
Figure 3.
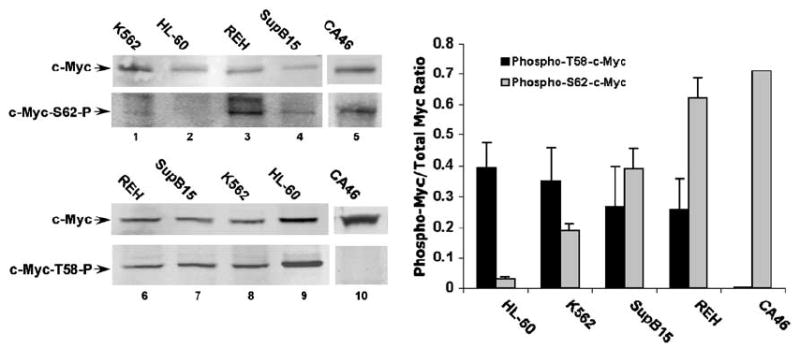
Leukemia cell lines with more stable c-Myc show increased levels of Serine 62 phosphorylation relative to a cell line with unstable c-Myc. Whole-cell lysates were prepared from REH, SupB15, K562 and HL-60 leukemia cell lines along with the CA46 Burkitt’s lymphoma cell line. Extracts were normalized for total c-Myc as measured from previous analyses. Gel electrophoresis was performed and Western blots were probed simultaneously with the C33 pan-c-Myc antibody (mouse) and the phospho-S62-c-Myc antibody (rabbit) (lanes 1–4) or the C33 antibody and phosho-T58-c-Myc antibody (rabbit) (lanes 6–9), using different IRDye secondary antibodies for detection (see Materials and methods). The Western blots shown are representative of three independent experiments. The amount of signal from each antibody was detected and quantitated using an Odyssey Infrared Imager and software from LI-COR Biosciences (see Materials and methods). The bar graph (with s.e.) shows the mean ratio from the three experiments of Theonine 58 phospho-specific signal to total c-Myc signal in black and the mean ratio of Serine 62 phospho-specific signal to total c-Myc signal in gray for the four leukemia cell lines. Phospho-Serine 62 and phospho-Threonine 58 signal in the CA46 Burkitt’s lymphoma with a known proline to serine mutation at position 57 is also shown (lanes 5 and 10, and graph).
We also observed high levels of Serine 62-phosphorylated c-Myc in the absence of Threonine 58 phosphorylation in the CA46 Burkitt’s lymphoma cell line (Figure 3, lanes 5 and 10, and graph), which has a known c-myc mutation at the codon for Proline 57 (Figure 1c). This Proline 57 to Serine mutation has previously been shown to prevent phosphorylation at Threonine 58, and to be associated with impaired c-Myc degradation in the CA46 cell line.29 Moreover, we and others have previously shown that Serine 62 phosphorylation is enhanced in a Threonine 58 to Alanine c-Myc mutant, and that a high degree of Serine 62 phosphorylation correlates with stabilized c-Myc protein.17,20,21,24 Similarly, in leukemia cell lines in this study, detection of high Serine 62 phosphorylation and lower Threonine 58 phosphorylation, relative to HL-60 cells, correlates with increased c-Myc stability. However, as we have shown that these leukemia cell lines do not have c-myc mutations at or near the Threonine 58 phosphorylation site (Figure 1c), the reason for increased Serine 62 phosphorylation is not apparent.
Activation of PI(3)K is important for aberrant stabilization of c-Myc in two ALL cell lines
In order to examine possible mechanisms underlying the altered phosphorylation and increased c-Myc stabilization we observe in some leukemia cell lines, we focused on the two ALL cell lines that showed the most dramatic change in Serine 62 and Threonine 58 phosphorylation: REH and SupB15. We evaluated the kinase pathways that have been reported to regulate the phosphorylation of Serine 62 and Threonine 58. Upon serum stimulation in normal cells, activation of the Ras/Raf/MEK/ERK cascade can phosphorylate Serine 62 and activation of the Ras/ PI(3)K/AKT pathway can inhibit GSK-3β, which would decrease phosphorylation at Threonine 58. Both Ras-activated pathways promote transient accumulation of c-Myc in murine fibroblasts.17 Examination of phosphorylated ERK and AKT in the REH and SupB15 cell lines revealed no increase in ERK or AKT activity that would explain the altered phosphorylation at Serine 62 and Threonine 58 (data not shown). However, both the REH and SupB15 cell lines are reported to have hemizygous deletion of the PTEN gene, which encodes the phosphatase that counteracts PI(3)K activity.39 PTEN protein levels were shown to be severely reduced in the REH and SupB15 cells, which should lead to hyperactivity of PI(3)K, although this was not associated with increased AKT activation,39 consistent with our results.
We examined whether activity of PI(3)K could contribute to increased c-Myc stability in the REH and SupB15 cell lines. We inhibited PI(3)K with the LY294002 compound and assessed affects on c-Myc accumulation in the REH and SupB15 cells, compared to the HL-60 cells, which do not have a PTEN gene deletion. After treating the cell lines with LY294002, we saw a significant decrease in c-Myc levels in the REH and SupB15 cell lines, but not in the HL-60 cell line (Figure 4a, lanes 1 and 2, all panels). This finding suggests that activity of PI(3)K is necessary to maintain high c-Myc expression in REH and SupB15 cells, but not in HL-60 cells. To ascertain whether the decrease in c-Myc levels upon PI(3)K inhibition is owing to an increase in c-Myc turnover, we simultaneously treated REH, SupB15 and HL-60 cells with LY294002 and the proteasome inhibitor, Lactacystin. The drop in c-Myc protein with PI(3)K inhibition was, at least to some degree, abrogated by concomitant proteasome inhibition in the REH and SupB15 cells (Figure 4a, lane 3). This finding shows that the full effect of PI(3)K inhibition on c-Myc levels requires proteasome-mediated degradation of c-Myc, and it suggests that increased activation of the PI(3)K pathway in REH and SupB15 cell lines plays a role in the aberrant stabilization of c-Myc.
Figure 4.
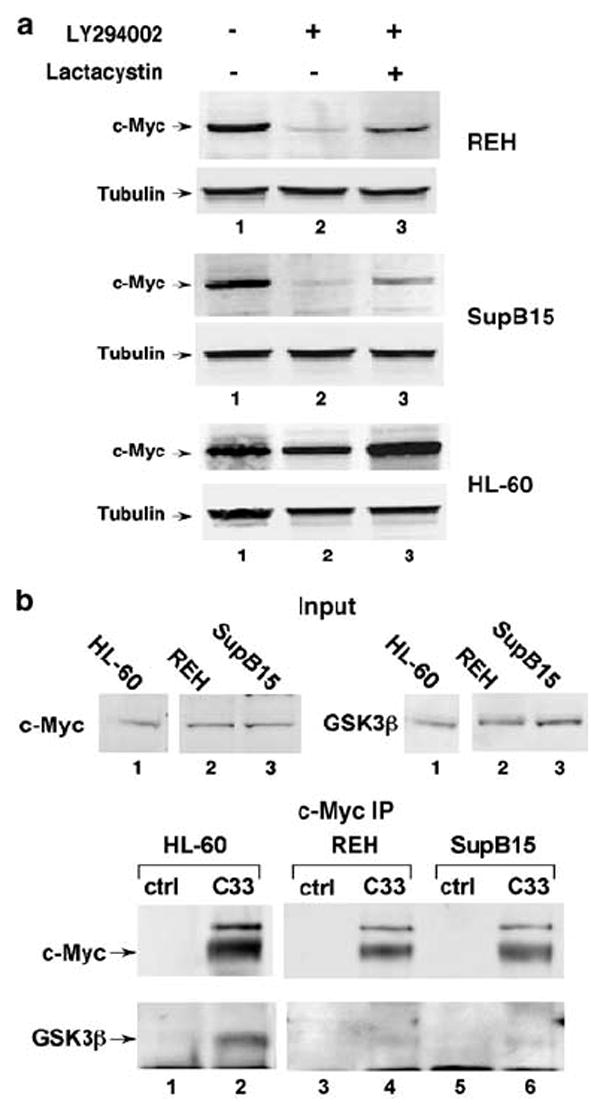
PI(3)K activity and inhibited GSK3β function towards c-Myc correlates with increased c-Myc stability in ALL cell lines. (a) PI(3)K inhibition decreases c-Myc protein levels in ALL cell lines with stabilized c-Myc; partial recovery by simultaneous proteasome inhibition. Leukemia cell lines were treated for 4 h with 20 μm LY 294002 with and without 10 μm lactacystin, as indicated. Cells were then harvested, and protein analyzed by gel electrophoresis and Western blot analysis using anti-c-Myc (N262) and anti-tubulin antibodies. (b) Co-IP shows decreased association between GSK3β and c-Myc in leukemia cell lines with stabilized c-Myc. Endogenous c-Myc and GSK3β levels in whole-cell lysates from each of the indicated cell lines are shown (Input). Immunoprecipitation (IP) was performed from these lysates using C33 (c-Myc) antibody-conjugated beads or control (ctrl) A + G agarose beads. Input and IP Blots were probed with C33 and GSK3β antibodies, as indicated.
Impaired c-Myc degradation correlates with decreased GSK3β interaction with c-Myc
We next assessed whether GSK3β activity toward c-Myc is altered in the REH and SupB15 ALL cells. As recent reports bring into question the role of Serine 9 phosphorylation on GSK3β as a marker of its activity toward targets not involved in insulin signaling,40,41 we chose not to base our assessment of GSK3β function on Serine 9 phosphorylation status. Rather, we directly assessed GSK3β activity toward c-Myc in the leukemia cell lines by examining the ability of GSK3β to associate with c-Myc. We performed c-Myc Co-IP assays from total cell lysates of equal numbers of HL-60, REH and SupB15 cells. As shown in Figure 4b, we observed a stable interaction between GSK3β and c-Myc that could be detected by Co-IP with the c-Myc antibody in the HL-60 cell line, where c-Myc is relatively unstable (lower panel, lane 2). In contrast, very little GSK3β was pulled down with c-Myc in the REH and SupB15 cells where c-Myc is stabilized (lower panel, lanes 4 and 6), even though these ALL cell lines had more GSK3β in their input cell lysates (upper right panel). Although GSK3β can clearly still phosphorylate c-Myc at Threonine 58 in the REH and SupB15 cells, the decreased, or unstable interaction of GSK3β with c-Myc in these cells could lead to the reduction in Threonine 58 phosphorylation that we observe (Figure 3). In addition, as Threonine 58 phosphorylation facilitates PP2A-mediated dephosphorylation of Serine 62,24 this defect in GSK3β function could allow for the observed accumulation of Serine 62-phosphorylated c-Myc in the REH and SupB15 ALL cell lines. Taken together, these results demonstrate that both activation of PI(3)K and decreased interaction with GSK3β contribute to increased c-Myc stability in the ALL cells, although the exact connection between PI(3)K signaling and GSK3β activity toward c-Myc remains unclear.
c-Myc is stabilized in primary ALL cells from pediatric patients
In order to determine whether c-Myc stabilization occurs in primary leukemia cells from patients, we obtained 10 banked bone marrow samples from pediatric patients with precursor B-cell or T-cell ALL and three banked normal bone marrow samples as controls. Sufficient cells were present in six of the ALL samples and three of the normal marrow samples to do pulse-chase analyses. In addition, four of the six ALL samples had enough cells to perform FISH to look for evidence of c-myc genetic changes. None of the previously frozen bone marrow samples proliferated in culture and no metaphase cells were observed in the presence of Colcemid. The cells were also not undergoing apoptosis or dying during our culturing period (data not shown). We measured c-Myc protein half-life in the six pediatric ALL bone marrow samples with sufficient material and the three normal bone marrow samples for comparison. 35S-methionine/cysteine pulse-chase experiments were performed on the ficolled bone marrow samples and the results are shown in Figure 5. We found c-Myc protein to be significantly stabilized in four of the six ALL samples (1 T cell and three pre-B cell; patient no.’s 1–4). The half-life of c-Myc in these samples, which ranged from 45 to 91 min, was significantly longer than in the control normal bone marrow samples, which showed c-Myc half-lives from 17 to 21 min in multiple independent experiments (Figure 5 and data not shown). Leukemia cells from patient no. 6 had a c-Myc half-life similar to that of normal marrow. The half-life of 31 min in cells from patient no. 5 was only slightly longer than what we found in normal marrow. The half-life of c-Myc in normal marrow was similar to that seen in the PBMC and JY control cells.
Figure 5.
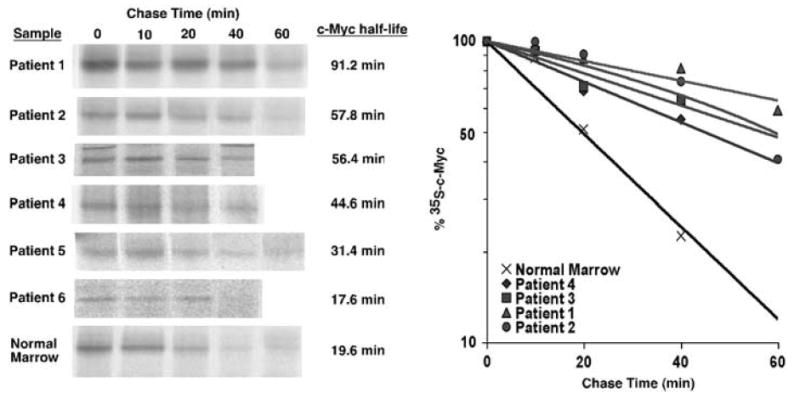
c-Myc protein half-life is prolonged in bone marrow samples from pediatric ALL patients. Stored diagnostic bone marrow samples from pediatric ALL patients were obtained after consent was received from the Oregon Health & Science University Institutional Review Board. Ficolled samples were thawed and maintained in media with 20% FBS for 16–20 h before analysis. Pulse-chase analyses were performed as described for Figure 2 and in Materials and methods. This figure shows results from the six samples in which sufficient c-Myc was detected for quantitation and assessment of half-life. Pulse-chase analyses were also performed on normal bone marrow samples as a control. The result for the ‘normal marrow’ sample shown is representative of three samples tested. c-Myc protein half-life is shown for each sample. The rate of c-Myc degradation over time is depicted in the graph for four samples with prolonged c-Myc half-life and for the representative normal marrow sample.
FISH analysis for c-myc was performed on interphase cells from four of the ALL samples that had sufficient material by the OHSU Cytogenetics Laboratory. As reported in Table 1, the normal two signals for c-myc were observed in leukemia cells from patient no.’s 2, 4 and 5. Patient no. 3 showed 57% of the cells having the normal two signals for c-myc and 39% of cells with three or four signals for c-myc indicating trisomy and tetrasomy for chromosome 8. Therefore, c-myc gene amplification was not apparent in any of these ALL bone marrow samples. FISH results along with patient characteristics including age, gender, immunophenotype of leukemia blasts and treatment outcome are shown in Table 1. These results indicate that aberrant stabilization of c-Myc protein can occur in some cases of pediatric ALL, and that protein stabilization does not correlate with any obvious c-myc genetic changes.
Table 1.
Clinical features of pediatric ALL cases evaluated for c-Myc protein stabilization
| Patient | Age (years) | Gender | Diagnosis | Outcome | c-Myc half-life (min) | FISH for c-myc |
|---|---|---|---|---|---|---|
| 1 | 6 | M | Pre-B ALL | CCR | 91.2 | IS |
| 2 | 3 | F | Pre-B ALL | BM Relapse→death | 57.8 | 2, c-myc; 2 CEP8 |
| 3 | 2 | F | Pre-B ALL | CCR | 56.4 | 2–4, c-myc; 2–4 CEP8a |
| 4 | 16 | M | T-cell ALL | N/A | 44.6 | 2, c-myc; 2 CEP8 |
| 5 | 2 | M | Pre-B ALL | CCR | 31.4 | 2, c-myc; 2 CEP8 |
| 6 | 4 | M | T-cell ALL | CCR | 17.6 | IS |
| Control | N/A | N/A | — | — | 19.6b | IS |
Abbreviations: ALL, acute lymphoblastic leukemia; BM relapse, bone marrow relapse; CCR, continuous complete remission; F, female; FISH, fluorescent in situ hybridization; IS, insufficient sample to perform FISH analysis; M, male; N/A, clinical information not available.
Notes: Age at diagnosis is listed in years. Immunophenotype was determined by flow cytometry at diagnosis for each patient. Measured c-Myc half-life is listed in minutes.
Twenty nine of the 100 cells had four signals for c-myc and four for CEP8, indicating tetrasomy 8, 10/100 cells showed trisomy 8 with three signals for c-myc and three for CEP8, 57/100 showed normal two c-myc, two CEP8 and 4/100 showed one c-myc and two CEP8.
Representative result of three samples tested (c-Myc half-life range: 17–21 min). (FISH) was performed on 100 interphase cells for each sample.
Discussion
Control of c-Myc expression occurs at multiple levels in normal cells. Transcriptional regulation, post-transcriptional stabilization of mRNA and post-translational control of protein stability are all known to play a role in determining c-Myc levels.36 Our previous work has revealed a complex pathway that controls c-Myc protein stabilization and degradation in response to cell proliferative signals and cell cycle entry.20,24 The data presented in this report shows that aberrant stabilization of c-Myc owing to perturbations of this complex degradation pathway occurs in some human leukemia cells.
Our experiments revealed that c-Myc protein is stabilized in a CML and several pre-B ALL cell lines as well as in primary pediatric pre-B- and T-cell ALL bone marrow samples. In contrast, the HL-60 AML cell line overexpressed c-Myc owing to gene amplification and not protein stabilization. It is intriguing that c-Myc degradation could occur at a normal rate in a cell line with c-myc gene amplification, where very high c-Myc protein levels might be expected to overwhelm the degradation machinery. Adaptation to high c-Myc expression could be an explanation. Although the HL-60 AML cell line did not have stabilized c-Myc, it is quite possible that c-Myc is stabilized in other AML cell lines or primary AML cells. Indeed, an initial analysis of the AML cell line, CTV-1, reveals a half-life for c-Myc of 147 min (data not shown). Moreover, three additional cell lines — MOLT-3, CCRF-CEM (T-cell ALLs) and HEL (erythroleukemia) — reveal half-lives for c-Myc ranging from 53 to 58 min (data not shown). While this report focuses on four leukemia cell lines and six pediatric ALL bone marrow samples, our results lay down an important framework that will set the stage for future analyses of c-Myc protein stabilization in a larger study of primary human leukemias and in other human cancers.
Previous reports have revealed that prolonged c-Myc half-life in certain Burkitt’s lymphoma cells is owing to mutations at or near the Threonine 58 phosphorylation site.28-30 We found no such mutations in any of the leukemia cell lines evaluated in this study. Our results in the REH, SupB15 and K562 leukemia cell lines represent the first report that we are aware of showing aberrant c-Myc protein stabilization in human cancer without a coding sequence mutation in the c-myc gene.
Assessment of phosphorylation at Serine 62 and Threonine 58 in leukemia cells with stabilized c-Myc as compared to cells with normal c-Myc degradation helped to delineate a potential mechanism underlying c-Myc stabilization. We know from previous experiments and other reports that Threonine 58 phosphorylation and subsequent Serine 62 dephosphorylation are important for ubiquitylation and proteasome-mediated degradation of c-Myc.20,22-25 However, the leukemia cell lines with stabilized c-Myc showed the inverse pattern of phosphorylation, with markedly higher levels of Serine 62-phosphorylated c-Myc and somewhat reduced levels of Threonine 58 phosphorylation.
In our analysis of the mechanisms contributing to c-Myc stabilization, we demonstrated a role for the activation of the PI(3)K pathway in REH and SupB15 ALL cells. These cell lines have hemizygous deletion of the PTEN gene, encoding the phosphatase that reverses PI(3)K function, which should lead to increased PI(3)K activity.39 Another recent study also described that increased PI(3)K activity, owing to reduced expression of PTEN, correlated with c-Myc overexpression in pancreatic cancer cells.42 In addition, our experiments showed a striking decrease in the association of GSK3β with c-Myc in the REH and SupB15 cells. Based on studies involving insulin signaling, AKT, which is activated by PI(3)K signaling, inhibits GSK3β by phosphorylating it on Serine 9.43 However, in our studies, REH and SupB15 cells did not show increased AKT activity (data not shown), consistent with the report describing hemizygous deletion of PTEN in these cells.39 It is possible that a highly homologous kinase, SGK, which is also activated by PI(3)K and can inactivate GSK3β, is responsible for the impaired GSK3β activity toward c-Myc in the ALL cell lines.44 On the other hand, recent reports have suggested that GSK3β activity on certain targets, such as β-catenin, is not affected by Serine 9 phosphorylation.40,41 Thus, while our data clearly implicate PI(3)K activity and GSK3β inhibition as mechanisms in c-Myc stabilization in the ALL cell lines, the exact link between PI(3)K and GSK3β in these leukemia cells remains to be elucidated.
Mechanistically, decreased association between GSK3β and c-Myc could explain the high Serine 62 phosphorylation and partial reduction in Threonine 58 phosphorylation observed in the ALL cell lines. Previous data shows that without Threonine 58 phosphorylation, PP2A cannot dephosphorylate Serine 62.24 Thus, a decrease in Threonine 58 phosphorylation could both increase Serine 62 phosphorylation levels and decrease SCFFbw7-mediated polyubiquitylation of c-Myc, resulting in the accumulation of stabilized c-Myc that is phosphorylated at Serine 62 in the ALL cell lines.
Along with finding evidence of impaired c-Myc degradation in leukemia cell lines and providing insight into the mechanism behind this impaired degradation, we show that c-Myc stabilization occurs in primary leukemia cells from pediatric patients with ALL. It would be of interest to expand this study in the future to look for the frequency of impaired c-Myc degradation and determine whether any correlation exists with clinical outcome. However, this would require a much larger cohort of leukemia patients. To our knowledge, this is the first report of aberrant c-Myc protein stabilization in primary human leukemia cells.
In summary, our findings reveal a previously unrecognized mechanism for overexpression of c-Myc in some human leukemias that relies on alteration in cellular factors other than c-Myc itself. Additionally, our analysis of the mechanism underlying c-Myc stabilization in certain leukemia cell lines suggests that the Threonine 58/Serine 62 phosphorylation/dephosphorylation pathway that controls c-Myc degradation can become deregulated in human leukemia, and that this pathway may constitute a novel therapeutic target in hematopoietic malignancies that have stabilized c-Myc.
Acknowledgments
We thank Maria Siri for technical assistance and Peter Kurre and Rachel Dresbeck for their generous help with editing the manuscript. We thank Grover Bagby for providing the JY control cell line. This work was supported by grants from the National Cancer Institute, R01 CA100855 and K01 CA086957 to RCS.
References
- 1.Cole MD. The myc oncogene: its role in transformation and differentiation. Annu Rev Genet. 1986;20:361–384. doi: 10.1146/annurev.ge.20.120186.002045. [DOI] [PubMed] [Google Scholar]
- 2.Prochownik EV, Kukowska J. Deregulated expression of c-myc by murine erythroleukaemia cells prevents differentiation. Nature. 1986;322:848–850. doi: 10.1038/322848a0. [DOI] [PubMed] [Google Scholar]
- 3.Spotts GD, Hann SR. Enhanced translation and increased turnover of c-myc proteins occur during differentiation of murine erythroleukemia cells. Mol Cell Biol. 1990;10:3952–3964. doi: 10.1128/mcb.10.8.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen HQ, Selvakumaran M, Liebermann DA, Hoffman B. Blocking c-Myc and Max expression inhibits proliferation and induces differentiation of normal and leukemic myeloid cells. Oncogene. 1995;11:2439–2444. [PubMed] [Google Scholar]
- 5.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Nat Rev Cancer. 2002;2:764–776. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 6.Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 7.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 8.Harris AW, Langdon WY, Alexander WS, Hariharan IK, Rosenbaum H, Vaux D, et al. Transgenic mouse models for hematopoietic tumorigenesis. Curr Top Microbiol Immunol. 1988;141:82–93. doi: 10.1007/978-3-642-74006-0_12. [DOI] [PubMed] [Google Scholar]
- 9.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 10.Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299:887–890. doi: 10.1126/science.1080280. [DOI] [PubMed] [Google Scholar]
- 11.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 12.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalla-Favera R, Wong-Staal F, Gallo RC. Onc gene amplification in promyelocytic leukaemia cell line HL-60 and primary leukaemic cells of the same patient. Nature. 1982;299:61–63. doi: 10.1038/299061a0. [DOI] [PubMed] [Google Scholar]
- 14.Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35(3 Part 2):603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 15.Jones TR, Cole MD. Rapid cytoplasmic turnover of c-myc mRNA: requirement of the 3’ untranslated sequences. Mol Cell Biol. 1987;7:4513–4521. doi: 10.1128/mcb.7.12.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luscher B, Eisenman RN. New light on Myc and Myb. Part I. Myc. Genes Dev. 1990;4:2025–2035. doi: 10.1101/gad.4.12a.2025. [DOI] [PubMed] [Google Scholar]
- 17.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- 18.Flinn EM, Busch CM, Wright AP. myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Mol Cell Biol. 1998;18:5961–5969. doi: 10.1128/mcb.18.10.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol. 1994;14:5510–5522. doi: 10.1128/mcb.14.8.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 25.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 26.Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat Genet. 1993;5:56–61. doi: 10.1038/ng0993-56. [DOI] [PubMed] [Google Scholar]
- 28.Smith-Sorensen B, Hijmans EM, Beijersbergen RL, Bernards R. Functional analysis of Burkitt’s lymphoma mutant c-Myc proteins. J Biol Chem. 1996;271:5513–5518. doi: 10.1074/jbc.271.10.5513. [DOI] [PubMed] [Google Scholar]
- 29.Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–2110. [PubMed] [Google Scholar]
- 30.Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol. 2000;20:2423–2435. doi: 10.1128/mcb.20.7.2423-2435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mata-Greenwood E, Cuendet M, Sher D, Gustin D, Stock W, Pezzuto JM. Brusatol-mediated induction of leukemic cell differentiation and G(1) arrest is associated with down-regulation of c-myc. Leukemia. 2002;16:2275–2284. doi: 10.1038/sj.leu.2402696. [DOI] [PubMed] [Google Scholar]
- 32.Blick M, Westin E, Gutterman J, Wong-Staal F, Gallo R, McCredie K, et al. Oncogene expression in human leukemia. Blood. 1984;64:1234–1239. [PubMed] [Google Scholar]
- 33.Bhatia K, Spangler G, Gaidano G, Hamdy N, Dalla-Favera R, Magrath I. Mutations in the coding region of c-myc occur frequently in acquired immunodeficiency syndrome-associated lymphomas. Blood. 1994;84:883–888. [PubMed] [Google Scholar]
- 34.Clark HM, Yano T, Otsuki T, Jaffe ES, Shibata D, Raffeld M. Mutations in the coding region of c-MYC in AIDS-associated and other aggressive lymphomas. Cancer Res. 1994;54:3383–3386. [PubMed] [Google Scholar]
- 35.Gaidano G, Pasqualucci L, Capello D, Berra E, Deambrogi C, Rossi D, et al. Aberrant somatic hypermutation in multiple subtypes of AIDS-associated non-Hodgkin lymphoma. Blood. 2003;102:1833–1841. doi: 10.1182/blood-2002-11-3606. [DOI] [PubMed] [Google Scholar]
- 36.Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004;3:1133–1137. [PubMed] [Google Scholar]
- 37.Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol. 1984;4:2486–2497. doi: 10.1128/mcb.4.11.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waters CM, Littlewood TD, Hancock DC, Moore JP, Evan GI. c-myc protein expression in untransformed fibroblasts. Oncogene. 1991;6:797–805. [PubMed] [Google Scholar]
- 39.Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, et al. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanism haematological malignancies. Hum Mol Genet. 1999;8:185–193. doi: 10.1093/hmg/8.2.185. [DOI] [PubMed] [Google Scholar]
- 40.Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–32481. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- 41.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–8580. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 43.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 44.Sakoda H, Gotoh Y, Katagiri H, Kurokawa M, Ono H, Onishi Y, et al. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J Biol Chem. 2003;278:25802–25807. doi: 10.1074/jbc.M301127200. [DOI] [PubMed] [Google Scholar]