

Abstract
Protein-tyrosine sulfation is a post-translational modification of an unknown number of secreted and membrane proteins mediated by two known Golgi tyrosylprotein sulfotransferases (TPST-1 and TPST-2). Tpst double knockouts were generated to investigate the importance of tyrosine sulfation in vivo. Double knockouts were born alive at the expected frequency, were normal in size, and their tissues do not synthesize sulfotyrosine. However, most pups die in the early postnatal period with signs of cardiopulmonary insufficiency. A combination of clinical, magnetic resonance imaging, and histological data indicated that lungs of Tpst double knockouts fail to expand at birth resulting in acute pulmonary hypertension, right-to-left shunting, and death by asphyxia in the early postnatal period. Some double knockouts survive the postnatal period, but fail to thrive and display delayed growth that is due in part to hypothyroidism. In addition, we find that Tpst2−/− mice have primary hypothyroidism, but that Tpst1−/− mice are euthyroid. This suggests that a protein(s) required for thyroid hormone production is sulfated and cannot be sulfated in the absence of TPST-2. Thus, Tpst1 and Tpst2 are the only Tpst genes in mice, tyrosine sulfation is required for normal pulmonary function at birth, and TPST-2 is required for normal thyroid gland function.
Keywords: protein-tyrosine sulfation, sulfotyrosine, lung maturity, hypothyroidism
INTRODUCTION
Protein-tyrosine sulfation is a common post-translational modification of secretory and membrane proteins that has been demonstrated to occur in an increasing number of animal and plant species (Huttner and Baeuerle, 1988; Monigatti et al., 2006; Moore, 2003). Protein-tyrosine sulfation involves the catalytic transfer of a sulfuryl group from the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the hydroxyl group of peptidyl-tyrosine to form a tyrosine O4-sulfate ester and 3′, 5′-ADP. Tyrosine sulfation is mediated by two known tyrosylprotein sulfotransferases, TPST-1 and TPST-2, that are localized in the trans Golgi network (Beisswanger et al., 1998; Ouyang et al., 1998; Ouyang and Moore, 1998). The amino acid sequences of human and mouse TPST-1 are ≈ 96% identical. Human and mouse TPST-2 have a similar high degree of identity and are ≈ 67% identical to their TPST-1 counterparts.
There is a growing consensus view that tyrosine sulfation promotes protein-protein interactions in the extracellular space similar to the well-accepted role of tyrosine phosphorylation in the intracellular space. At this time, 3–4 dozen tyrosine-sulfated proteins have been identified in man, many of which play important roles in inflammation, hemostasis, immunity, and other processes. These include certain adhesion molecules, G-protein coupled receptors, coagulation factors, serpins, extracellular matrix proteins, hormones, and others (Kehoe and Bertozzi, 2000; Moore, 2003). A growing number of tyrosine-sulfated proteins have been described in which sulfation is required for optimal protein function (Moore, 2003). For example, tyrosine sulfation of P-selectin glycoprotein ligand-1 and endoglycan is required for binding of selectins, sulfation of platelet gp1bα is required for thrombin binding, factor VIII sulfation is required for VWF binding, and sulfation of factor V, factor VIII, and progastrin are required for optimal proteolytic processing. In addition, tyrosine sulfation of the N-terminal domains of several G-protein coupled receptors has been shown to be required for optimal ligand binding in vitro. However, a functional role for tyrosine sulfation is not known for many proteins that are known to contain sulfotyrosine.
To assess the importance of tyrosine sulfation in vivo our laboratory has developed Tpst1−/−and Tpst2−/− mice using gene targeting (Borghei et al., 2006; Ouyang et al., 2002). Our studies revealed that both Tpst1−/− and Tpst2−/− mice were viable, appear healthy, and have normal lifespans. However, Tpst1−/− and Tpst2−/− mice have distinct phenotypes. Tpst1−/− mice have normal body weights at weaning, but beyond weaning are ≈ 5% lower in average body weight compared to wild type mice. In addition, although fertility of Tpst1−/− males and females per se is normal, Tpst1−/− females have significantly smaller litters due to fetal death between 8.5 and 15.5 days post coitum (Ouyang et al., 2002). In contrast, Tpst2−/− mice are smaller than wild type mice at weaning, have a moderate pubertal growth delay, but appear healthy and attain normal body weight by 10 weeks of age. Furthermore, in contrast to Tpst1−/− males, Tpst2−/− males are infertile. Our analysis of the infertility in Tpst2−/− males shows that these animals are eugonadal and have normal spermatogenesis, but Tpst2−/− sperm are severely defective in their ability to fertilize eggs (Borghei et al., 2006). Furthermore, we have published evidence that certain sperm/epididymal proteins are undersulfated in Tpst2−/− mice (Hoffhines et al., 2006). This genetic and biochemical evidence strongly supports the conclusion that TPST-1 and TPST-2 have differences in macromolecular substrate specificities and demonstrate conclusively that TPST-1 and TPST-2 have distinct biological roles.
To further explore the importance of protein-tyrosine sulfation in vivo, we have generated Tpst double knockout (DKO) mice. In this study, we report biochemical evidence that Tpst1 and Tpst2 are the only Tpst genes in the mouse genome, that protein-tyrosine sulfation is required for normal pulmonary function at birth, and that the absence of TPST-2 causes primary hypothyroidism.
MATERIALS AND METHODS
Tpst Double Knockout Mice
Tpst1 null (Tpst1tm1Klm, MGI:2183366) and Tpst2 null (Tpst2tm1Klm, MGI:3512111) mice were generated and characterized as previously described (Borghei et al., 2006; Ouyang et al., 2002). The generation of Tpst DKO mice was described previously (Hoffhines et al., 2006). Briefly, Tpst1−/− males were mated with Tpst2−/− females to generate obligate in trans compound heterozygotes. Male and female offspring from this cross were mated and their offspring screened by PCR for the presence of the wild type and mutant alleles at both loci as described (Borghei et al., 2006; Ouyang et al., 2002). Male and female Tpst1−/− Tpst2+/− offspring were then interbred to generate Tpst DKO. Cages were inspected each morning to determine the date of birth of litters and to retrieve any dead pups. Genotypes were determined by PCR using DNA prepared from tissues collected from dead pups or a toe sample collected from surviving pups at post-natal day 14 (P14). All experiments reported here were performed on mice in the inbred 129S6/SvEvTac background. Animals were mated, housed, and fed as previously described (Ouyang et al., 2002) and animal procedures complied with all relevant federal guidelines and were approved by the Institutional Animal Care and Use Committee at the Oklahoma Medical Research Foundation.
Sulfotyrosine Analysis
Liver, spleen, skin, and lymph node taken from Tpst DKO and wild type mice at P20 were metabolically labeled in sulfate-free Joklik modified Eagle’s medium containing 2% dialyzed fetal bovine serum and 0.15 mCi/ml carrier-free 35NaSO4. Sulfoamino acid analysis was performed on conditioned media and detergent extracts of the tissue explants as previously described (Borghei et al., 2006).
Cesarean Sections
Timed matings between male and female Tpst1−/− Tpst2+/− or wild type mice were set up and females were checked the next morning for the presence of copulatory plugs. The morning that plugs were detected was considered embryonic day 0.5 (E0.5). At E19.5, pregnant females were euthanized and fetuses were removed from the uterus and placed in 35 mm Petri dishes warmed to 37°C. Pups were gently prodded to stimulate breathing and then continuously observed for 3 h. Observational endpoints included; whether pups were alive at birth, the time after birth when pups became acyanotic, and the time of death. After the 3 h observation period, the pups were euthanized, weighed, tissue samples were collected for genotyping and the pups were fixed in 10% phosphate buffered zinc formalin for histological analysis.
Magnetic Resonance Imaging
In some cases, magnetic resonance imaging (MRI) was performed immediately after Cesarean birth. Pups were introduced into a 7T Bruker 730 USR 30 cm horizontal bore small animal MRI system and body temperature was maintained at 37°C. Transverse images were collected using a multi slice-multi echo standard pulse sequence (TR = 1200 ms, TE = 15 ms, 4 repetitions). Image segments were collected using a 15 mm × 15 mm field of view and a 128 × 128 matrix resulting in an in-plane resolution of 150 μm and an acquisition time of 10 min. Images were analyzed using ImageJ software.
Thyroid Function Testing
Serum total T3, total T4, and thyroid-stimulating hormone (TSH) assays were performed by Anilytics Inc. (Gaithersburg, MD). T3 and T4 assays were performed using Coat-A-Count® RIA kits (Diagnostic Products Corporation, Los Angeles, CA) and TSH assays were performed using a double antibody radioimmunoassay procedure (Schneider et al., 2001). In some cases, total T4 was measured in house as described above.
Histology
All tissues were fixed in 10% phosphate buffered zinc formalin and processed for histology. Light microscope sections were cut at 5 μm, stained with hematoxylin and eosin. Slides were viewed using a Nikon Eclipse E800 microscope and images were captured using a Nikon DXM 1200 digital camera.
RESULTS
Characterization of Tpst Double Knockout Mice
Tpst1−/− Tpst2+/− males and females mated over a 1-year period yielded 5.0 ± 2.0 pups per litter (mean ± SD, n = 82 litters) for a total of 406 offspring. Statistical analysis showed that DKOs were born at the expected Mendelian ratio (X2 = 3.888, p = 0.143). Excluding pups for which genotypes could not be obtained (n = 24) the genotypes of the offspring from these matings were; 28% Tpst1−/− Tpst2+/+, 45% Tpst1−/− Tpst2+/−, and 27% Tpst1−/− Tpst2−/−. However, 63% of the DKO pups were found dead on the morning after birth (P1), 94% were dead by P5 and none survived beyond P31 (Fig. 1). All Tpst DKO pups found dead at P1 were normal in gross appearance and size. At necropsy, a number of the dead P1 Tpst DKO pups had milk in their stomachs, indicating that at least some were able to feed in the early postnatal period.
Figure 1.
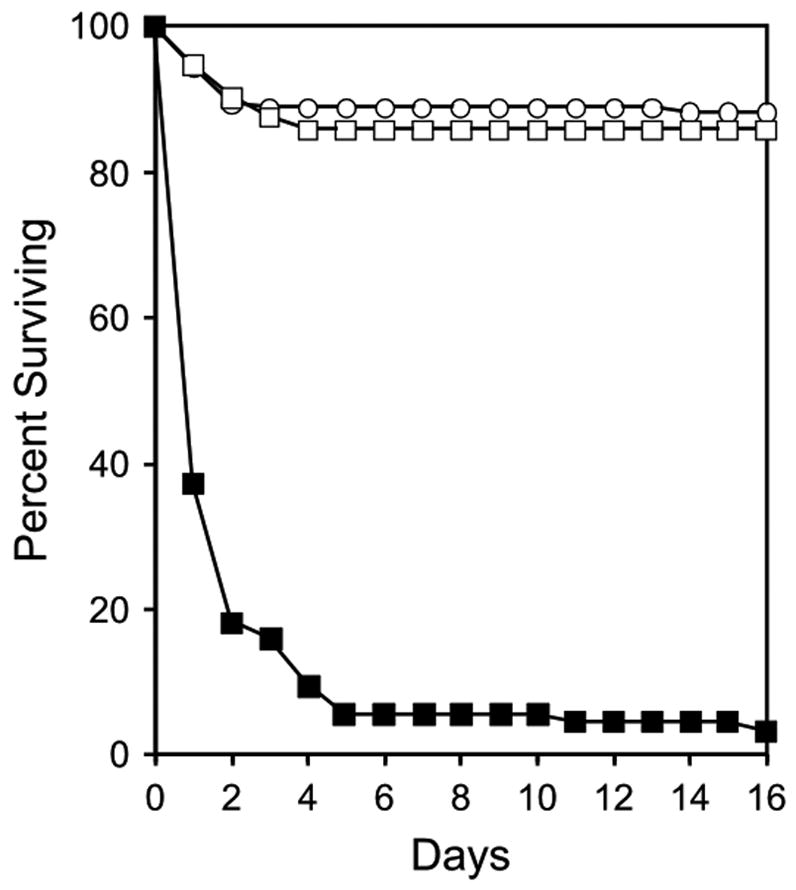
Postnatal survival of Tpst DKO mice. Mating pairs of male and female Tpst1−/− Tpst2+/− mice were set up and cages were checked each morning. All offspring were genotyped as described in Materials and Methods. This dataset includes all litters born over a 1-year period from 20 different mating pairs (5.0 ± 2.0 pups/litter, mean ± SD, n = 82 litters) and consisted of 112 Tpst1−/− Tpst2+/+ mice (open squares), 177 Tpst1−/− Tpst2+/− mice (open circles), and 94 Tpst1−/− Tpst2−/− mice (closed squares).
Tpst DKO pups that survived the postnatal period had delayed in-growth of hair, appeared less active, and were obviously smaller than their littermates. The mean body weight of Tpst DKO survivors sacrificed between P14 and P31 was 6.4 ± 1.4 gm while that of age- and sex-matched wild type mice was 10.0 ± 1.3 gm (mean ± SD, n = 14, p = 2.4 × 10−6, paired two-tailed t test). On several occasions Tpst DKO mice were found dead prior to P14 and intra-abdominal and/or subcutaneous hemorrhage was evident at necropsy. Whether this occurred pre- or postmortem is uncertain. On a couple of other occasions, Tpst DKO mice were found dead and signs of bleeding were evident in the cage the day after toe clipping at P14. Finally, one animal that was sacrificed at P20 was found to have a 1–2 mm intra-cerebral hemorrhage at necropsy. These later findings suggest that Tpst DKO animals have a hemorrhagic diathesis.
Sulfotyrosine Analysis
To confirm that Tpst DKO mice lack the ability to sulfate peptidyltyrosine, tissues were collected from Tpst DKO and age-matched wild type animals. Tissue explants were metabolically labeled with Na35SO4 and secreted and cellular proteins were subjected to alkaline hydrolysis and [35S]sulfoamino acid analysis as described in Materials and Methods. Our analysis showed that [35S]sulfotyrosine was readily detectable in alkaline hydrolysates of proteins from wild type E15.5 fetal liver, but was undetectable in samples from Tpst DKO fetal liver (Fig. 2A). The same result was obtained in analyses of tissues at P20, including skin (Fig. 2B), liver, spleen, and lymph node (data not shown). These data show that Tpst1 and Tpst2 are the only functionally active Tpst genes in the fetal liver at E15.5 and in several mouse tissues at P20. These data support the conclusion that Tpst1 and Tpst2 are the only two Tpst genes in the mouse genome.
Figure 2.
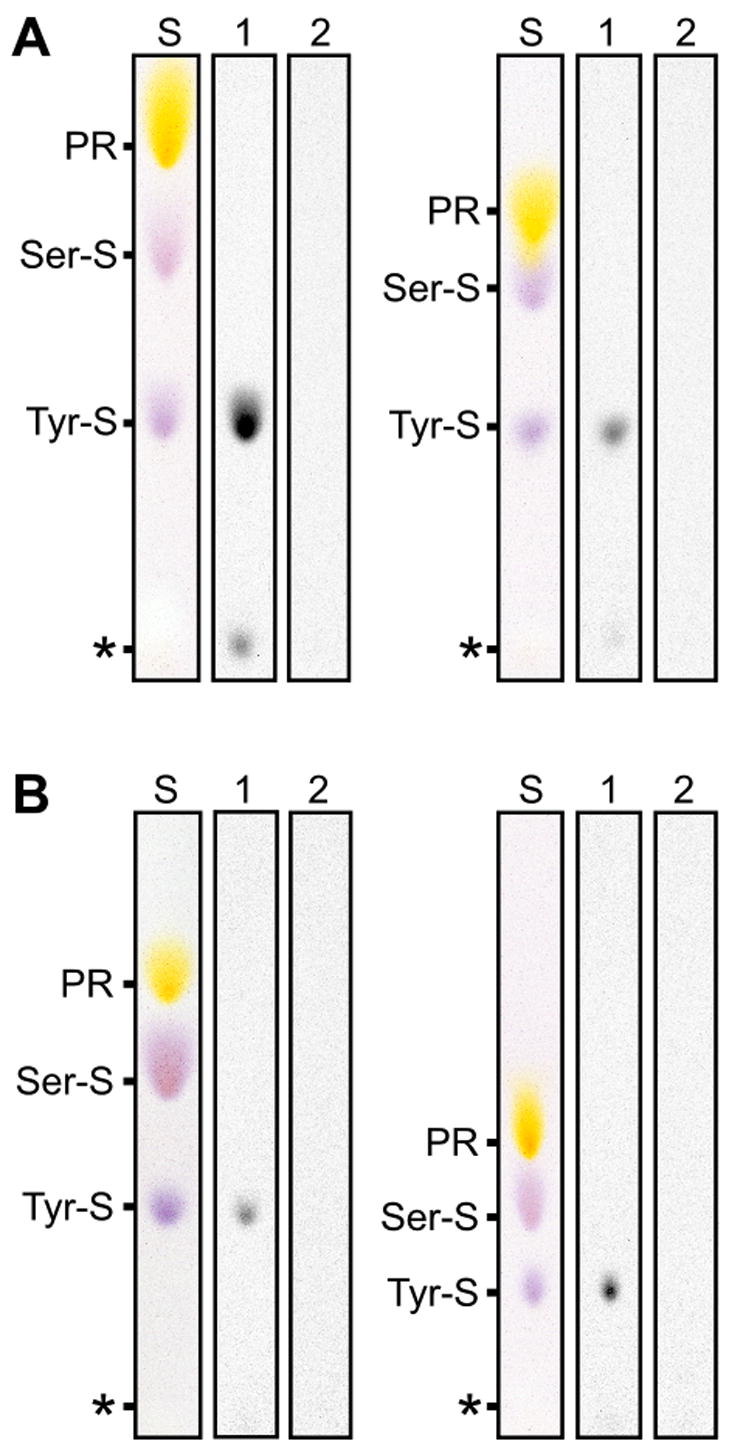
Sulfoamino acid analysis of protein synthesized by Tpst DKO tissues. Tissue explants were metabolically labeled with Na35SO4 and sulfoamino acid analysis was performed as described in Materials and Methods. A. Analysis of secreted proteins (left) and total cellular protein (right) from fetal livers at E15.5. B. Analysis of secreted proteins (left) and total cellular protein (right) from skin at P20. [35S]sulfotyrosine from wild type (lanes 1) and Tpst DKO (lanes 2) were visualized by autoradiography. Internal standards were visualized with ninhydrin (S). The origin is indicated by an asterisk. PR = phenol red.
Cesarean Sections
Given that 63% of the Tpst DKO pups are found dead on P1, we sought to assess if death occurred before or after birth and, if possible, to gain insight into the possible cause(s) of death. Timed matings between male and female Tpst1−/− Tpst2+/− and control wild type animals were performed and pups were delivered by Cesarean section at E19.5 as described in Materials and Methods. All pups from matings between Tpst1−/− Tpst2+/− mice (Tpst1−/−Tpst2+/+, n = 6; Tpst1−/− Tpst2+/−, n = 26; Tpst1−/− Tpst2−/−, n = 11) and control wild type animals (n = 12) were alive at the time of Cesarean delivery. Immediately after birth all pups appeared cyanotic and would gasp for breath upon tactile stimulation. Following delivery, pups were closely observed for a 3-hour period during which the time that each pup became acyanotic, the time at which each had vigorous spontaneous movement, and the time of death of each pup were recorded. Results for these endpoints are shown in Figure 3. As a group, wild type pups became acyanotic at 28.3 ± 14.3 min compared to 101.0 ± 78.2 min for Tpst DKOs (p = 0.012). Likewise, wild type pups had vigorous spontaneous movement by 23.9 ± 6.7 min compared to 85.1 ± 75.5 min for Tpst DKOs (p = 0.023).
Figure 3.
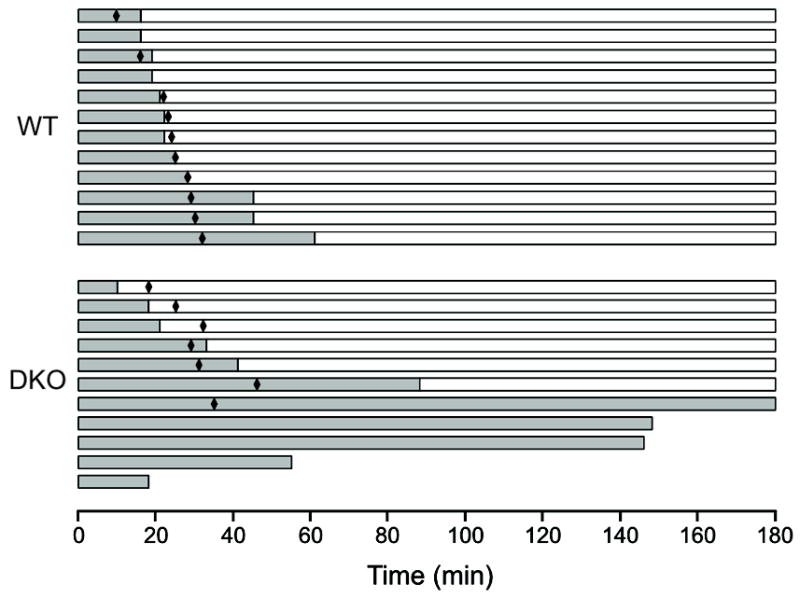
Cesarean sections. Pups were delivered by Cesarean section at E19.5 from timed matings between male and female wild type and Tpst1−/− Tpst2+/− animals and pups were observed for 3 hrs as described in Materials and Methods. Data for wild type (n = 12) and Tpst DKO (n = 11) animals are each represented by a horizontal bar. The change in the color of the bar from gray to white indicates the time at which the pup became acyanotic, the length of each bar indicates the survival time and black diamonds indicate when the pups began vigorous spontaneous movement. All pups were euthanized at 180 min.
Most notably, 4 of 11 (36%) of the Tpst DKO pups died during the 3-hour observation period at 18, 55, 145, and 148 minutes after delivery, while all wild type pups survived. One other Tpst DKO never developed a regular unlabored breathing pattern, never began to have vigorous spontaneous movement, and remained cyanotic over the entire 3-hour observation period. The body weights and crown-to-rump lengths of Tpst DKO pups were indistinguishable from both their littermates and wild type newborns (data not shown). These observations strongly indicate that cardiopulmonary insufficiency is the primary mechanism of death of Tpst DKO newborn mice.
Magnetic Resonance Imaging
To further investigate the cardiopulmonary insufficiency in Tpst DKO mice, pups were examined by MRI shortly after Cesarean delivery as described in Materials and Methods (Fig. 4). All Tpst DKO pups (n = 4) displayed obvious abnormalities in the lungs and heart that were not observed in wild type pups (n = 8). In all Tpst DKO pups, the lungs were poorly aerated and patchy to extensive areas of tissue density with air bronchograms were apparent (Fig. 4D and 4F). In 3 of 4 Tpst DKO pups we also observed marked enlargement of the right atrium (Fig. 4E, 4F, and data not shown). In addition, dependent edema was clearly evident in 2 of 4 Tpst DKO pups (Fig. 4D and 4F) that was not observed in the wild type pups. Despite these abnormalities, the lung volumes of Tpst DKO pups were not different than wild type controls (data not shown). All 8 wild type pups imaged were alive and were fully responsive after the imaging sequence. However, only 1 of 4 Tpst DKO pups survived the imaging sequence.
Figure 4.
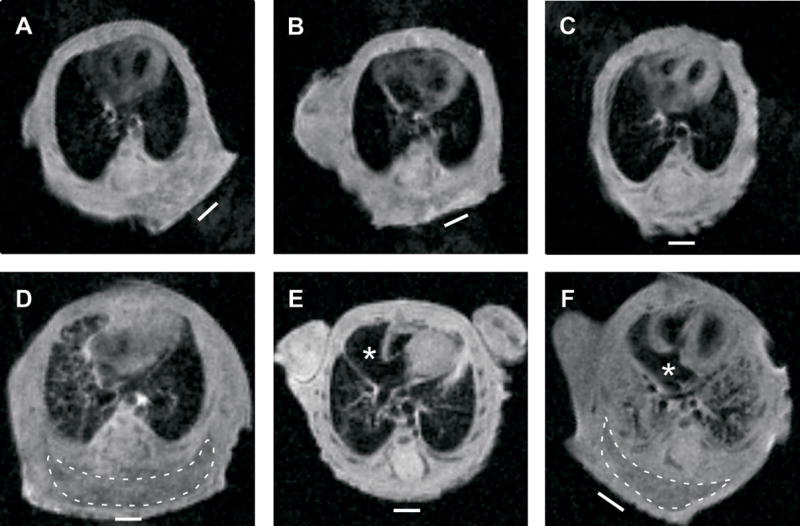
Magnetic resonance imaging. MRI imaging on wild type (n = 8) and Tpst DKO (n = 4) was performed on pups delivered by Cesarean section at E19.5 as described in Materials and Methods. Coronal images at the level of the entry of the inferior vena cava into the right atrium from three wild type (panels A–C) and three Tpst DKO (panels D–F) pups are shown. Images from the fourth Tpst DKO pup are not shown but look nearly identical to the image in panel E. The right atria are marked by an asterisk and areas of dependent edema are surrounded by a dashed line. The white bar indicates the horizontal plane during the imaging sequence.
Histology
To further assess the anatomical abnormalities visualized by MRI, Tpst DKO pups were subjected to a detailed histological examination after imaging. This analysis was also conducted on Tpst DKO animals that survived to P21. Brain, eye, thymus, diaphragm, liver, stomach, spleen, pancreas, kidney, small and large intestine, rectum, skeletal muscle, and skin from Tpst DKOs at E19.5 and P21 were histologically indistinguishable from age-matched wild type controls. However, distinct abnormalities were discernable in heart, lungs, and thyroid gland.
Histological examination of the heart and lungs confirmed that marked enlargement of the right atrium and vena cava was present in the same three Tpst DKO pups examined by MRI (Fig. 5B, 5C, and not shown). In addition, at necropsy a markedly dilated vena cava was apparent in three Tpst DKO mice sacrificed at P21 (data not shown). In contrast to wild type controls (Fig. 5D), the lungs of Tpst DKOs at E19.5 showed moderate to severe abnormalities. In the less severe cases, alveoli were poorly inflated with thickened and highly cellular alveolar septa (Fig. 5E). In the worst case, it appeared that no lung expansion had occurred and as a result well-defined alveolar structures were absent (Fig. 5F). We also examined Tpst DKO embyros at E17.5 and E18.5. However, the lung and heart of Tpst DKO were histologically indistinguishable from wild type embryos (data not shown).
Figure 5.
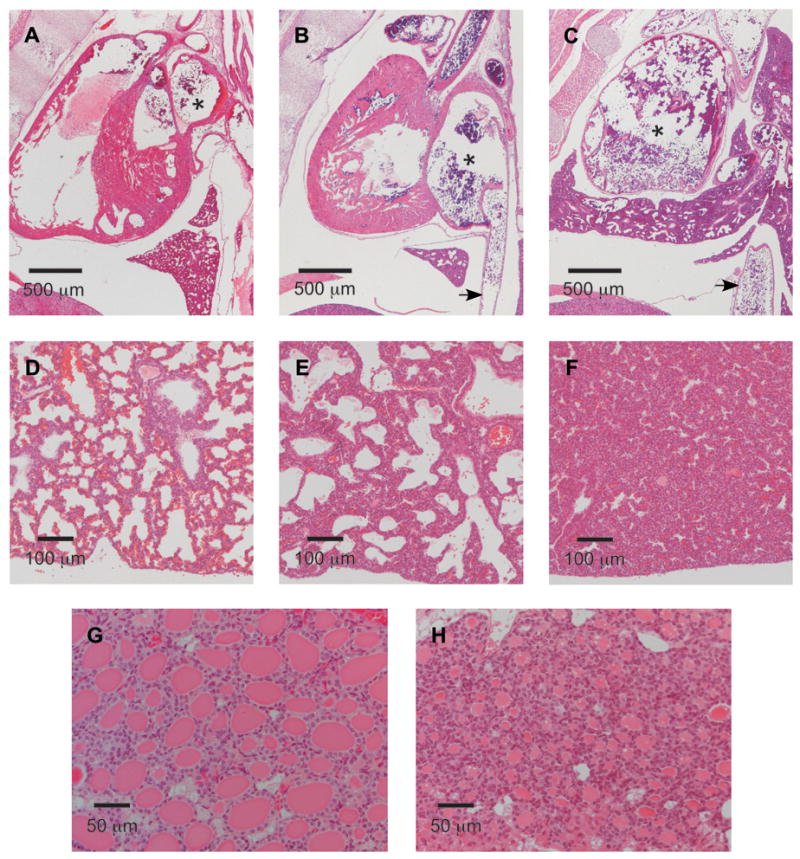
Histology. Hematoxylin and eosin staining of paraffin-embedded tissues from Tpst DKO and wild type mice. A. Midline sagittal section of a wild type pup at E19.5. B. Midline sagittal section of a Tpst DKO pup at E19.5. C. Right para-sagittal section of a second Tpst DKO pup at E19.5. The right atrium is indicated by an asterisk and the inferior vena cava is indicated by an arrow. D. Section of lung at E19.5 from the wild type pup shown in Fig. 4A. E. Section of lung at E19.5 from the same Tpst DKO pup shown in Fig. 4D. F. Section of lung at E19.5 from the Tpst DKO pup shown in Fig. 4F. G. Wild type thyroid gland at P21. H. Tpst DKO thyroid gland at P21.
The thyroid glands of 3 Tpst DKO mice at E19.5 and 4 Tpst DKO mice at P21 were normally positioned, of similar size compared to wild type mice, and overall follicular architecture was normal. However, in contrast to wild type glands, follicles were devoid of colloid at E19.5 (data not shown) and contained significantly less colloid at P21 (Fig. 5G and 5H). These findings suggested that Tpst DKO mice might be hypothyroid.
Thyroid Function Testing
To assess thyroid status of Tpst DKO mice, total T4 was determined in serum from three surviving Tpst DKO mice at P21. In these mice, total T4 was <0.57 μg/dl, significantly below that of wild type controls (see Fig. 6). Unfortunately, the amount of serum was insufficient to measure TSH levels. Nevertheless, this data coupled with the abnormal thyroid histology observed in the Tpst DKO mice prompted us to examine the thyroid function of Tpst1−/− and Tpst2−/− mice. Serum total T3, total T4, and TSH levels were performed on a group of Tpst1−/−, Tpst2−/−, and age- and sex-matched wild type controls. T3, T4, and TSH levels in wild type and Tpst1−/− mice were indistinguishable. In contrast, T3 and T4 levels were significantly reduced, whereas TSH levels were elevated in Tpst2−/−mice compared to both wild type and Tpst1−/− mice (Fig. 6).
Figure 6.
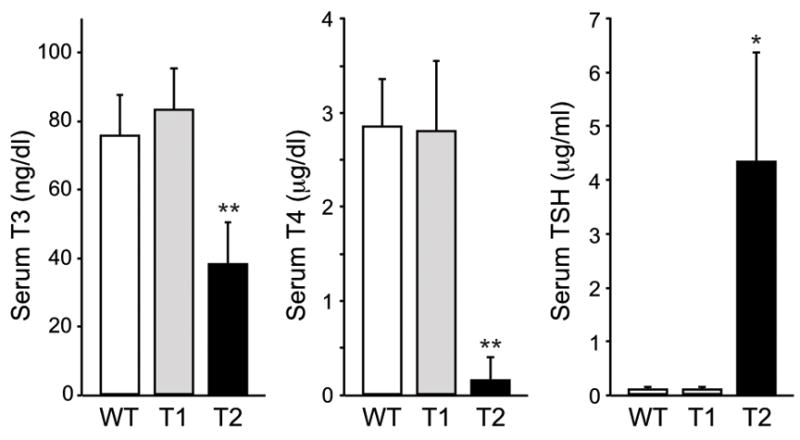
Thyroid function tests. Total T3, total T4, and TSH assays were performed on sera collected from adult males at between 12 and 17 weeks of age as described in Materials and Methods. Results are expressed as mean ± SD of wild type (n = 12), Tpst1−/− (n = 5), and Tpst2−/− (n = 5) samples. Statistical differences between groups were tested using a two-tailed Student’s t test with unequal sample variance. T3, T4, and TSH levels in wild type and Tpst1−/− mice are not statistically different. T3, T4, and TSH levels in Tpst2−/− mice are statistically different than wild type and Tpst1−/− mice. * p < 0.01, ** p < 0.001.
This finding prompted us to perform a morphometric analysis of thyroid follicles in Tpst2−/−mice. The internal diameters of 30 follicles were measured in 3 Tpst2−/− and 3 wild type mice. We found that the internal diameter of follicles in Tpst2−/− mice (132 ± 10 μm) was significantly smaller than in wild type controls (190 ± 17 μm) (Mean ± SD, n = 3, p = 0.007, 2-tailed t test). These data demonstrate that Tpst2−/− mice, and a priori Tpst DKO mice, have primary hypothyroidism.
DISCUSSION
Protein-tyrosine sulfation is a post-translational modification of an unknown number of secreted and membrane proteins that is mediated by two known Golgi tyrosylprotein sulfotransferases (TPST-1 and TPST-2). To date only two closely related Tpst genes have been identified in the human and mouse genome. However, it is possible that other more distantly related Tpst genes might exist. Therefore, we first sought to determine if Tpst DKO cells retain some ability to synthesize sulfotyrosine. Our analysis failed to detect [35S]sulfotyrosine in alkaline hydrolysates of proteins synthesized in several tissues from Tpst DKO mice at E15.5 and P20. Thus, TPST-1 and TPST-2 appear to be the only TPSTs expressed in the mouse at E15.5 and P20. This strongly suggests that Tpst1 and Tpst2 are the only Tpst genes in the mouse genome.
Tpst DKO mice are born alive at the expected Mendelian ratio, but combined deficiency of TPST-1 and TPST-2 results in severely impaired postnatal viability. To gain insight into potential causes of this early postnatal mortality, we performed a detailed clinical and pathological assessment of Tpst DKO pups born by Cesarean delivery at E19.5. Our observations show that although all Tpst DKO pups are born alive, about a third die within 3 hrs of Cesarean delivery and only 27% survive to P1 after natural birthing. Our clinical observations suggested cardiopulmonary insufficiency as the likely mechanism of death of the Tpst DKO pups. First, Tpst DKO pups displayed signs of pulmonary insufficiency at birth (i.e. prolonged cyanosis and irregular breathing pattern). Second, MRI imaging showed that Tpst DKO mice have obvious heart and lung abnormalities, including pulmonary infiltrates, marked dilatation of the right atrium and vena cava, and dependent edema, findings that were confirmed by histological analyses. However, we observed no discernable histological abnormalities in the heart or lungs prior to birth.
At birth a transition from the fetal to neonatal pattern of circulation must occur rapidly to ensure efficient O2/CO2 exchange and survival. This transition is triggered by expansion of the lungs and is associated with a rapid drop in pulmonary vascular resistance, a marked increase in pulmonary blood flow, and closure of the ductus arteriosus and foramen ovale. Taken together, our observations indicate that this transition is severely impaired in Tpst DKO pups and are consistent with the clinical scenario that at birth the lungs of Tpst DKO mice fail to expand resulting in acute pulmonary hypertension, right-to-left shunting, and death by asphyxia in the early postnatal period.
The clinical phenotype that we observe in Tpst DKO mice is reminiscent of that observed in mice with targeted disruption of a number of proteins involved in basement membrane assembly which are associated with various defects in lung development. For example, combined deficiency of nidogen-1 and -2 results in postnatal fatality associated with delayed saccular and alveolar maturation of the E17.5–E18.5 lung and impaired heart morphogenesis (Bader et al., 2005). Likewise, ablation of the nidogen binding site in the laminin γ1 chain results in impaired lung development and early postnatal death as well as renal agenesis (Willem et al., 2002). In addition, deficiency of the N-deacetylase/N-sulfotransferase-1, an enzyme required for heparan sulfate modification, results in delayed lung development leading to severe respiratory difficulties and death at birth (Fan et al., 2000; Ringvall et al., 2000). Although we observe no histological abnormalities in the lungs of Tpst DKO mice prior to birth, more detailed analyses is required to rule out defects in late lung development and maturation in Tpst DKO mice. Nevertheless, our data clearly demonstrate that protein-tyrosine sulfation of as of yet unknown protein(s) plays a significant role in normal lung function at birth. It is interesting to note that nidogen-1 has been shown to be tyrosine-sulfated in the mouse, most likely at Tyr290 and Tyr295 located in the G1 domain that binds fibulin-2 (Aratani and Kitagawa, 1988; Mann et al., 1989; Paulsson et al., 1985; Ries et al., 2001). However, it is not known if tyrosine sulfation of nidogen-1 is important for interactions with fibulin-2 or any other of its many binding partners.
Another interesting and unexpected finding in our study was abnormal thyroid gland histology that suggested Tpst2−/− and Tpst DKO mice might be hypothyroid. Although detailed thyroid function testing on Tpst DKO mice was not possible due to their scarcity and limited viability, we show that total T4 is very low in Tpst DKO mice. This finding prompted us to perform thyroid function testing on Tpst1−/− and Tpst2−/− mice that clearly showed the Tpst2−/−mice have moderate to severe primary hypothyroidism while Tpst1−/− mice are euthyroid.
In the mouse and rat, thyroid hormone affects lung epithelial and mesenchymal differentiation, lung branching morphogenesis, and postnatal alveolar maturation (Archavachotikul et al., 2002; Massaro et al., 1986; Volpe et al., 2003). However, Tpst2−/− mice have normal post-natal viability (Borghei et al., 2006) and their lungs are histological normal at P1 and beyond (data not shown) despite the presence of severe hypothyroidism. This would argue that hypothyriodism per se is unlikely to be a significant contributing factor in the pulmonary insufficiency and post-natal death of Tpst DKO mice.
As to possible mechanism(s) for the hypothyroidism, two proteins involved in thyroid hormone synthesis are known to be tyrosine-sulfated, the thyroid-simulating hormone receptor (TSH-R) (Bonomi et al., 2006; Costagliola et al., 2002) and thyroglobulin (Tg) (Herzog, 1986). TSH-R is a member of a subfamily of the G protein-coupled receptor superfamily that also includes the luteinizing hormone and follicle-stimulating hormone receptors. These receptors have large (359–414 residues) N-terminal extracellular domains followed by a rhodospin-like transmembrane domain. The extracellular domains of TSH-R, LH-R, and FSH-R are composed of two cysteine-rich clusters flanking a series of leucine-rich repeats and are responsible for high affinity binding of TSH, LH, or FSH, respectively. Previous studies demonstrated that the human TSH-R is tyrosine-sulfated in the extracellular domain downstream of the second cysteine-rich cluster and 27-residues upstream from the first transmembrane helix (Costagliola et al., 2002). Sulfation occurs at two sites within a YDY motif that is conserved in mouse TSH-R as well as many other species. Sulfation of the first tyrosine (Y385) within the YDY motif is required for high affinity binding of TSH and receptor activation (Costagliola et al., 2002).
Sasaki et al recently reported that a mutation in the Tpst2 gene is responsible for an autosomal recessive form of primary hypothyroidism in the growth-retarded grt/grt mouse (Sasaki et al., 2007). The mutation they identified (C798G) results in a H266Q substitution in the catalytic domain of TPST-2. They showed that TSH-R signaling was restored by over expression of wild type, but not mutant TPST-2, in grt/grt fibroblasts expressing the TSH-R. They further showed that recombinant wild type TPST-2, but not wild type TPST-1 or the H266Q TPST-2 mutant, could sulfate a peptide modeled on the tyrosine sulfation site in the TSH-R in vitro, suggesting that TPST-2, but not TPST-1, can efficiently sulfate TSH-R. These data are consistent with our observations that Tpst2−/− mice are hypothyroid while Tpst1−/− mice are euthyroid. Thus, undersulfation of TSH-R may be one mechanism for the primary hypothyroidism we observe in Tpst2−/− and Tpst DKO mice. However, it remains to be determined if the native TSH-R is undersulfated in Tpst2 mice in vivo.
Thyroglobulin is the most abundant protein in the thyroid gland and the scaffold for thyroid hormone biosynthesis and is tyrosine-sulfated in multiple species, including mice (Baumeister and Herzog, 1988; Herzog, 1986). Studies in porcine thyrocytes indicate that the stoichiometry of Tg tyrosine sulfation falls in response to TSH stimulation (Nlend et al., 1999b), that in vitro production of thyroid hormone from Tg is decreased when sulfation is inhibited pharmacologically (Nlend et al., 1999a), and that Tyr5, a hormonogenic tyrosine residue in porcine Tg, can be tyrosine-sulfated. However, the precise role of Tg tyrosine sulfation in thyroid hormone synthesis remains unclear. Mutations in the thyroglobulin gene in both man and mice (i.e., tgcog/tgcog mice) result in severe congenital hypothyroidism with colloid-deficient goiter (Beamer et al., 1987; Kim et al., 1998). The growth phenotype of tgcog/tgcog mice also resembles that observed in Tpst2−/− mice, but Tpst2−/− and Tpst DKO mice do not have congenital goiter. Thus, although the hypothyroid phenotype in Tpst2−/− mice is more consistent with a dysfunctional TSH-R, studies are required to determine if sulfation of Tg is affected in Tpst2−/− mice.
In summary, our studies of Tpst DKO mice provide novel insight into the biological role of protein-tyrosine sulfation. Our findings provide the first evidence for an important role for protein-tyrosine sulfation in normal lung function at birth and firmly support recent observations for a role of tyrosine sulfation in TSH-R function in vitro and in vivo (Bonomi et al., 2006; Costagliola et al., 2002; Sasaki et al., 2007). Finally, our data support the conclusion that Tpst1 and Tpst2 are the only two Tpst genes in the mouse genome.
Acknowledgments
This work was supported in part by NIH Grant HL074015 and Grant HR07-068 from the Oklahoma Center for the Advancement of Science and Technology (K.L.M.). The MRI imaging work was supported in part by Grant fMRI-002 from the Oklahoma Center for the Advancement of Science and Technology.
We thank Dr. Rheal Towner, Preeti Kshirsagar, Yasvir Tesiram, Andrew Abbott, and Rebecca Cranford for help with MRI imaging and Martin Lansdale for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aratani Y, Kitagawa Y. Tyrosine sulfation is not the last modification of entactin before its secretion from 3T3-L1 adipocytes. FEBS Lett. 1988;235:129–32. doi: 10.1016/0014-5793(88)81247-x. [DOI] [PubMed] [Google Scholar]
- Archavachotikul K, et al. Thyroid hormone affects embryonic mouse lung branching morphogenesis and cellular differentiation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L359–69. doi: 10.1152/ajplung.00400.2000. [DOI] [PubMed] [Google Scholar]
- Bader BL, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. 2005;25:6846–56. doi: 10.1128/MCB.25.15.6846-6856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister FA, Herzog V. Sulfation of thyroglobulin: a ubiquitous modification in vertebrates. Cell Tissue Res. 1988;252:349–58. doi: 10.1007/BF00214377. [DOI] [PubMed] [Google Scholar]
- Beamer WG, et al. Inherited congenital goiter in mice. Endocrinology. 1987;120:838–40. doi: 10.1210/endo-120-2-838. [DOI] [PubMed] [Google Scholar]
- Beisswanger R, et al. Existence of distinct tyrosylprotein sulfotransferase genes: Molecular characterization of tyrosylprotein sulfotransferase-2. Proc Natl Acad Sci U S A. 1998;95:11134–9. doi: 10.1073/pnas.95.19.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi M, et al. Structural differences in the hinge region of the glycoprotein hormone receptors: evidence from the sulfated tyrosine residues. Mol Endocrinol. 2006;20:3351–63. doi: 10.1210/me.2005-0521. [DOI] [PubMed] [Google Scholar]
- Borghei A, et al. Targeted disruption of tyrosylprotein sulfotransferase-2, an enzyme that catalyzes post-translational protein tyrosine O-sulfation, causes male infertility. J Biol Chem. 2006;281:9423–31. doi: 10.1074/jbc.M513768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costagliola S, et al. Tyrosine sulfation is required for agonist recognition by glycoprotein hormone receptors. EMBO J. 2002;21:504–13. doi: 10.1093/emboj/21.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, et al. Targeted disruption of NDST-1 gene leads to pulmonary hypoplasia and neonatal respiratory distress in mice. FEBS Lett. 2000;467:7–11. doi: 10.1016/s0014-5793(00)01111-x. [DOI] [PubMed] [Google Scholar]
- Herzog V. Secretion of sulfated thyroglobulin. Eur J Cell Biol. 1986;39:399–409. [PubMed] [Google Scholar]
- Hoffhines AJ, et al. Detection and purification of tyrosine-sulfated proteins using a novel anti-sulfotyrosine monoclonal antibody. J Biol Chem. 2006;281:37877–87. doi: 10.1074/jbc.M609398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Baeuerle PA. Protein sulfation on tyrosine. Mod Cell Biol. 1988;6:97–140. [Google Scholar]
- Kehoe JW, Bertozzi CR. Tyrosine sulfation: a modulator of extracellular protein-protein interactions. Chem Biol. 2000;7:R57–61. doi: 10.1016/s1074-5521(00)00093-4. [DOI] [PubMed] [Google Scholar]
- Kim PS, et al. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc Natl Acad Sci U S A. 1998;95:9909–13. doi: 10.1073/pnas.95.17.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K, et al. Amino acid sequence of mouse nidogen, a multidomain basement membrane protein with binding activity for laminin, collagen IV and cells. Embo J. 1989;8:65–72. doi: 10.1002/j.1460-2075.1989.tb03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaro D, et al. Postnatal development of pulmonary alveoli: modulation in rats by thyroid hormones. Am J Physiol. 1986;250:R51–5. doi: 10.1152/ajpregu.1986.250.1.R51. [DOI] [PubMed] [Google Scholar]
- Monigatti F, et al. Protein sulfation analysis-A primer. Biochim Biophys Acta. 2006;1764:1904–1913. doi: 10.1016/j.bbapap.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Moore KL. The biology and enzymology of protein tyrosine O-sulfation. J Biol Chem. 2003;278:24243–6. doi: 10.1074/jbc.R300008200. [DOI] [PubMed] [Google Scholar]
- Nlend MC, et al. Sulfated tyrosines of thyroglobulin are involved in thyroid hormone synthesis. Biochem Biophys Res Commun. 1999a;262:193–7. doi: 10.1006/bbrc.1999.1173. [DOI] [PubMed] [Google Scholar]
- Nlend MC, et al. Thyrotropin regulates tyrosine sulfation of thyroglobulin. Eur J Endocrinol. 1999b;141:61–9. doi: 10.1530/eje.0.1410061. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, et al. Reduced body weight and increased postimplantation fetal death in tyrosylprotein sulfotransferase-1-deficient mice. J Biol Chem. 2002;277:23781–7. doi: 10.1074/jbc.M202420200. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, et al. Tyrosylprotein sulfotransferase: Purification and molecular cloning of an enzyme that catalyzes tyrosine O–sulfation, a common posttranslational modification of eukaryotic proteins. Proc Natl Acad Sci U S A. 1998;95:2896–901. doi: 10.1073/pnas.95.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Moore KL. Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in Caenorhabditis elegans. J Biol Chem. 1998;273:24770–4. doi: 10.1074/jbc.273.38.24770. [DOI] [PubMed] [Google Scholar]
- Paulsson M, et al. Nature of sulphated macromolecules in mouse Reichert’s membrane. Evidence for tyrosine O-sulphate in basement-membrane proteins. Biochem J. 1985;231:571–9. doi: 10.1042/bj2310571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries A, et al. Recombinant domains of mouse nidogen-1 and their binding to basement membrane proteins and monoclonal antibodies. Eur J Biochem. 2001;268:5119–28. doi: 10.1046/j.0014-2956.2001.02437.x. [DOI] [PubMed] [Google Scholar]
- Ringvall M, et al. Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J Biol Chem. 2000;275:25926–30. doi: 10.1074/jbc.C000359200. [DOI] [PubMed] [Google Scholar]
- Sasaki N, et al. A mutation in Tpst2 encoding tyrosylprotein sulfotransferase causes dwarfism associated with hypothyroidism. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0040. [DOI] [PubMed] [Google Scholar]
- Schneider MJ, et al. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. 2001;15:2137–48. doi: 10.1210/mend.15.12.0740. [DOI] [PubMed] [Google Scholar]
- Volpe MV, et al. Thyroid hormone affects distal airway formation during the late pseudoglandular period of mouse lung development. Mol Genet Metab. 2003;80:242–54. doi: 10.1016/j.ymgme.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Willem M, et al. Specific ablation of the nidogen-binding site in the laminin gamma1 chain interferes with kidney and lung development. Development. 2002;129:2711–22. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]