

Summary
A critical step in mRNA degradation is the removal of the 5' cap structure, which is catalyzed by the Dcp1-Dcp2 complex. The crystal structure of a S. pombe Dcp1p-Dcp2n complex combined with small-angle X-ray scattering analysis (SAXS) reveals that Dcp2p exists in open and closed conformations, with the closed complex being, or closely resembling the catalytically more active form. This suggests that a conformational change between these open and closed complexes might control decapping. A bipartite RNA binding channel containing the catalytic site and Box B motif is identified with a bound ATP located in the catalytic pocket in the closed complex, suggesting possible interactions that facilitate substrate binding. Dcp1 stimulates the activity of Dcp2 by promoting and/or stabilizing the closed complex. Notably, the interface of Dcp1 and Dcp2 is not fully conserved, explaining why the Dcp1-Dcp2 interaction in higher eukaryotes requires an additional factor.
Introduction
RNA degradation plays an important role in control of gene expression and the elimination of aberrant mRNAs. In eukaryotes, two major pathways are utilized to degrade mRNA and both are preceded by deadenylation of the 3’ poly(A) tail (Parker and Song, 2004; Meyer et al., 2004). In the 5’→3’ decay pathway, the 5’ cap is removed by a decapping reaction following deadenylation, thereby exposing the 5’ end for exonucleolytic digestion. Decapping is a critical step in mRNA turnover since it catalyzes the bulk of mRNA turnover, at least in yeast, and functions in specialized decay pathways such as nonsense-mediated decay (NMD), AU-rich element (ARE)-mediated decay, and the turnover of some mRNAs promoted by miRNAs (Coller and Parker, 2004; Behm-Ansmant et al., 2006; Rehwinkel et al., 2005; Fenger-Gron et al., 2005).
Removal of the 5’ cap structure is catalyzed by the decapping complex consisting of at least two subunits Dcp1p and Dcp2p (Coller and Parker, 2004). Decapping also correlates with the formation of a decapping mRNP containing the Dcp1p-Dcp2p holoenzyme and both general and mRNA-specific activators of mRNA decapping that can accumulate in cytoplasmic foci referred to as P-bodies (Parker and Sheth, 2007; Anderson and Kedersha, 2006; Eulalio et al., 2007). Activators of mRNA decapping appear to function both upstream and downstream of the formation of this decapping mRNP. For example, in yeast the decapping activators Pat1p and Dhh1p, appear to function, at least in part, to promote translation repression and assembly of the decapping mRNP for bulk mRNA turnover (Coller and Parker, 2005). In contrast, in yeast the Lsm1-7p complex appears to activate decapping of bulk mRNA after assembly of a decapping mRNP that can accumulate in P-bodies (Teixeira and Parker, 2007). Similarly, in the process of NMD, the yeast Upf2 and Upf3 proteins appear to activate decapping after the formation of an mRNP containing the decapping enzyme and Upf1p (Sheth and Parker, 2006). These observations suggest that the process of decapping includes an unknown and critical step following assembly of a decapping mRNP that enhances the rate of decapping.
In yeast, Dcp2p and Dcp1p form a stable holoenzyme with Dcp2p and Dcp1p as the catalytic and regulatory subunits (Beelman et al., 1996; Sakuno et al., 2004; Steiger et al., 2003). The crystal structure of free Dcp2p from S. pombe (She et al., 2006) reveals that a conserved N-terminal region forms a bi-lobed structure with a N-terminal α-helical domain, which interacts with Dcp1p, preceding a Nudix domain (She et al., 2006). Dcp2p recognizes capped mRNA substrates to cleave the pyrophosphate bond of the m7GpppN cap, releasing m7GDP as product. Dcp2p prefers longer RNA substrates suggesting the presence of spatially separated RNA-binding and catalytic sites on the Nudix domain, but the nature and site of the RNA binding domain of Dcp2p has not been determined (LaGrandeur and Parker, 1998; Piccirillo et al., 2003; Steiger et al., 2003; Wang et al., 2002). Dcp1p is a small protein containing an EVH1 domain (She et al., 2004), which is generally a protein-protein interaction module (Ball et al., 2002). Despite the conservation of Dcp1p and its requirement for decapping, how Dcp1p interacts with and activates Dcp2p remains elusive.
To gain insight into how Dcp1p interacts with and influences Dcp2p activity, we determined the crystal structure of Dcp1p in complex with a truncated S. pombe Dcp2p, (designated as Dcp2n in She et al., 2006) consisting of an α-helical domain and a Nudix domain. Crystal structure combined with SAXS and mutagenesis analyzes, reveals the Dcp1p-Dcp2n complex can exist in closed and open conformations with the closed form having increased catalytic activity. The structure also reveals the nature of the Dcp1p-Dcp2p interaction, thereby explaining why the binding of Dcp1 to Dcp2 in higher eukaryotes requires an additional protein factor (Xu et al., 2006; Fenger-Gron et al., 2005).
RESULTS AND DISCUSSION
Overall structure of the Dcp1p-Dcp2n complex
The crystal structure of Dcp1p complexed with the N-terminal helical domain of Dcp2p (residues 1–95; Dcp1p-Dcp2NTD) was solved first (Figure S1), and served as the search model for structural determination of a larger complex containing Dcp1p and Dcp2n (designated as Dcp1p-Dcp2n) by molecular replacement method. The final model of the Dcp1p-Dcp2n complex contains four complexes in the asymmetric unit (AU; Figure S2), which can be divided into two groups by the non-crystallographic symmetry (NCS) axis with each group containing two types of complexes corresponding to an extended or “open” conformation and a "closed" conformation of Dcp2n (Figures 1A and 1B).
Figure 1. Overall structure and structural comparison of the Dcp1p-Dcp2n complex.
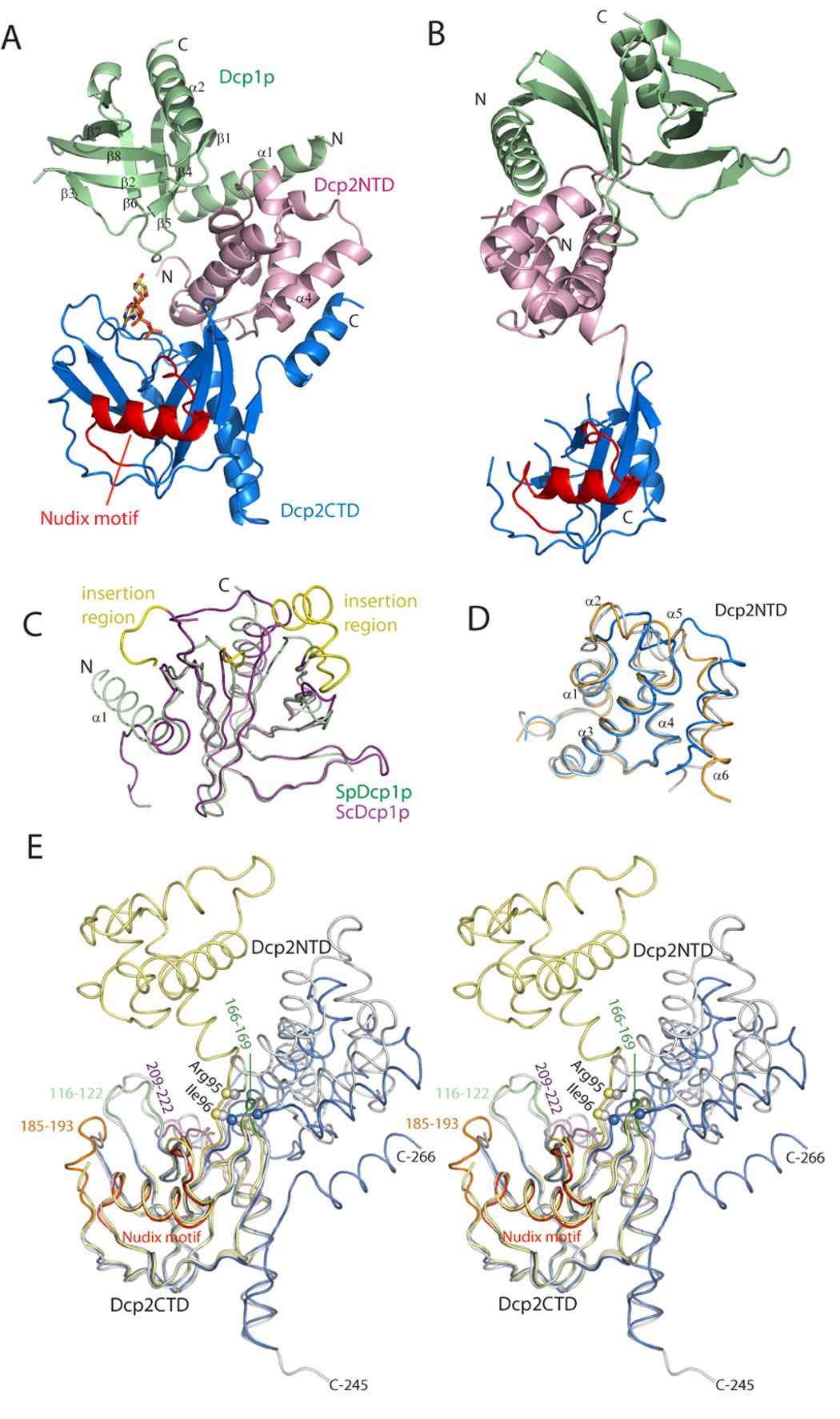
A. Ribbon diagram of the Dcp1p-Dcp2n complex in the closed conformation co-crystallized with one ATP molecule (sticks). Dcp1p is shown in green, the N-terminal domain (NTD) of Dcp2n in pink, the C-terminal Nudix domain of Dcp2n (CTD) in blue with the Nudix motif (residues 129–151) highlighted in red.
B. Ribbon diagram of the Dcp1p-Dcp2n complex in the open conformation with the same coloring scheme and Dcp2CTD oriented as in A.
C. Superposition of the structures of S. pombe Dcp1p (green) and S. cerevisiae Dcp1p (purple, PDB code: 1Q67). The EVH1 domain core structures are highly similar and the major differences lay in the N-terminal extension α1, except for the ScDcp1p specific insertions (yellow).
D. Superposition of the structures of Dcp2NTD in three conformations: open (orange), closed (blue) and free (grey, PDB code: 2A6T). Helices α5 and α6 are variable among three structures.
E. Stereo view of the superimposed structures of SpDcp2n in three conformations: open (yellow), closed (blue) and free (grey) with Dcp2CTD domains fixed in the same orientation. The Cα atoms of residues Arg95 and Ile96 in the hinge region are shown as spheres. In the closed form of Dcp2CTD, the peripheral loops in the Nudix domain that undergo conformational changes are highlighted and labeled.
In the closed complex, both Dcp1p and Dcp2NTD are in close proximity to the Nudix domain of Dcp2n (residues 96–266) to form a compact structure (Figure 1A). Moreover, an ATP molecule used as a crystallization additive is found to be situated in the catalytic center. In the extended conformation, the α-helical and Nudix domains of Dcp2n do not interact and are separated by a flexible linker, thereby making the whole molecule resemble a dumb-bell (Figure 1B). No ATP molecule is bound to the active site in the extended form. The model in the extended form of the complex is less complete than the closed form with substantial loop regions disordered, especially in the Nudix domain of Dcp2n (Figure 1B; also see Experimental Procedures).
Dcp1p in both the Dcp1p-Dcp2NTD and Dcp1p-Dcp2n complexes contains two α-helices and eight β-strands (Figures 1A). Strands β1 to β8 constitute the concave β-sheet, forming the canonical EVH1 domain together with α2 in the C-terminus. The N-terminal region of Dcp1p forms a long helix α1, which packs against one face of the β sheet and is perpendicular to α2. In both the extended and closed forms, Dcp1p interacts with the N-terminal domain of Dcp2n in the same manner as that in the Dcp1p-Dcp2NTD complex (see below).
Structural comparison
The overall structure of SpDcp1p in both the Dcp1p-Dcp2NTD complex and the Dcp1p-Dcp2n complex is similar to S. cerevisiae Dcp1p (ScDcp1p; She et al., 2004; r.m.s.d of ~1.2 Å for all the equivalent Cα atoms), although ScDcp1p has a species-specific insertion (residues 161–181; Figure 1C). More importantly, there is a marked conformational difference between SpDcp1p and ScDcp1p in the N-terminal region. In the ScDcp1p structure alone, this region is a 310-helix plus an extended polypeptide, whereas in SpDcp1p this region is a long α-helix that packs against Dcp2NTD (Figure 1C). Given the high sequence similarity in the N-terminus between SpDcp1p and ScDcp1p, this region of ScDcp1p could adopt a similar structure in complex with Dcp2p.
The individual domain structures of Dcp2n among the free, open and closed forms are similar. Superposition of the N-terminal α-helical domain of free Dcp2n with those in both the open and closed forms gives an r.m.s.d of 1.2 Å (Figure 1D). When the Nudix domain is superimposed, the r.m.s.d for Cα atoms is 0.66Å and 0.68Å for free vs. closed form and free vs. open form, respectively (Figure 1E). Despite these similarities, the relative orientations of Dcp2NTD with respect to the Nudix domain are quite different among these three forms. When the Nudix domain is superposed, the orientations of Dcp2NTD differ by 92°, 41° and 109° for free vs. open form, free vs. closed form and open vs. closed form, respectively (Figure 1E). These differences indicate that Dcp2p contains a flexible hinge between the α-helical and Nudix domains that allows for these two regions to adopt different conformations relative to each other.
Apart from the distinct overall structural differences in all three forms of Dcp2n, there are local structural changes as well. In Dcp2NTD, α5, α6 and the loop between them rearrange in the closed form (Figure 1D). Since α6 is connected to the hinge (residues 92–97) linking the α-helical and Nudix domains of Dcp2n, the larger positional shift for this region may be caused by the hinge angle changes induced by Dcp1p and ATP binding (see below). In the Nudix domain, loops 209–222 and 166–169 display large positional shifts, and loop 185–193 is disordered in both the free and the open form and becomes ordered in the closed form (Figure 1E). The conformational changes of these loops may be attributed to ATP and/or Dcp1p binding.
Nucleotide binding to the active site
In the closed form, an ATP molecule is located in a cavity formed mainly by loops 166–169, 209–222, 185–193 and the Nudix motif in the Nudix domain of Dcp2n, and clad by part of the α-helical domain (Figures 2A and 2B). The ATP molecule may be mimicking the interaction of the substrate with Dcp2p and the adenosine base could represent either the m7G moiety or the first base of the RNA substrate. The adenine ring of ATP points towards the interior of the protein and the phosphate groups extend to the vicinity of the catalytic site (Figures 2A and 2B). ATP makes contacts with several residues from the loops 166–169 and 185–193 in the Nudix domain, but does not directly contact the α-helical domain or Dcp1p. Notably, the adenine base stacks with the side-chain of Tyr220 with the ribose and first phosphate moieties interacting with the side chains of Arg167 and Gln169 respectively, through hydrogen bonding. Lys129 is located in close proximity to the β-phosphate of ATP. The side chain of Glu192, a residue critical for decapping (She et al., 2006) points to the bound ATP. Analogous to the role Glu150 plays in the nuclear decapping protein X29, Glu192 is likely to be involved in coordination of the catalytic divalent metal ions in Dcp2p (Figure 2C).
Figure 2. The ATP molecule in the active site.
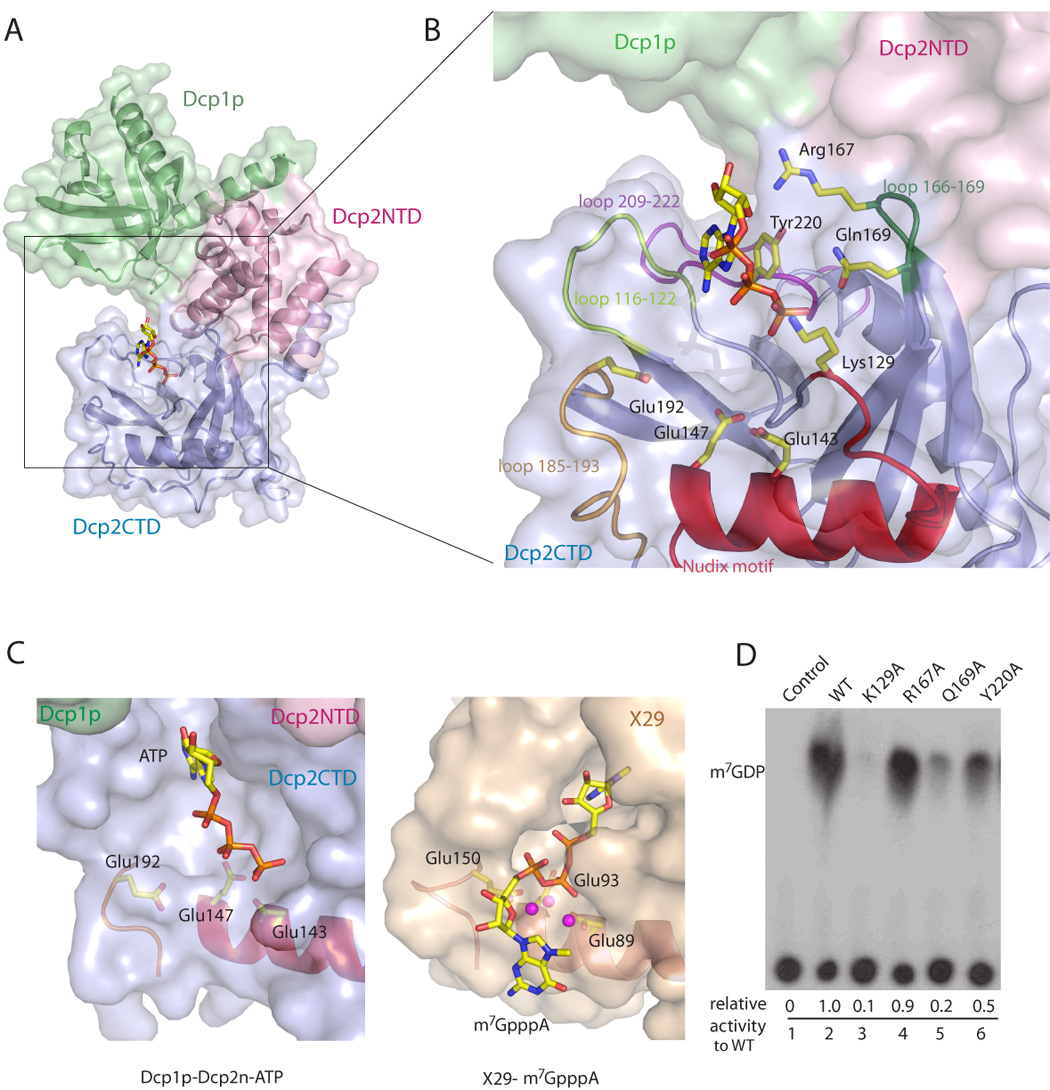
A. The ATP molecule co-crystallized with the closed form of the Dcp1p-Dcp2n complex is located in the pocket formed by the Dcp2CTD (blue) and clad by Dcp1p (green) and Dcp2NTD (pink).
B. Close-up view of the ATP-binding pocket. ATP is close to the active site of the Nudix motif and makes contact with residues from the peripheral loops.
C. Structural comparison of the Dcp1p-Dcp2n-ATP and X29-m7GpppA (PDB code: 2A8T) complexes. Partial Nudix motif and peripheral loop that harbor the three glutamate residues critical for decapping are shown and the side chains of glutamate residues are labeled. Three core Mn2+ ions from X29-m7GpppA complex are shown as spheres.
D. The in vitro decapping assay of Dcp2n mutants with the ATP-binding residues substituted by alanine. The relative decapping activities are averaged from duplicated experiments.
To examine whether the residues that interact with ATP are required for decapping, we generated single alanine-substituted mutants for Lys129, Arg167, Gln169, and Tyr220 and tested the decapping efficiency of these mutant proteins in vitro (Figure 2D). The activity of Q169A was strongly reduced, and that of Y220A was slightly reduced, while R167A had comparable activity to wild type Dcp2n. Markedly, the K129A mutant almost completely lost its activity. Moreover, mutation of the equivalent residue Lys135 to alanine in ScDcp2p abolished decapping activity in vivo (data not shown). These results showed that Lys129, Gln169 and Tyr220 are involved in mRNA decapping. Interestingly, mutations in this binding pocket in the ScDcp2p Nudix domain can have different or multiple effects on both the Km and Kcat of Dcp2p (Deshmukh et al., accompanying article). This suggests that this cavity in the Dcp2p surface both interacts with the substrate and stabilizes its binding as well as providing functional groups that contribute to catalysis.
The RNA-binding channel
Dcp2p is an RNA-binding protein and shows enhanced activity on longer substrates (LaGrandeur and Parker, 1998; Piccirillo et al., 2003; Steiger et al., 2003; Wang et al., 2002). One conserved region in the Nudix domain, termed Box B, has been implicated in RNA binding (Piccirillo et al., 2003). Consistent with this possibility, electrostatic potential mapping on the molecular surface of Dcp2n showed that this region is rich in positively charged residues (Figures 3A and 3B). Interestingly, two bulky electron densities were located between two NCS-related Dcp2n molecules in the closed form, associating with the positively charged side chains of Lys230 and Lys234 from one molecule and Lys231 and Lys235 from the other in the Box B region (Figure S2), suggesting that these electron densities could correspond to the triphosphate moiety of the additional ATP molecules.
Figure 3. The RNA-binding site of Dcp2n.

A. Electrostatic potential surface representation of the Dcp1p-Dcp2n complex in the closed form. The positively-charged surface is colored in blue and negatively-charged surface is in red. The figure is generated by ABPS (Baker et al., 2001), with the electrostatics value ranging from -6KT (red) to 6KT (blue). The ATP molecule is shown as sticks. Four clustered lysine residues in the Dcp2CTD are labeled.
B. Surface view of the closed form of the Dcp1p-Dcp2n complex in the same orientation as in A showing the putative RNA binding channel. The ribbon diagram of Box B region is colored in purple. A 12-mer poly(A) mRNA is modeled to show the proposed path of the RNA body bound to this channel.
C. The SPR experiments showing the RNA-binding affinity of wild type (WT) and mutant Dcp2n proteins with single or double lysine residues substituted by alanine in the Box B region.
D. The in vitro decapping assay of WT and mutant Dcp2n proteins. The relative decapping activities are averaged from duplicate experiments.
To test if this region of Dcp2p was involved in RNA binding, we introduced single or double alanine mutations into the lysine residues implicated in RNA binding, and checked the RNA binding affinity of wild-type and mutant Dcp2n proteins by surface plasmon resonance (SPR). As shown in Figure 3C, Dcp2n binds to the single-stranded RNA with fast association and dissociation rates, giving rise to a kD of about 1.0 µM. All four single mutations of K230A, K231A, K234A and K235A showed reduced resonance unit (RU) by four-fold. Double mutants of K230A K231A and K234A K235A diminished RNA binding further, with RU only one-eighth of the wild type level. Accordingly, the decapping assay carried out with single lysine to alanine mutants showed slightly decreased activity and double mutants had modest defects in decapping (Figure 3D). These results confirm the previous notion that Box B is required for RNA binding. More direct evidence for RNA binding to this region has come from NMR chemical shift perturbation experiments that mapped a related set of residues important for RNA binding to this region in the Nudix domain of ScDcp2p (Deshmukh et al., accompanying article).
Nature of the Dcp1-Dcp2 interaction
Dcp1p associates with the N-terminal α-helical domain of Dcp2n through extensive hydrophobic and hydrogen bonding interactions, burying a solvent accessible surface of 1016Å2 (Figure 4A). Superposition of the Dcp1p-Dcp2NTD complex with the same portion in both the extended and closed forms of the Dcp1p-Dcp2n complex gives a r.m.s.d of ~1.3 Å, indicating that the interface between Dcp1p and Dcp2p is similar in both the short and longer complexes. Three conserved patches identified previously in Dcp1p with functional roles in decapping are not involved in Dcp2p binding (Figure 4A). This is consistent with the findings that mutations in these regions of Dcp1p affected decapping but not Dcp2p binding (She et al., 2004). Instead, SpDcp1p mainly uses residues that are only highly conserved in yeast species located in helix α1 and the loop between β5-β6 to interact with Dcp2NTD (Figures 4B and 4C). Dcp2NTD interacts with Dcp1p via a previously identified patch consisting of three invariant residues Arg18, Phe19 and Phe44 (Figures 4B and 4D), therefore strongly supporting the previous findings that these residues are crucial in Dcp1p binding, and for Dcp2p to be stimulated by Dcp1p in vitro (She et al., 2006).
Figure 4. The Dcp1p-Dcp2n interface.
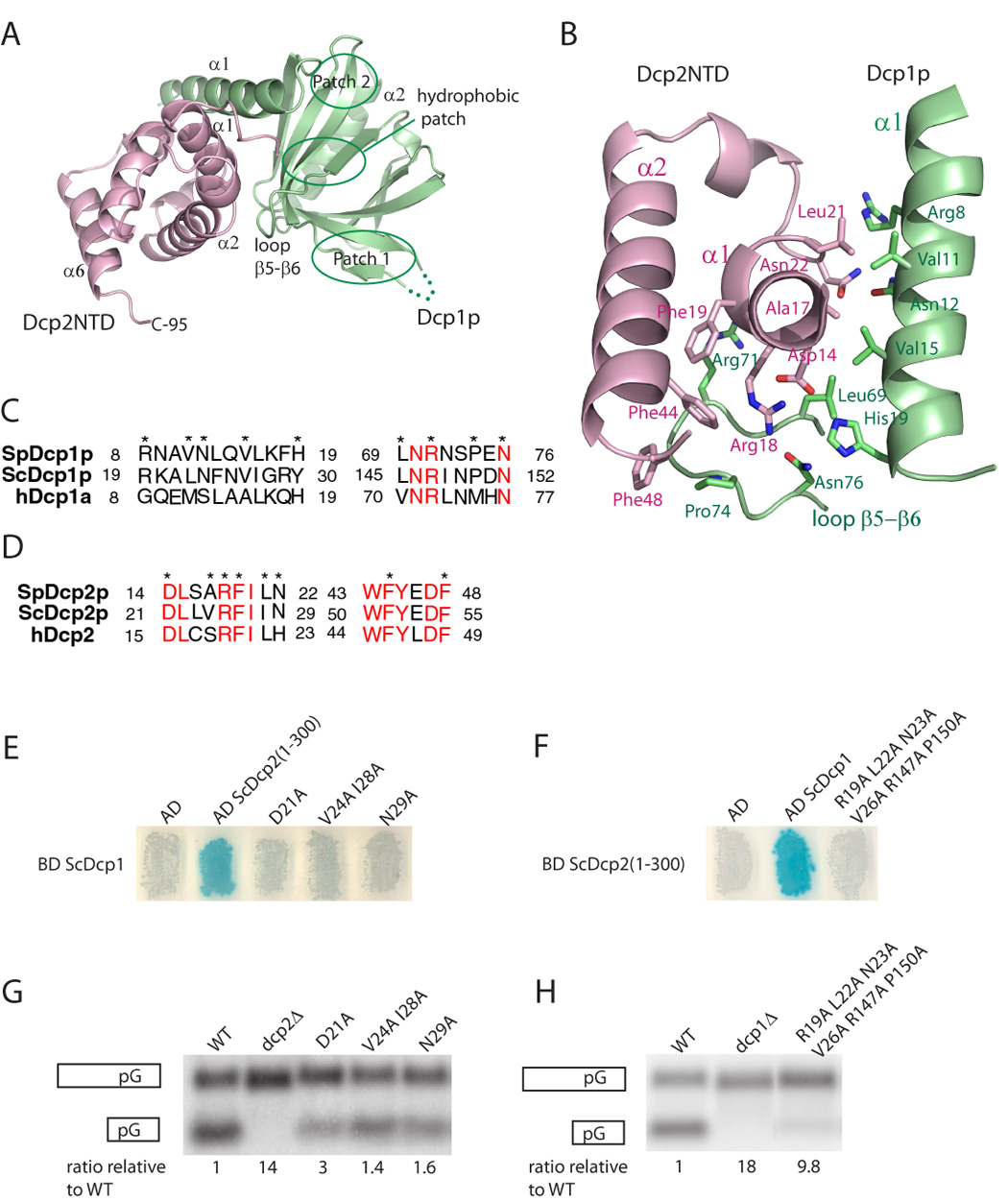
A. The ribbon diagram of Dcp1p (green) in complex with Dcp2NTD (pink). The positions of the conserved patch 1, patch 2 and hydrophobic patch on Dcp1p are indicated.
B. The protein-protein interface of Dcp1p-Dcp2NTD. The side-chains of residues involved in binding are shown as sticks.
C&D. The sequence alignment of Dcp2-binding region in SpDcp1p, ScDcp1p and hDcp1a and Dcp1-binding region in SpDcp2p, ScDcp2p and hDcp2. The invariant residues are highlighted in red and the residues in the protein-protein interface are marked with asterisks.
E. Two hybrid analysis of the interaction between ScDcp1p fused to the GAL4 DNA binding domain and residues 1–300 of ScDcp2p fused to the GAL4 activation domain. AD, activation domain alone; AD ScDcp2(1–300), wild-type 1–300 residues of ScDcp2p fused to the activation domain; remaining lanes, AD Dcp2(1–300) carrying the indicated amino acid changes. Analysis of LacZ activity is shown.
F. Two hybrid analysis of the interaction between residues 1–300 of ScDcp2p fused to the GAL4 DNA binding domain and ScDcp1p fused to the GAL4 activation domain. AD, activation domain alone; AD ScDcp1, wild-type Dcp1 fused to the activation domain; remaining lane, AD Dcp1 carrying the indicated amino acid changes. Analysis of LacZ activity is shown.
G & H. The effect of point mutations in ScDcp1p and ScDcp2p on decapping in vivo. Northern analysis of steady-state levels of full-length MFA2 mRNA containing a poly(G) tract in its 3’UTR and its decay intermediate produced by decapping and 5’ to 3’ exonucleolytic digestion. The full-length mRNA and the decay intermediate are illustrated on the left of each panel. The ratio of full-length MFA2 mRNA to the decay intermediate in each mutant strain compared to the ratio in the wild-type strain is indicated below each lane. G. Analysis in a S. cerevisiae dcp2Δ strain expressing wild-type ScDcp2p (WT), no Dcp2p (dcp2Δ) or Dcp2p with the indicated amino acid changes. H. Analysis in a S. cerevisiae dcp1Δ strain expressing wild-type ScDcp1p (WT), no Dcp1p (dcp1Δ) or Dcp1p with the indicated amino acid changes.
Consistent with the N-terminal region of Dcp1p being important for interaction with Dcp2p, some mutations in ScDcp1p that affect decapping in vivo are mapped to the N-terminal helix of Dcp1p (Tharun and Parker, 1999). Moreover, two temperature sensitive (ts) mutants isolated in S. pombe, Spdcp1-P74S and Spdcp1-L69S, are defective in mRNA decay at the non-permissive temperature (Sakuno et al., 2004). Interestingly, Pro74 and Leu69 are located in the β5-β6 loop of SpDcp1p (Figure 4B) mediating its interaction with SpDcp2NTD, suggesting that the phenotypes observed for these ts mutants could be due to their defects in Dcp2p binding.
To test the role of the interface between yeast Dcp1 and Dcp2 proteins in vivo, we mutated residues in the interface between the ScDcp1p and ScDcp2p proteins and examined the effect of those mutations on decapping in vivo and on their interactions using a two-hybrid assay. The ScDcp2p mutations D21A, N29A, and the double mutant V24A I28A, interfered with binding to ScDcp1p in the two hybrid assay (Figure 4E) and all caused a partial defect in mRNA decapping in vivo (Figure 4F) as assessed by the ratio of full length MFA2pG mRNA to its decay fragment which is a function of decapping rate (Cao and Parker, 2001). Similarly, a sextuple mutant in ScDcp1p (R19A, L22A, V26A, N23A, R147A, P150A) disrupted binding to ScDcp2p (Figure 4E), and strongly inhibited decapping in vivo (Figure 4F). These results document that the ScDcp1p N-terminus is required for interaction with ScDcp2p in vivo and that the Dcp1p-Dcp2p interaction is required for decapping in vivo.
Three aspects of the Dcp1p-Dcp2p interaction are worth noting. First, this mode of extensive protein-protein interaction has not been observed in other EVH1 proteins thus revealing a new type of interaction for this protein family. Second, the binding of Dcp2NTD on SpDcp1p does not shield the putative proline rich binding site (PRS; referred to as patch 1 in She et al., 2004) of the EVH1 domain in Dcp1p (Figure 4A). This suggests that yeast Dcp1p may use patch 1 to associate with other proteins affecting decapping in vivo. Finally, the SpDcp1p residues that are involved in Dcp2p association are more variable in higher eukaryotes as compared to yeast (Figures 4C and 4D). This suggests that the Dcp1p-Dcp2p interaction may be different or weaker in higher eukaryotes. This provides a structural explanation for why direct interactions are not observed between Dcp1p and Dcp2p with human or nematode proteins in vitro (Lykke-Andersen, 2002; Cohen et al., 2005), and why the H. sapiens Hedls protein or its homolog VARICOSE in A. thaliana, are required to bridge the interactions between Dcp1 and Dcp2 in humans or plants (Fenger-Gron et al., 2005; Xu et al., 2006). This additional complexity of the decapping complex in higher eukaryotes may reflect additional modes of regulation of decapping in these organisms.
The closed conformation is, or closely resembles, a more active form of the enzyme
The crystal structure revealed both open and closed forms of the Dcp1p-Dcp2n complex. In addition, the presence of ATP, which could mimic the 5' end of the mRNA substrate, in the active site of the closed complex suggested that ligand binding to the active site might promote closure of the complex. To determine if two forms also exist in solution, and whether formation of the closed complex was influenced by ligand binding, small-angle X-ray scattering (SAXS) was employed. The scattering patterns from the wild type complex without and with ATP are presented in Figure 5A and the overall parameters are summarized in Table 1. The radius of gyration Rg and the maximum size Dmax of the complex without ATP agree well with those of the open crystal form (Rg=33.5 Å, Dmax=105 Å). Addition of ATP leads to a significant compaction of the complex (Rg decreases by 5 Å and Dmax by about 10 Å as illustrated by the distance distribution functions in Figure 5A insert), and the sizes become compatible with those of the closed crystal form (Rg=25.5 Å, Dmax=85 Å). The low resolution shapes of the complex with and without ATP reconstructed from the scattering patterns by an ab initio program GASBOR (Svergun et al., 2001) agree well with the closed and open crystal structures, respectively, as illustrated in Figure 5B,C and Table 1. Moreover, the experimental data in the presence of ATP is neatly matched by the scattering pattern computed from the closed model using the program CRYSOL (Svergun et al., 1995) but not by that computed from the open model (Figure 5A and Table 1). The scattering computed from the open and closed forms display systematic deviations with the measured scattering from the ATP-free complex (green and blue curves, respectively, in the upper plot in Figure 5A). The open form, however, still provides a better fit (Table 1), matching the scattering at the smallest angles responsible for the overall particle size, whereas at higher angles the computed scattering oscillates around the measured pattern. This suggests that the structure in solution may be flexible, leading to a smearing of the scattering profile compared to that computed from the open crystal structure. Such a conformational mobility would not be surprising given the flexible hinge region in the rather extended open structure. These results indicate that the open and closed forms of the Dcp1p-Dcp2n complex do exist in solution, demonstrating that both forms are functionally relevant.
Figure 5. SAXS data and shape reconstructions.
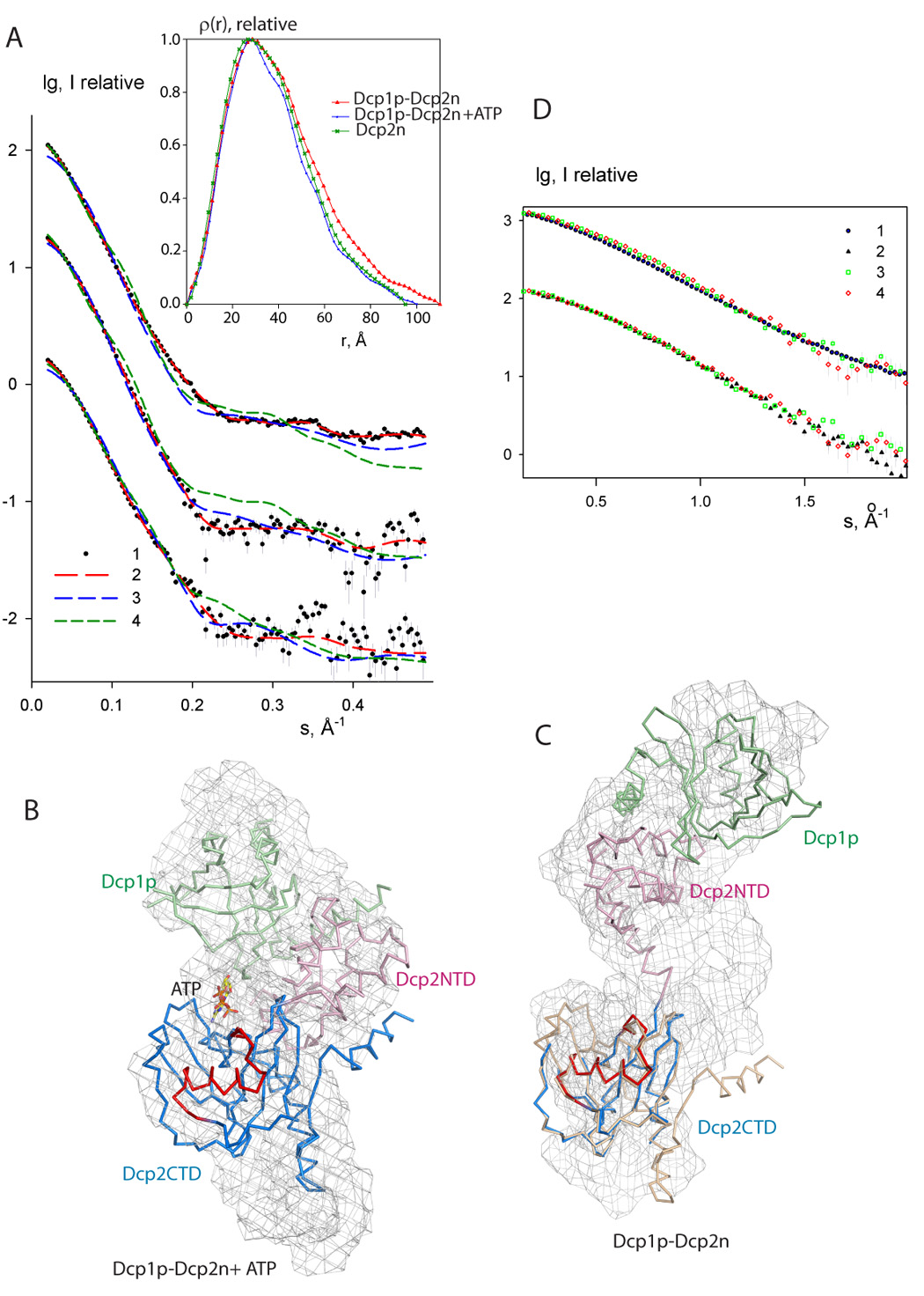
A. Experimental and computed scattering data. From top to bottom: Dcp1p-Dcp2n complex without and with ATP, and free Dcp2n without ATP (addition of ATP to Dcp2n yields the curve coinciding within the errors). The logarithm of the scattering intensity is plotted against the momentum transfer s = 4π sinθ/λ, where 2θ is the scattering angle and λ = 1.5 Å is the X-ray wavelength. The plots are displaced along the ordinate for better visualization. Dots with error bars: experimental scattering; dashed lines: scattering from the models. Red: ab initio GASBOR models; green: open crystal complex, blue closed crystal complex (for Dcp2n, green and blue are extended Dcp2n as in the open crystal complex and compact free Dcp2n in the crystal, respectively). Insert: distance distribution functions computed from the data using GNOM. The associated errors in the data points are computed from the counting statistics of the detected scattering patterns and appropriately propagated over the processing steps following standard procedures.
B. The Dcp1p-Dcp2n complex with ATP. The backbone of the crystal structure of the closed Dcp1p-Dcp2n complex is superimposed onto the shape construction from the ab inito model (shown as mesh). Dcp1p is colored in green, Dcp2NTD in pink and Dcp2CTD in blue. The Nudix motif is in red and ATP is shown as sticks.
C. The Dcp1p-Dcp2n complex without ATP. Superimposed is the crystal structure of the open Dcp1p-Dcp2n complex shown as backbone onto the shape reconstruction. The coloring scheme is the same as in panel B. Dcp2CTD from the closed Dcp1p-Dcp2n complex is superimposed onto the open Dcp1p-Dcp2n backbone and colored in wheat.
D. Comparison of the effects of ATP, m7GpppA cap and the pAA oligonucleotide on the scattering from Dcp1p-Dcp2n complex. Experimental pattern (1) corresponds to the scattering from the wild type Dcp1p-Dcp2n, patterns (2–4) are measured after addition of ATP, m7GpppA cap and pAA, respectively. The least squares overlaps of (1) with (3,4) and (2) with (3,4), are shown separately. The initial portion of the scattering data up to the resolution of about 30Å responsible for the overall conformation is displayed and the two comparisons are displaced along the logarithmic axis for clarity.
Table 1.
Overall parameters derived from SAXS data and computed from the models
| Construct | Rgexp Å | Dmexp Å | χ | χop | χcl | NSD op | NSD cl |
|---|---|---|---|---|---|---|---|
| Dcp1-Dcp2 | 33±1 | 110±10 | 0.98 | 2.8 | 3.1 | 1.37 | 1.45 |
| Dcp1-Dcp2+ATP | 28±1 | 100±10 | 1.0 | 2.3 | 1.3 | 1.37 | 1.30 |
| Dcp1-Dcp2+m7GpppA | 29±1 | 100±10 | - | 2.1 | 1.2 | - | - |
| Dcp1-Dcp2+pAA | 28±1 | 100±10 | - | 1.8 | 1.1 | - | - |
| Dcp1-Dcp2 (R95P) | 32±1 | 110±10 | - | 1.5 | 2.2 | - | - |
| Dcp1-Dcp2 (R95P)+ATP | 34±1 | 110±10 | - | 2.2 | 2.5 | - | - |
| Dcp1-Dcp2 (I96P) | 33±1 | 110±10 | - | 2.5 | 2.9 | - | - |
| Dcp1-Dcp2 (I96P)+ATP | 35±1 | 110±10 | - | 2.1 | 3.3 | - | - |
| Dcp2 | 27±1 | 100±10 | 1.0 | 1.7 | 2.5 | 1.53 | 1.76 |
| Dcp2+ATP | 26±1 | 100±10 | - | 1.9 | 2.6 | - | - |
| Dcp2+m7GpppA | 28±1 | 100±10 | - | 1.8 | 2.3 | - | - |
| Dcp2+pAA | 27±1 | 100±10 | - | 1.6 | 2.2 | - | - |
Rg, radius of gyration, Dmax, maximum size, superscript op and cl points to the open/closed crystal structure for Dcp1-Dcp2 complex, or to extended/compact Dcp2 configuration in the crystal. χ denotes discrepancy between the experimental data and the scattering from the model (no superscript: ab initio GASBOR model, superscript op and cl discrepancy to the above opened/closed high resolution models computed by CRYSOL). NSDop/cl denotes the normalized spatial discrepancy between the typical ab initio model and the high resolution open/closed models. 10mM ATP, 10mM m7GpppA or 5mM pAA were used in those SAXS measurements where one of these ligands was included in the protein solution.
To determine if ATP mimics the effects of the 5' end of the mRNA substrate, SAXS experiments were performed on Dcp1p-Dcp2n solutions upon incubation with the m7GpppA cap and with pAA, a 5’ phosphorylated di-adenine-nucleotide. The scattering patterns from the Dcp1-Dcp2n complex obtained in the presence of m7GpppA or pAA were identical with that obtained with ATP within the experimental errors (Figure 5D, lower overlap), and display distinct deviations from the Dcp1p-Dcp2n pattern without ATP (upper overlap). Moreover, the overall parameters and the fits to the open and closed models for the samples with m7GpppA cap and pAA are fully compatible with those obtained for the ATP-containing sample (Table 1), suggesting that formation of the closed complex is enhanced by binding of these ligands to the active site. Consistent with the SAXS data, ATP, the m7GpppA cap and pAA at high concentration (>1mM) inhibited the decapping activity of the Dcp1p-Dcp2n complex (Figure S3).
We have also measured SAXS from Dcp2n alone (Figure 5A and Table 1). Contrary to the complex, addition of neither ATP, m7GpppA nor pAA revealed a measurable change in the scattering pattern. Interestingly, the overall parameters and the low resolution shape of free Dcp2n agree well with the extended Dcp2n conformation extracted from the open crystallographic model of the complex (Rg=27.5 Å, Dmax=90 Å) but not with the compact crystallographic model of free Dcp2n in the crystal (Rg=24.0 Å, Dmax=80 Å) (She et al., 2006). The results that ATP, m7GpppA and pAA failed to induce the formation of a closed form of Dcp2n in the absence of Dcp1p seem at odds with the observation that these ligands inhibited the decapping activity of Dcp2n if provided at high concentration (>1mM) (Figure S3). The simple explanation for this discrepancy is that these ligands bind to the active site (outcompete the RNA substrate in the decpapping assay) and transiently induce the formation of the closed form of Dcp2n, which is not stable enough for SAXS detection. Altogether, these results indicate that Dcp2n exists in equilibrium between conformations in solution and that Dcp1p binding drives the equilibrium towards the closed ligand-bound state of Dcp2n.
Several features of the closed form suggest that Dcp2p is most active for catalysis when it is in a closed structure. First, the closed form brings residues in Dcp2NTD close to the active site and they contribute to forming a binding pocket for the substrate. Residues Glu39, Trp43, Asp47 and Phe48 in Dcp2NTD that have been identified as affecting Dcp2p function in vitro (She et al., 2006), are positioned in the closed form at the domain interface (Figure 6A). This positioning is stabilized by a network of hydrogen bonds between the α-helical and Nudix domains and the hinge region (residues 92–97). Specifically, while in both the free and open forms the linker is extended, in the closed form, the hinge bends ~45° through a series of Phi-Psi changes that are distributed across residues 94 to 96 (values reported in Table S1; Figure 1E), enabling salt bridges between Glu39 with Lys93 and Arg95 and contacts with the Nudix domain (Asn170; Figure 6B). This provides a structural explanation for why residues in Dcp2NTD affect the catalytic properties of Dcp2p (She et al., 2006) and argues that the closed form is the more active structure. Additional evidences that the closed complex is the more active complex are the presence of an ATP bound in the active site (see above), and the observations that pAA and m7GpppA, like ATP, can induce formation of a closed form of the complex and inhibit decpaping (Figure S3), consistent with substrate preferentially binding to the closed form of the complex. Moreover, the closed form has enhanced order in the catalytic domain consistent with being the active form. For example, Glu192, which is required for Dcp2p activity both in vivo and in vitro (She et al., 2006), is oriented towards the active site (Figure 2B) due to the ordering of loop 185–193. Similarly, loop 209–222 undergoes a significant positional shift and loop 166–169 tilts towards active site in the closed form (Figure 1E and Figure 2B). These changes all are likely to contribute to substrate recognition, as residues that associate with the ATP molecule are all located in these loops (see above).
Figure 6. The closed and open conformations of the Dcp1p-Dcp2n complex.
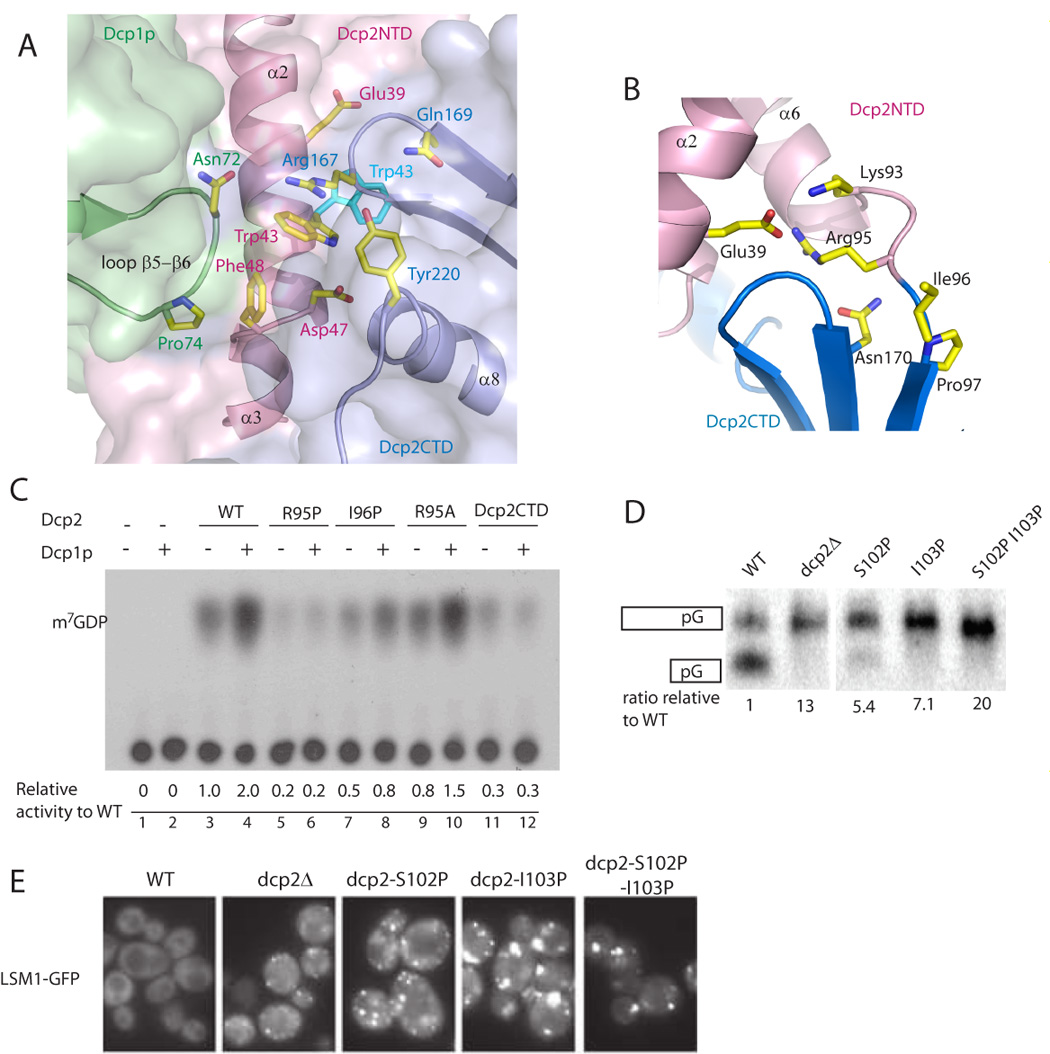
A. The figure depicts the role of Dcp1p in promoting and/or stabilizing the closed form of the Dcp1p-Dcp2n complex. The side-chains of residues involved in stabilizing the interaction are shown as sticks. The side-chain positions of Trp43 (cyan) are identical in the free form Dcp2p (PDB code: 2A6T) and the open conformation of the Dcp1p-Dcp2n complex.
B. Ribbon diagram of the hinge region (residue 92–96) between Dcp2NTD and Dcp2CTD in the closed Dcp1p-Dcp2n complex. Residues in the hinge region interact with both domains. The side chains of related residues are shown in sticks.
C. In vitro decapping assay of Dcp2n WT and variants with mutations in the hinge region.
D. In vivo decapping assay of S. cerevisiae Dcp2 WT and variants with mutations in the hinge region.
E. Microscopic visualization of cells during mid-log phase of LSM1-GFP in dcp2Δ strains containing either wild-type (WT), vector (dcp2Δ), or dcp2 hinge region variants.
To directly test whether closure of the hinge between the α-helical and Nudix domains of Dcp2p would enhance catalysis, we created mutations that would prevent the formation of the closed complex by mutating the residues Arg95 and Ile96 to proline in the hinge region. Substitution of Arg95 to proline strongly reduced Dcp2n decapping activity to the extent comparable to the deletion of the whole N-terminal domain (Figure 6C). More importantly, R95P mutant could not be stimulated by Dcp1p, although it is able to form a 1:1 complex with Dcp1p (data not shown). As expected, replacing Arg95 with alanine restored the activity and capacity to be stimulated by Dcp1p (Figure 6C). Another single mutation in the linker region, I96P, had moderately reduced activity. And could only be partially stimulated by Dcp1p (Figure 6C). SAXS analysis showed that both R95P and I96P in complex with Dcp1p adopted an open conformation even in the presence of ATP (Table 1). Note that the weaker phenotype of the I96P mutation is probably because its side-chain points to the solvent region, therefore proline substitution would restrict backbone flexibility but could still allow formation of a closed form. Moreover, the R95P lesion might have a stronger phenotype since it both restricts movement and disrupts a salt bridge formed in the closed complex between Arg95 and Glu39. These observations argue that efficient stimulation of Dcp2p by Dcp1p requires formation of the closed complex.
Consistent with the in vitro results, proline substitutions of Ser102 or Ile103 in ScDcp2p, which correspond to Arg95 and Ile96 respectively, showed strong defects in decapping in vivo while a double mutant with both Ser102 and Ile103 substituted by proline showed no decapping activity at all (Figure 6D). These results indicate that prolines in the hinge region disrupt decapping in vivo and in vitro and suggest that this is due to a failure to form the closed form of the complex.
Implications of conformational flexibility in Dcp2p
The observation that Dcp2p can form both open and closed complexes with Dcp1p suggests that control of this transition would influence the catalytic rate of Dcp2p since in the open form Dcp2p would be a poor catalyst. Strikingly, recent observations suggest that there is a critical step in activating decapping after interaction of the decapping enzyme with the mRNA. For example, yeast strains lacking Lsm1p, which is a key component of the decapping activator complex Lsm1-7p, show reduced decapping rates, but allow the formation of an mRNP containing the decapping enzyme that accumulates in P-bodies. These observations suggest the Lsm1-7p complex is required for activating a step in decapping following interaction of the decapping enzyme with the mRNA (Teixeira and Parker, 2007). Similarly, in the process of NMD in yeast cells, the Upf2 and Upf3 proteins are required to promote decapping at a similar late stage of degradation (Sheth and Parker, 2006). This suggests that an open form of the Dcp1p-Dcp2p complex might be sufficient to interact with the mRNA and form an mRNP capable of accumulating in P-bodies, but actual decapping would require formation of the closed form of the complex, whose formation might be promoted by either the Lsm1-7p complex or Upf2/Upf3p.
Consistent with the formation of the closed Dcp1p-Dcp2p complex being required at a late stage of decapping, we observed that Saccharomyces cerevisae strains expressing the S102P, I103P proline hinge mutants, which are analogous to the S. pombe alleles that cannot form the closed complexes, accumulate large P-bodies (Figure 6E). Since the formation of large P-bodies requires an assembly function of Dcp2p (Teixeira and Parker, 2007), and the S102P, I103P alleles inhibit decapping (Figure 6D), these observations indicates that the S102P, I103P proline hinge mutants are capable of assembling an mRNP that can accumulate in P-bodies, but are unable to promote catalysis. Based on these results, we suggest that formation of the closed complex might be an important step in the process of decapping occurring at a late stage of the reaction and might be controlled by additional protein interactions that facilitate the formation of the closed complex in cells.
Dcp1p activates Dcp2p by stabilizing a closed conformation of the complex
Previous experiments have demonstrated that Dcp1p is required for decapping in vivo (Beelman et al., 1996; Sakuno et al., 2004) and can stimulate the activity of Dcp2p in vitro (Steiger et al., 2003; She et al., 2006). However, an unresolved issue is how Dcp1p activates Dcp2p to increase decapping. The observation that Dcp1p can associate with Dcp2NTD independently of the presence or the orientation of the Nudix domain suggested that Dcp1p activates the decapping activity of Dcp2p indirectly, possibly through promoting the binding of RNA to the enzyme. However, RNA binding to Dcp2n alone and to a Dcp1p-Dcp2n complex measured by isothermal titration calorimetry (ITC) indicated that Dcp2n and the Dcp1p-Dcp2n complex bind to RNA with similar affinities (Figure S4), arguing that Dcp1p does not increase Dcp2p activity by increasing the affinity for RNA. Consistent with this interpretation, kinetic analysis of the Dcp1p-Dcp2p complex as compared to Dcp2p alone indicates that Dcp1p affects the chemistry of the reaction and not substrate binding (Deshmukh et al., accompanying article)
The ability of Dcp2p to adopt open and closed conformations in the presence of Dcp1p suggests that Dcp1p might activate Dcp2p by promoting and/or stabilizing the formation of the closed complex. Direct evidence supporting that Dcp1p promotes and stabilizes a closed form of the complex comes from the SAXS analysis. In solution, free Dcp2n adopts an open conformation independent of the presence or absence of ATP, m7GpppA or pAA (Table 1). In contrast, the Dcp1p-Dcp2n complex adopts a closed form in the presence of ATP or m7GpppA or pAA while it exhibits an open conformation in the absence of these ligands (Figures 5B and 5C). These results argue that Dcp1p can promote the formation of a closed complex, which is stimulated by the presence of substrate in the active site.
The exact structural details by which Dcp1p promotes the formation of the closed form remain to be elucidated. However, in the Dcp1p-Dcp2n complex, the binding of Dcp1p to Dcp2NTD alters several residues that can then interact with the Nudix domain. For example, the loop connecting β5 and β6 of Dcp1p interacts with Trp43 of Dcp2p, whose side-chains flips in the closed form from its position in both the free and open forms of Dcp2p to stack with the side-chain of Arg167, which is hydrogen-bonded to the ribose ring of ATP (Figure 6A). By promoting this conformational change Dcp1p may act to stabilize the stacking interaction between Trp43 and Arg167, which might favor binding of the substrate.
CONCLUDING REMARKS
The experiments described in this work have revealed important features of the Dcp1p-Dcp2p complex. First, we have determined the nature of the interaction between these two proteins. Second, we have documented by both X-ray crystal structures and SAXS that the Dcp1-Dcp2p complex contains a flexible hinge region and can exist in both open and closed forms. Third, we have provided evidence that Dcp1p enhances Dcp2p function by promoting the formation of the closed complex which has higher catalytic activity. Interestingly, we also observed that ATP is bound to the active site, and both ATP and pAA as well as m7GpppA enhanced the formation of the closed complex and inhibited decapping. Finally, we have identified a region involving in RNA binding located at some distance from the active site.
Our structural analyses and the new biochemical data suggest a possible mechanism by which Dcp2p recognizes its substrate. In this model, the substrate is bound in a channel on the surface of Dcp2p with the cap structure in the active site and then the body of the RNA wraps across the Nudix domain and then down the channel along the Box B helix (Figure 3B). The notion that this channel is for RNA binding is supported by the observations that residues in Dcp2NTD important for decapping (She et al. 2006) are located along this channel (Figure 6A), and thereby might stabilize substrate binding. This model provides a possible structural explanation for why the decapping enzyme prefers longer RNA substrates as a modeled RNA would need to be at least 12 residues long to bind to both the active site and the Box-B region on Dcp2p (Figure 3B). The increased activity of Dcp2p on even longer RNA substrates (Steiger et al., 2003) might be explained by Dcp2p being able to bind RNA anywhere along the substrate, followed by a one-dimensional diffusion along the body of the RNA to the 5' cap structure. Since the formation of the closed complex is stimulated by the presence of a cap analogue or a substrate mimic in the active site, a likely possibility is that the open form of the complex could bind a capped RNA, then diffuse along the RNA structure until reaching the 5' cap structure. When the 5’ end of the mRNA binds to the catalytic site of the Nudix domain, a closed form will be formed whereby cap recognition and catalysis would take place. Since cap recognition occurs during the catalysis step and requires the presence of RNA body (Deshmukh et al., accompanying article), a high resolution crystal structure of the Dcp1-Dcp2n complex complexed with a capped RNA will be required to elucidate the process of cap recognition. Another important area of future research will be in determining if various regulators of decapping in vivo influence the transition between the open and closed forms of the complex, and thereby control decapping.
Experimental Procedures
Protein purification and Crystallization
The constructs for expressing GST-fusion recombinant S. pombe Dcp2NTD (1–95 residues), Dcp2n (1–266 residues) and Dcp1p proteins were from previous studies (She et al., 2006). Single mutation or double mutation was introduced into the plasmid using the Site-Directed Mutagenesis Kit (QIAGEN).
To get the Dcp1p-Dcp2NTD complex, purified Dcp1p and Dcp2NTD were mixed at 1:1 molar ratio for 2 hours at 4 °C before loaded onto the Superdex S75 gel filtration column. Fractions containing the Dcp1p-Dcp2NTD complex were pooled and concentrated to 25 mg/ml in buffer (20 mM HEPES pH 7.3, NaCl 150 mM, and 2 mM DTT). For the Dcp1p-Dcp2n complex, similar procedure was followed and the protein complex was concentrated to 10 mg/ml in storage buffer (10 mM HEPES pH 7.0, NaCl 150 mM, and 2 mM DTT). The formation of 1:1 complex was confirmed by both SDS-PAGE and dynamic light scattering. Individual mutant protein was purified to homogeneity by a combination of GST affinity and ion-exchange chromatography.
The crystal of the Dcp1p-Dcp2NTD complex was grown in crystallization buffer (0.1 M Ammonium Acetate and 5% PEG3350) and soaked in mother liquor supplemented with 2mM trimethyl lead acetate (TMLA) for 12 hours. The Pb-derivative crystal was frozen in a buffer containing 0.1 M Ammonium Acetate, 5% PEG3350, 25% Ethylene Glycol, and 2mM TMLA. The crystal of the Dcp1p-Dcp2n complex was obtained in crystallization buffer (8% Ethylene Glycol, 100 mM HEPES pH 7.0, 11.6% MPD, 4.4% PEG 4000 and 10 mM ATP) at 15 °C and was flash-frozen in cryo-buffer (6% Ethylene Glycol, 100 mM Tris-HCl pH 8.0, 18% MPD, 6% PEG 4000 and 10 mM ATP).
Structure determination
The diffraction data for the crystals of the Dcp1p-Dcp2NTD complex were collected using in-house RIGAKU FR-E High Brilliance X-Ray Generator. The TMLA-soaked crystal belongs to space group P212121 with one molecule in the asymmetric unit.One Pb site was found by SnB (Miller R et al., 1994) and phase determination was done using SHARP (De la Fortelle and Bricogne, 1997) by single-wavelength anomalous dispersion (SAD) method at 2.33Å. Partial model was traced and modified with O(Jones et al., 1991). Model refinement was carried out with CNS (Brunger et al., 1998) and Refmac5 (CCP4, 1994) with a working R factor and a free R factor of 22.7% and 25.9%, respectively (Table 2). Residues 39–44 in Dcp1p and residues 1, 92–95 in Dcp2p are disordered.
Table 2.
Data collection and crystallographic refinement statistics
| Dcp1p-Dcp2NTD | Dcp1p-Dcp2n | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.5418 | 0.9792 |
| Space group | P212121 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 42.7, 49.7, 115.0 | 68.3, 161.4, 91.4 |
| α, β, γ (°) | 90, 90, 90 | 90, 97.4, 90 |
| Resolution range (Å) | 20-2.33 | 90-2.8 |
| Ramerge(%) | 8.1 (34.5) | 7.9 (32.3) |
| I/σ | 7.6 (2.2) | 5.9 (2.2) |
| Completeness(%) | 99.9 (99.5) | 99.9 (99.9) |
| Redundancy | 9.9(9.5) | 3.6 (3.6) |
| Refinement | ||
| Resolution range (Å) | 20-2.33 | 20.0-2.8 |
| Number of reflections | 10,420 | 45,695 |
| Number of atoms | ||
| Protein | 1778 | 11684 |
| Water | 41 | 108 |
| Ligand | - | 2 × ATP |
| Rb work(%) | 22.7 | 23.4 |
| Rc free(%) | 25.9 | 28.7 |
| R.m.s.deviations | ||
| Bond lengths (Å) | 0.010 | 0.009 |
| Bong angles (°) | 1.3 | 1.2 |
Values in parentheses indicate the specific values in the highest resolution shell.
Rmerge = ∑|Ij−<I>|/∑Ij, where Ij is the intensity of an individual reflection, and <I> is the average intensity of that reflection.
Rwork = ∑||Fo| − |Fc||/∑|Fc|, where Fo denotes the observed structure factor amplitude, and Fc denotes the structure factor amplitude calculated from the model.
Rfree is as for Rwork but calculated with 5.0% of randomly chosen reflections omitted from the refinement.
The diffraction data of the Dcp1p-Dcp2n complex were collected at beamline ID29 of ESRF (Grenoble, France). The structure was determined by molecular replacement with program PHASER (CCP4, 1994) using the structures of S. pombe Dcp2p (She et al., 2006) and the Dcp1p-Dcp2NTD complex as searching models. The initial model was built and modified manually with program O (Jones et al., 1991) and refined with CNS (Brunger et al., 1998) and Refmac5 (CCP4, 1994) to 2.8Å resolution. There are four Dcp1p-Dcp2n complexes and two ATP molecules in the final model, with a working R factor of 23.4% and a free R factor of 28.7% (Table 2). In the Dcp1p-Dcp2n complex, there are more disordered residues in the open complex than in the closed complex. Specifically, disordered residues are: Dcp1p, chain A:40–43; Dcp1p, chain C: 123–127; Dcp2n, chain D: 73–75, 189–195, 115–122, 207–219, 234–266; Dcp1p, chain E:40–43; Dcp2n, chain F: 211–214; Dcp2n, chain H: 73–76, 115–121, 176–195, 207–220, 236–266.
Surface Plasmon Resonance
Surface plasmon resonance was carried out on a BIACORE 2000 instrument. 20 µl of 100 nM 5’ biotin-labeled single-stranded RNA ligand (TGTCATTCGAGTACAGTCTGTTCAGCTAGTCTCC, from CureVac) was captured on the streptavidin-coated sensorchip SA (Biacore). After ligand immobilization on the analyte cell, both the analyte and the reference cells were blocked with 100 µl of 1mg/ml biotin and washed with the RNA binding buffer (20 mM HEPES, pH7.0, 50 mM NaCl, 2mM MgCl2, 2mM DTT, 5% Glycerol and 0.005% v/v Tween20). Typically, 100 µl of 80 nM protein sample was injected across the chip. Binding proteins were removed by 1.5 M NaCl after each dissociation cycle. The data were fitted to a 1:1 binding mode with mass transfer effect using the BIAevaluation 3.1 software (BIAcore).
Decapping Assay
The preparation of P32-labeled capped mRNA is described as before (Zhang et al., 1999; She et al., 2006). The decapping buffer contained 50 mM Tris-HCl, pH 7.9, 30 mM (NH4)2SO4 and 1 mM MgCl2. Typically, 3 pmole wild type or mutant Dcp2n was assayed in a 13.5 µl reaction at 37°C for 30 minutes. For detection of the Dcp1p stimulation effect, 10 pmole Dcp1p was incubated with 3 pmole Dcp2n proteins in the reaction buffer on ice for 30 minutes prior to mRNA decapping.
SAXS data collection, analysis, and shape reconstruction
Dcp2n or Dcp1p-Dcp2n proteins were prepared in the storage buffer at 10, 5 and 2mg/ml concentrations. For measurements with ligands, 10mM ATP, 10mM m7GpppA or 5mM pAA were supplemented in the storage buffer respectively. The synchrotron radiation X-ray scattering data from solutions of the Dcp1-Dcp2 complex and Dcp2n were collected on the X33 camera of the EMBL on the storage ring DORIS III (DESY, Hamburg, Germany) (Roessle et al., 2007). Using a MAR345 image plate detector at a sample-detector distance of 2.7m, the range of momentum transfer 0.01 < s < 0.5 Å−1 was covered. To monitor for the radiation damage, two successive two-minute exposures of protein solutions were compared and no significant changes were observed. The data were processed using standard procedures and extrapolated to infinite dilution using PRIMUS (Konarev et al., 2003).
The forward scattering I(0) and the radius of gyration Rg were evaluated using the Guinier approximation (Guinier, 1939) assuming that at very small angles (s < 1.3/Rg) the intensity is represented as Iexp(s) = I(0) exp(−(sRg)2/3). These parameters were also computed using GNOM (Svergun, 1992), providing the pair distribution function of the particle p(r) and the maximum size Dmax. The molecular mass of the solutes was evaluated by comparison of the I(0) values with that from a reference solution of bovine serum albumin, and all constructs appeared monomeric. The scattering patterns from the high resolution models were calculated by CRYSOL (Svergun et al., 1995).
The low resolution ab initio models were constructed by using GASBOR (Svergun et al., 2001) representing the protein as an assembly of dummy residues. Starting from a random distribution the program employs simulated annealing to construct a model minimizing discrepancy:
| (1) |
where N is the number of experimental points, c is a scaling factor and Icalc(s) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively.
Repetitive GASBOR runs were performed yielding superimposable models, which were averaged to determine the common structural features using the programs DAMAVER (Volkov and Svergun, 2003) and SUPCOMB (Kozin and Svergun, 2001). The latter program aligns two arbitrary low or high resolution models represented by ensembles of points by minimizing a quantitative dissimilarity measure called normalized spatial discrepancy (NSD). Generally, NSD values close to unity indicate that the two models are similar. In the comparisons and Figure 5, most typical models as indicated by DAMAVER comparison, are presented.
Coordinates
The coordinates and structure-factor amplitudes for the Dcp1p-DcpNTD complex and the Dcp1p-Dcp2n complex have been deposited in the Protein Data Bank with accession codes 2QKL and 2QKM respectively.
Supplementary Material
Acknowledgements
We would like to thank the beamline scientists at ID29 (ESRF, France) for assistance and access to synchrotron radiation facilities. This work is financially supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research)(H.S), and by the Howard Hughes Medical Institute (R.P). A.R and D.S acknowledge financial support from the EU Framework 6 Programme (Design Study SAXIER, RIDS 011934).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LJ, Jarchau T, Oschkinat H, Walter U. EVH1 domains: structure, function and interactions. FEBS Lett. 2002;513:45–52. doi: 10.1016/s0014-5793(01)03291-4. [DOI] [PubMed] [Google Scholar]
- Beelman CA, Stevens A, Caponigro G, LaGrandeur TE, Hatfield L, Fortner DM, Parker R. An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature. 1996;382:642–646. doi: 10.1038/382642a0. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cao D, Parker R. Computational modeling of eukaryotic mRNA turnover. RNA. 2001;7:1192–1212. doi: 10.1017/s1355838201010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Mikhli C, Jiao X, Kiledjian M, Kunkel G, Davis RE. Dcp2 Decaps m2,2,7GpppN-capped RNAs, and its activity is sequence and context dependent. Mol Cell Biol. 2005;25:8779–8791. doi: 10.1128/MCB.25.20.8779-8791.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for Multiple Isomorphous Replacement and Multiwavelength Anomalous Diffraction method. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Guinier A. La diffraction des rayons X aux tres petits angles; application a l'etude de phenomenes ultramicroscopiques. Ann. Phys. (Paris) 1939;12:161–237. [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001;34:33–41. [Google Scholar]
- LaGrandeur TE, Parker R. Isolation and characterization of Dcp1p, the yeast mRNA decapping enzyme. EMBO J. 1998;17:1487–1496. doi: 10.1093/emboj/17.5.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S, Temme C, Wahle E. Messenger RNA turnover in eukaryotes: pathways and enzymes. Crit Rev Biochem Mol Biol. 2004;39:197–216. doi: 10.1080/10409230490513991. [DOI] [PubMed] [Google Scholar]
- Miller R, Gallo SM, Khalak HG, Weeks CM. SnB: crystal structure determination via shake-and-bake. J Appl Cryst. 1994;27:613–621. [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct. Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA. 2005;11:1640–1647. doi: 10.1261/rna.2191905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessle MW, Klaering R, Ristau U, Robrahn B, Jahn D, Gehrmann T, Konarev PV, Round A, Fiedler S, Hermes S, Svergun DI. Upgrade of the Small Angle X-ray scattering Beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 2007;40:s190–s194. [Google Scholar]
- Sakuno T, Araki Y, Ohya Y, Kofuji S, Takahashi S, Hoshino S, Katada T. Decapping reaction of mRNA requires Dcp1 in fission yeast: its characterization in different species from yeast to human. J Biochem (Tokyo) 2004;136:805–812. doi: 10.1093/jb/mvh190. [DOI] [PubMed] [Google Scholar]
- She M, Decker CJ, Chen N, Tumati S, Parker R, Song H. Crystal structure and functional analysis of Dcp2p from Schizosaccharomyces pombe. Nat Struct Mol Biol. 2006;13:63–70. doi: 10.1038/nsmb1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Sundramurthy K, Liu Y, Chen N, Parker R, Song H. Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat Struct. Mol Biol. 2004;11:249–256. doi: 10.1038/nsmb730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 2006;125:1095–1109. doi: 10.1016/j.cell.2006.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. Analysis of recombinant yeast decapping enzyme. RNA. 2003;9:231–238. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. Determination of the regularization parameter in indirect transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- Svergun DI, Barberato C, Koch MHJ. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995;28:768–773. [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MHJ. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Parker R. Analysis of P-Body Assembly in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2274–2287. doi: 10.1091/mbc.E07-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun S, Parker R. Analysis of mutations in the yeast mRNA decapping enzyme. Genetics. 1999;151:1273–1285. doi: 10.1093/genetics/151.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small angle scattering. J. Appl. Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yang JY, Niu QW, Chua NH. Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell. 2006;18:3386–3398. doi: 10.1105/tpc.106.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Williams CJ, Wormington M, Stevens A, Peltz SW. Monitoring mRNA decapping activity. Methods. 1999;17:46–51. doi: 10.1006/meth.1998.0706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.