

SUMMARY
miR-17~92, miR-106b~25 and miR-106a~363 belong to a family of highly conserved miRNA clusters. Amplification and overexpression of miR-17~92 is observed in human cancers, and its oncogenic properties have been confirmed in a mouse model of B cell lymphoma. Here we show that mice deficient for miR-17~92 die shortly after birth with lung hypoplasia and a ventricular septal defect. The miR-17~92 cluster is also essential for B cell development. Absence of miR-17~92 leads to increased levels of the pro-apoptotic protein Bim and inhibits B cell development at pro-B to pre-B transition. Furthermore, while ablation of miR-106b~25 or miR-106a~363 has no obvious phenotypic consequences, compound mutant embryos lacking both miR-106b~25 and miR-17~92 die at mid-gestation. These results provide key insights into the physiologic functions of this family of microRNAs, and suggest a link between the oncogenic properties of miR-17~92 and its functions during B lymphopoiesis and lung development.
INTRODUCTION
Vertebrate embryonic development is a carefully orchestrated process that requires execution of tightly regulated gene expression programs. MicroRNAs (miRNAs) constitute a class of small (21–24 nucleotide) non-coding RNAs that reduce translation and stability of target mRNAs through sequence-specific interaction with their 3’-untranslated region (Bagga et al., 2005; Bartel, 2004; Lim et al., 2005). It is becoming increasingly clear that miRNAs play a crucial role in vertebrate development, as attested by the early embryonic lethality of mice with defects in the miRNA biogenesis pathway (Abbott et al., 2005; Bernstein et al., 2003; Liu et al., 2004). MicroRNAs could act either by regulating a specific set of critical developmental genes or by reinforcing gene-expression programs through suppression of transcriptional noise (Bartel and Chen, 2004; Hornstein and Shomron, 2006; Yekta et al., 2004).
A close link exists between deregulation of developmental programs and tumorigenesis, and a growing body of evidence indicates that altered expression of miRNAs is involved in the pathogenesis of human cancers (Costinean et al., 2006; Croce and Calin, 2005; Esquela-Kerscher and Slack, 2006; Hammond, 2006; Johnson et al., 2005; Kumar et al., 2007; Lu et al., 2005; Mayr et al., 2007; Voorhoeve et al., 2006). In this context, studies in mouse and human cells have led to the identification of the miR-17~92 cluster (also known as Oncomir-1) as a potential oncogene. It consists of six miRNAs that are processed from a common precursor transcript and can be grouped in four families based on their “seed” sequence (Figure 1A and B). A role for miR-17~92 in the pathogenesis of human cancers is suggested by its frequent amplification and overexpression in diffuse large B cell lymphomas (Ota et al., 2004) and in other tumor types, including small cell lung cancer (Hayashita et al., 2005; Matsubara et al., 2007). Additionally, miR-17~92 has been shown to cooperate with c-Myc in a mouse model of B cell lymphoma (He et al., 2005) and to stimulate proliferation of the lung epithelium (Lu et al., 2007). Further evidence indicates that miR-17~92 can also affect tumor angiogenesis (Dews et al., 2006) and the expression of members of the E2F family of oncogenic transcription factors (O'Donnell et al., 2005; Sylvestre et al., 2006; Woods et al., 2006). Very little is known, however, regarding the physiologic functions of this cluster in vertebrates.
Figure 1. Generation of targeted deletions in miR-17~92, miR-106b~25 and miR-106a~363.
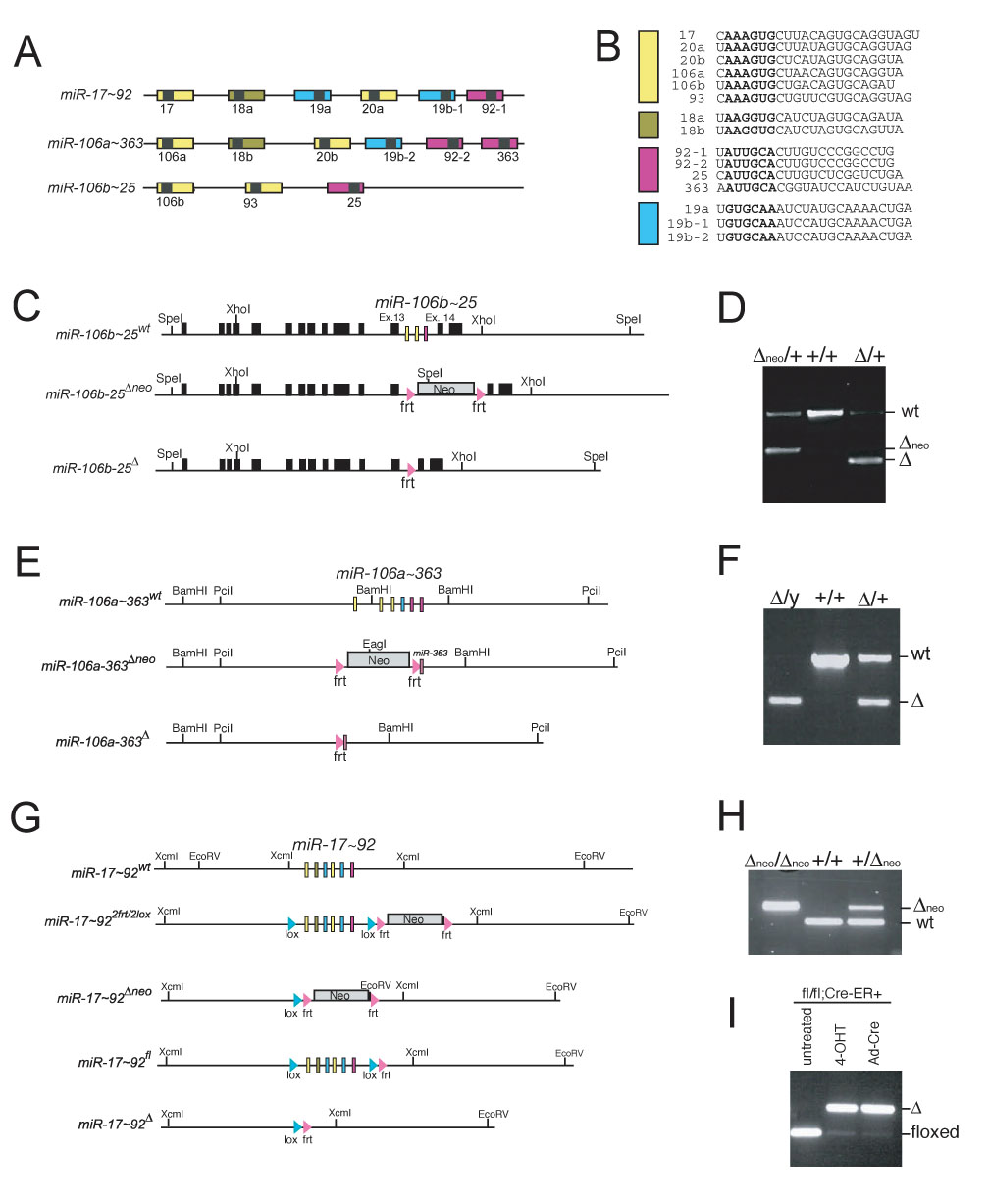
(A) Schematic representation of the three miRNA clusters. Pre-miRNAs are indicated as color-coded boxes. Black boxes correspond to the mature miRNA. The color code identifies miRNAs with the same seed sequence. (B) Sequence comparison of miRNAs expressed from the three miRNA clusters. MiRNAs with the same seed sequence (bold) are grouped together and color-coded according to panel (A). (C) Targeting scheme for the deletion of miR-106b~25. Mcm-7 exons are represented by black boxes. The mature miRNAs are represented by colored boxes. (D) Genotyping PCR performed on tail DNA showing germline transmission of the targeted miR-106b~25 alleles. (E) Targeting scheme for the deletion of miR-106a~363. (F) Genotyping PCR performed on tail DNA showing germline transmission of the targeted miR-106a~363 allele. (G) Targeting scheme for the deletion of miR-17~92. (H) Genotyping PCR performed on DNA extracted from E13.5 embryos showing germline transmission of the miR-17~92-Δneo allele. (I) Genotyping PCR performed on DNA extracted from miR-17~92fl/fl;Cre-ERT2+ mouse embryo fibroblasts. Cre-mediated deletion of miR-17~92 was assessed 5 days after infection with Cre-expressing recombinant Adenoviruses (Ad-Cre) or after treatment with 4-Hydroxytamoxifen.
The functional analysis of miR-17~92 is complicated by the existence of two paralogs: miR-106a~363 and miR-106b~25. It appears likely that the three clusters originated via a series of duplication and deletion events during early vertebrate evolution (Tanzer and Stadler, 2004). The miR-106a~363 locus maps to chromosome X in humans and mice and consists of six miRNAs (Figure 1A). The miR-106b~25 cluster is located on mouse chromosome 5 (chromosome 7 in humans) in the 13th intron of the DNA replication gene Mcm7 and consists of three miRNAs (Figure 1A). Because miR-106a~363 and miR-106b~25 contain miRNAs that are highly similar, and in some cases identical, to those encoded by miR-17~92 (Figure 1B), it is possible that they regulate a similar set of genes and have overlapping functions. Here we report the generation and initial characterization of mice deficient for each of these three miRNA clusters.
RESULTS
Expression pattern of miR-17~92 and its two paralogs
To determine the expression patterns of the miR-17~92, miR-106b~25 and miR-106a~363 clusters we performed RNAse protection assays (RPA) in a panel of murine tissues and cells. This technique was preferred to Northern blots because it has higher sensitivity and is capable of discriminating between similar miRNAs (Figure 2B, 2G and Figure S1). These experiments revealed that members of the miR-17~92 and miR-106b~25 clusters have similar expression pattern in the adult mouse, being detectable in most tissues (Figure S1). In contrast, we have not been able to detect expression of members of the miR-106a~363 cluster in the tissue tested (Figure S1). Expression of members of this cluster was undetectable by RPA even in murine embryonic stem (ES) cells and in mid-gestation embryos, where miR-17~92 and miR-106b~25 are abundantly expressed (Figure S1 and data not shown).
Figure 2. Deletion of miR-106a~363 or miR-106b~25 results in viable and fertile mice.
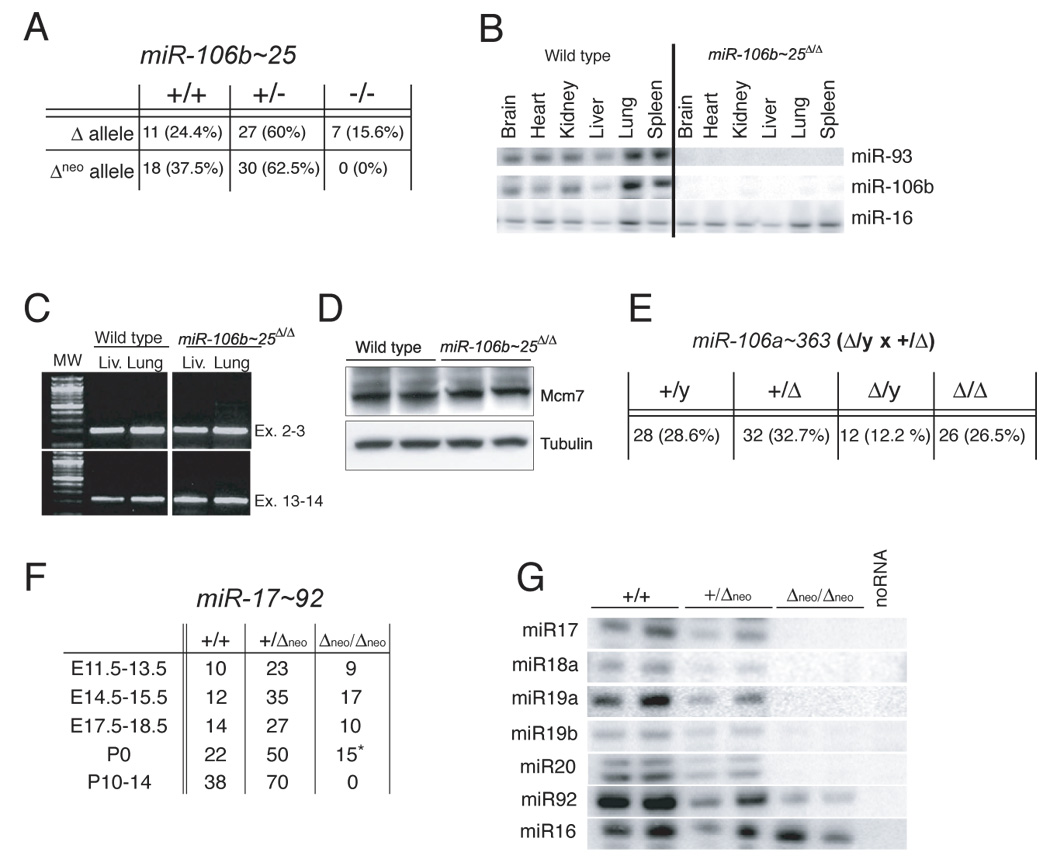
(A) Genotypes of mice from miR-106b~25+/Δneo and miR-106b~25+/Δ intercrosses. (B) RPAs on total RNA extracted from wild-type and miR-106b~25Δ/Δ adult tissues. (C) RT-PCR on RNA extracted from miR-106b~25Δ/Δ and wild-type lungs and livers. Primers designed to amplify the junctions of exons 13–14 were used to determine whether the deletion of miR-106b~25 from intron 13 affects splicing between these two exons. Amplification of the junction between exons 2 and 3 was used as control. (D) Mcm-7 Western blot on whole cell lysates from wild type and miR-106b~25 Δneo/Δneo mouse embryo fibroblasts. (E) Genotypes of mice from miR-106a~363Δ/Y × miR-106a~363+/Δ crosses. (F) Genotypes of mice from miR-17~92+/Δneo intercrosses determined at the indicated gestation stages. The asterisk indicates that all miR-17~92Δneo/Δneo newborn mice died soon after birth. (G) RPAs on total RNA from E13.5 embryos with the indicated genotypes.
Generation and characterization of miR-106b~25 deficient mice
The miR-106b~25 cluster is located in the 13th intron of the protein coding gene Mcm7. To reduce the risk of interfering with transcription and processing of the surrounding gene, the targeting construct was designed to replace the miR-106b~25 cluster with a neomycin-resistance (Neo) cassette flanked by frt sites (miR-106b~25Δneo) (Figure 1C). This strategy allowed the subsequent removal of the Neo cassette by crossing the chimeric animals to flpe-expressing transgenic mice (Rodriguez et al., 2000), thus generating the miR-106b~25Δ allele (Figure 1C). MiR-106b~25+/Δ and miR-106b~25Δ/Δ mice were viable, fertile and showed no obvious abnormalities (Figure 2A). In contrast, we did not obtain any mutant animal homozygous for the miR-106b~25Δneo allele (Figure 2A). This observation suggests that if left in place the Neo cassette interferes with the expression of Mcm7, and highlights the importance of carefully designing the targeting strategy for intronic miRNAs. All results presented hereafter derive from the use of the miR-106b~25Δ allele. Successful deletion of miR-106b~25 was confirmed by RPA and qPCR performed on total RNA extracted from wild-type and mutant mid-gestation embryos and adult tissues (Figure 2B and data not shown). Importantly, the expression and splicing of Mcm7 RNA was identical in wild-type and miR-106b~25Δ/Δ animals (Figure 2C, D).
Generation and characterization of miR-106a~363-deficient mice
A similar strategy was employed to delete miR-106a~363 on the X chromosome (Figure 1E, F). This allele was generated before the identification of miR-363 as a member of the cluster and, as such, part of this miRNA is still present in the targeted allele. However, because the 5’ flanking region of miR-363 is deleted, it is unlikely that this miRNA will be successfully processed from the targeted allele. Unfortunately, because miR-106a~363 expression is undetectable by either RPA or qPCR we have not been able to verify whether this is indeed the case.
Mice lacking the miR-106a~363 cluster were viable, reproduced efficiently and had no obvious abnormalities (Figure 2E). Mice homozygous or hemizygous for the miR-106a~363Δ or the miR-106a~363Δneo alleles were phenotypically indistinguishable (data not shown).
Generation of miR-17~92-deficient mice
To delete miR-17~92, a construct was designed in which this cluster is flanked by loxP sites and followed by a frt-Neo-frt cassette (Figure 1G, miR-17~92 2f/2l). After successful homologous recombination in ES cells and germ line transmission of the targeted allele, the cluster was deleted in vivo by crossing to a Cre-deletor mouse strain in which the Cre-recombinase is expressed under the control of the human beta actin promoter (Lewandoski et al., 1997). The resulting allele will be referred to hereafter as miR-17~92 Δneo (Figure 1G, H). An additional allele, miR-17~92Δ (Figure 1G, H), was generated by removing the Neo-cassette from the miR-17~92 Δneo allele via flpe-mediated recombination. Mice mutant for the miR-17~92Δneo or the miR-17~92Δ allele were phenotypically indistinguishable. This targeting strategy also allowed the generation of a conditional (“floxed”) miR-17~92 allele (miR-17~92fl; Figure 1G, I) via flpe-mediated deletion of the Neo cassette from miR-17~922frt/2lox mice. In mice and cells carrying the miR-17~92fl allele, Cre-expression leads to efficient deletion of miR-17~92 (Figure 1I and Supplementary Figure 2). As predicted, miR-17~92fl/fl mice were born at the expected Mendelian ratio, showed no obvious abnormalities (data not shown), and expressed normal levels of miR-17~92 (Supplementary Figure 2 and data not shown).
Early postnatal lethality of miR-17~92-deficient mice
MiR-17~92+/Δneo mice were born at the expected Mendelian ratio, were viable, fertile and only slightly smaller than their wild-type littermates (Figure 2F, Figure 3B and data not shown). In contrast, intercrosses between miR-17~92+/Δneo mice failed to produce viable miR-17~92 Δneo/Δneo animals. Viable miR-17~92 Δneo/Δneo embryos were recovered throughout gestation at about the expected Mendelian frequency and the majority of them reached birth (Figure 2F). However, miR-17~92 Δneo/Δneo newborns invariably died within minutes after birth, and no miR-17~92 deficient mice were observed at later time points (Figure 2F).
Figure 3. Characterization of miR-17~92-deficient mice.
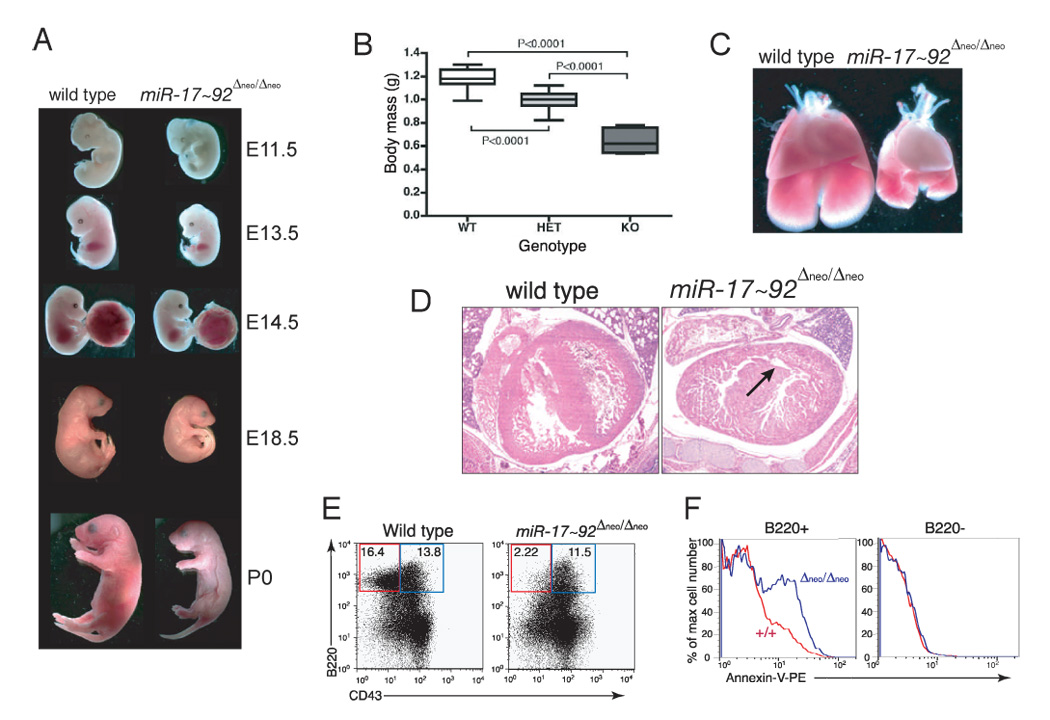
(A) Macroscopic appearance of wild-type and miR-17~92-deficient embryos at various developmental stages. (B) Body mass of wild-type (WT), heterozygous (HET) and miR-17~92-null (KO) E18.5 embryos. The boxes indicate the 25th–75th percentile range. The horizontal lines represent the median, and the error bars indicate the lowest and highest values. P values were calculated using the two-tailed t test. (C) Macroscopic appearance of wild-type and miR-17~92-deficient lungs and hearts from E17.5 embryos. Notice the markedly hypoplastic lungs in miR-17~92-null embryos. (D) Hematoxylin-eosin staining of comparable transverse sections through the hearts of E18.5 wild-type and miR-17~92-null embryos. The arrow indicates a ventricular septal defect. (E) Flow cytometry plots of fetal liver cells from E18.5 wild-type and mutant embryos showing a marked reduction of B220+;CD43−;IgM− pre-B cells in mice lacking miR-17~92. The results are representative of three independent experiments. (F) Annexin-V staining showing increased apoptosis in miR-17~92-deficient fetal liver B cell progenitors (blue) compared to wild-type control (red). The right panel shows the same analysis performed on B220-negative cells.
Deletion of miR-17~92 was verified by RPA and qPCR on total RNA from wild-type and mutant E13.5 embryos (Figure 2G and data not shown). As predicted, miR-17-5p, miR-18a, miR-20a, miR-19a and miR-19b-1 were undetectable in miR-17~92 Δneo/Δneo embryos (Figure 2G), and their expression in heterozygous mutant embryos was roughly halved compared to wild-type controls. Interestingly, the signal for miR-92 miRNA was greatly reduced, but not abolished, in the miR-17~92 Δneo/Δneo embryos. Because these embryos lack the entire miR-17~92 locus, this observation is most likely explained by hybridization of the probes to identical or nearly identical microRNAs expressed from one of the paralogs.
Lung hypoplasia and ventricular septal defect in miR-17~92-deficient mice
Macroscopically, homozygous mutant embryos were easily distinguished from wild-type littermates by embryonic day 13.5 (E13.5) because of their smaller size (Figure 3A). At E18.5, the weight of miR-17~92-null embryos was ~60% that of wild-type littermates (Figure 3B).
To determine the cause of the postnatal lethality, miR-17~92-null late-gestation embryos (E18.5-P0) were subjected to necropsy and histological examination. Macroscopically, the most significant phenotype in the miR-17~92-null embryos was the presence of severely hypoplastic lungs (Figure 3C). Despite the smaller size, histological examination did not reveal any specific branching defect or other obvious developmental abnormalities (data not shown). Interestingly, it has been recently reported that over-expression of miR-17~92 in the developing lung can increase proliferation and impair differentiation of the lung epithelium (Lu et al., 2007).
Although the hearts of miR17~92-deficient and wild-type embryos were of comparable size, mutant E18.5 embryos presented a clear ventricular septal defect (Figure 3D). This phenotype occurred in all mutant embryos (n=4) that were subjected to serial sectioning. Other organ systems examined, including the placenta, appeared histologically normal.
miR-17~92 is essential for fetal B cell development
The role of miR-17~92 in the pathogenesis of B cell lymphomas led us to investigate its function in normal B cell development. Mice deficient for miR-106a~363 or miR-106b~25, as well as miR-17~92 +/Δneo mice, had normal numbers of circulating B cells and bone marrow B cell progenitors (Figure S3). We next examined the effects of deletion of miR-17~92 on B cell development. Because by mid-gestation the liver is the primary hematopoietic organ in mice, we performed flow cytometry analysis of fetal liver cells from control and miR-17~92 Δneo/Δneo embryos. Although homozygous mutant fetal livers contained fewer total cells than controls at E14.5, the frequency of hematopoietic stem cells was comparable to wild-type controls and the frequency of early B cell progenitors was only slightly reduced (Figure S4). In contrast, at E18.5, miR-17~92-deficient fetal livers had a greatly reduced percentage and absolute number of pre-B cells (Figures 3E). This reduction in pre-B cells in the mutant embryos was associated with increased apoptosis, as measured by staining with Annexin-V (Figure 3F). Importantly, the increase in apoptosis was specific to the B cell compartment, since levels of apoptosis in non-B cells was comparable in mutant and control fetal livers (Figure 3F). Higher levels of apoptosis were not detected in other organs of the embryos (data not shown).
miR-17~92 is essential for adult B cell development
To study adult B cell development and determine whether the defect in B cell development was cell autonomous, we reconstituted the hematopoietic system of lethally irradiated mice with fetal liver cells from wild-type or miR-17~92 Δneo/Δneo E14.5 embryos (Figure 4A). Eight to ten weeks after transplantation, donor red blood cells, granulocytes and monocytes were present at similar frequencies in the blood of mice reconstituted with wild-type or miR-17~92 Δneo/Δneo hematopoietic cells (Figure 4B, 4C). Similar numbers of granulocytes and monocytes were also present in the spleens and bone marrows from both groups of recipient mice (Figure S5). In contrast, the number of circulating lymphocytes was greatly reduced in mice reconstituted with miR-17~92-deficient hematopoietic cells due to a significant and specific reduction of circulating B cells (Figure 4C–F). The percentage and absolute number of splenic B cells were also significantly reduced in mice reconstituted with miR-17~92-deficient hematopoietic cells (Figure 4G). Interestingly, in all mice, splenic B cells had a comparable cell surface phenotype and we observed similar relative (but not absolute) numbers of transitional and mature B cells (Figure 4H). Splenic marginal zone, follicular, and newly-formed B cells, as well as peritoneal B1a and B1b cells, were all reduced by miR-17~92-deficiency (Figure S6).
Figure 4. Deletion of miR-17~92 impairs adult B cell development.
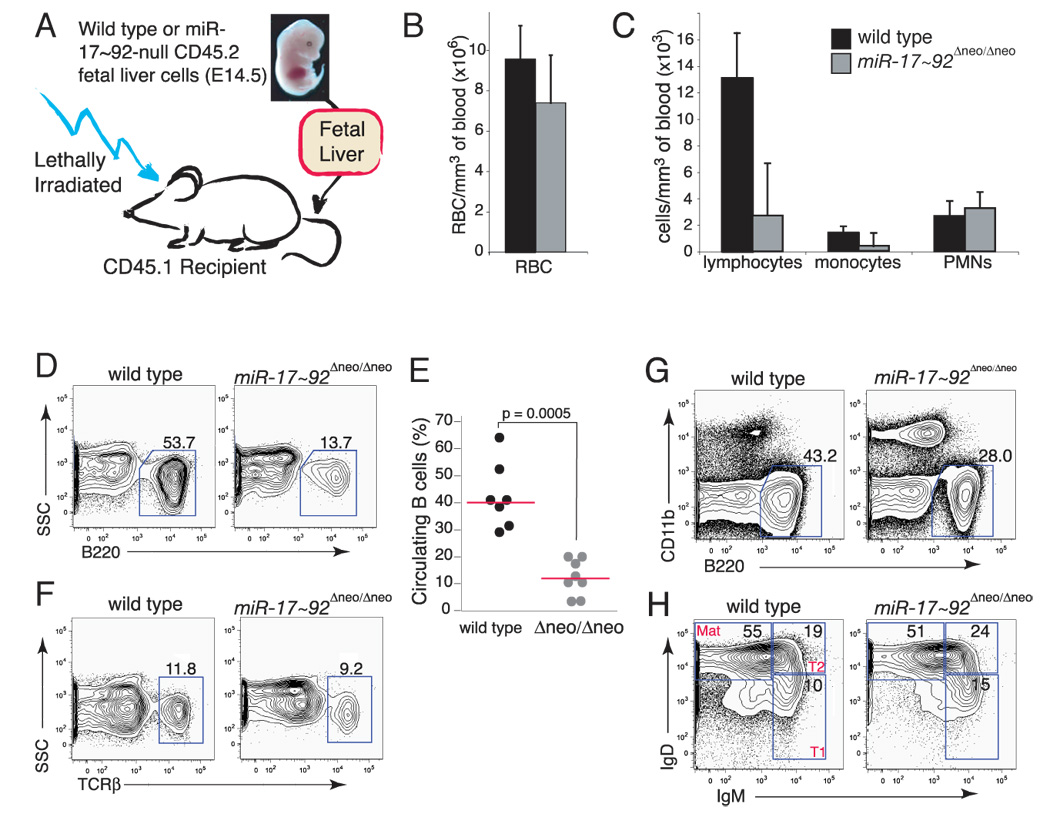
(A) Schematic of the reconstitution experiment. (B) Total number of circulating RBC per mm3 of blood in mice reconstituted with wild-type (black) or miR-17~92-deficient (gray) fetal livers 8 weeks post transplantation. Error bar indicates +1 S.D. The average of seven wild-type and eight miR-17~92-deficient mice is shown. (C) Differential white blood cells counts in mice reconstituted with wild-type (black) and miR-17~92-deficient (gray) fetal livers. PMNs: polymorphonucleates. Error bar is +1 S.D. n=7 wild-type and 8 miR-17~92 mutants. (D) Representative flow cytometry plot showing reduction of B220+ B cells in the blood of mice reconstituted with miR-17~92-deficient fetal liver cells. (E) Dot plot showing the number of circulating B cells in reconstituted mice. Horizontal bars in red indicate the median value for each genotype. (F) Flow cytometry plot of T cells in the periphery of reconstituted mice. On representative example per genotype is shown. (G) Flow cytometry plot of spleen cells stained for CD11b and B220, showing a reduction in splenic B cells in mice reconstituted with miR-17~92-deficient cells. (H) Flow cytometry plot on B-220-positive splenic B cells stained with IgM and IgD to determine the relative fraction of Transition (T1 and T2) and Mature B cells.
miR-17~92 regulates B cell survival
To characterize the effect of miR-17~92 deficiency on early B cell development more fully, we analyzed the bone marrow from mice reconstituted with mutant or wild-type cells. B cells develop through well-characterized stages that are distinguished by expression of specific cell surface markers (Fractions A to F, (Hardy et al., 1991)). B220+CD43+ pro-B cells (Fr. A through C) were only slightly reduced in the bone marrow of mice transplanted with miR-17~92-deficient fetal liver cells (Figure 5A and B). The subsets of pro-B cells were also largely unaffected by the absence of miR-17~92 (Figure 5C). In contrast, the percentages of pre-B cells (B220+CD43−IgM−) and later stages (Fraction E and F) were substantially reduced in mice transplanted with miR-17~92-null fetal liver cells (Figure 5A and B).
Figure 5. miR-17~92 regulates early B cell survival.
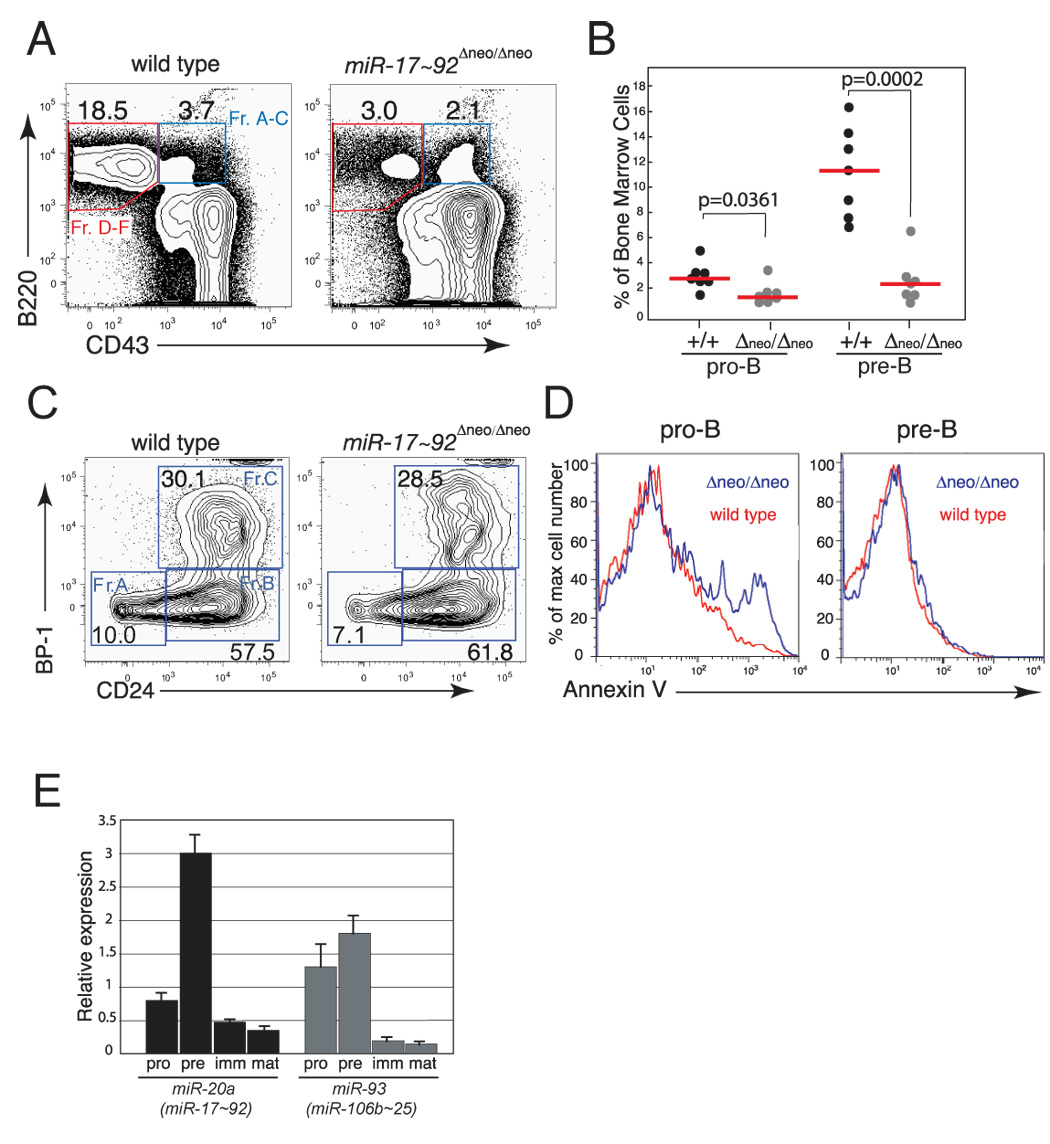
(A) Representative flow cytometry plots of bone marrow cells from mice reconstituted with wild-type or miR-17~92-deficient fetal liver cells. (B) Percentage of pro-B and pre-B cells in reconstituted mice. The median (red bars) and the p-values are indicated. (C) Representative flow cytometry plots showing relative proportion of fraction A–C pro-B cells in the bone marrow of reconstituted mice. (D) Histogram overlays of Annexin-V staining of pro-B (left panel) and pre-B cells (right panel), showing increased apoptosis of pro-B cells in the bone marrow of mice reconstituted with miR-17~92-deficient fetal liver cells. The analysis was performed on the bone marrow of mice reconstituted with wild-type (red line) or miR-17~92-deficient (blue line) fetal livers. (E) qPCR analysis of miR-17~92 and miR-106b~25 expression during B cell development. Pro-, pre-, immature and mature B cells were sorted and their RNA assayed for miR-20a and miR-93 expression. Data are normalized to sno-142 expression.
To confirm the role of miR-17~92 in early B cell development, we took advantage of the conditional miR-17~92 allele. To achieve conditional ablation of miR-17~92 in hematopoietic cells in vivo, miR-17~92fl/fl mice were crossed to Mx-Cre transgenic mice (Kuhn et al., 1995). In these mice the Cre transgene is expressed under the control of the inducible Mx1 promoter, which can be activated in the hematopoietic compartment in response to treatment with polyinosinic-polycytidylic acid (pI:pC) double-stranded RNA. Two-month-old mice were injected intra-peritoneally (i.p.) with pI:pC and examined 4 weeks later. As shown in Figure S2, efficient deletion of miR-17~92 was accompanied by a marked reduction of pre-B cells in the bone marrow of miR-17~92fl/fl;Mx-Cre-positive mice but not in control animals.
The reduced number of pre-B cells could be due to either enhanced cell death or to reduced proliferation of miR-17~92-deficient pro- or pre-B cells. Cell cycle analysis of the various stages of B cell development showed no differences in the cell cycle profiles of pro- and pre-B cells (Figure S7). In contrast, pro-B cell death was increased in the absence of miR-17~92, as determined by DAPI-exclusion and Annexin-V staining (Figure 5D and data not shown). The increase in apoptosis was specific to the early stages of B cell development, as the viability of immature B cells and mature B cells in the bone marrow was not significantly affected by the deletion of miR-17~92 (Figure 5D and data not shown). Analysis of immunoglobulin heavy chain rearrangement in control and miR-17~92-null pro-B cells showed that DH to JH recombination occurs normally, while the next step (VH to DJH rearrangement) is somewhat less efficient than in controls (data not shown). These results indicate that miR-17~92 plays a critical role in promoting the survival of early B cell progenitors. Consistent with this interpretation, the expression of miRNAs belonging to the miR-17~92 cluster appears to be tightly regulated during B cell development, reaching a maximum at the pre-B cell stage and then rapidly returning to basal levels at later stages (Figure 5E).
Regulation of the pro-apoptotic protein Bim by miR-17~92
The data presented above suggest the possibility that members of the miR-17~92 cluster regulate survival of early B cell progenitors by repressing the expression of pro-apoptotic genes at the pro- to pre-B cell transition. We therefore surveyed the list of putative miR-17~92 target genes predicted by the TargetScan software (http://www.targetscan.org/) to identify genes that could mediate this phenotype. TargetScan predicts miRNA targets based on a series of criteria that include sequence complementarity between the miRNA and the 3’UTR, evolutionary conservation, position of the binding site within the 3’UTR and local “AU” content (Grimson et al., 2007; Lewis et al., 2003). Several known pro-apoptotic genes, including PTEN, E2F1 and Bcl2l11/Bim are predicted targets of miR-17~92. PTEN and E2F1 have been previously validated (Novotny et al., 2007; O'Donnell et al., 2005). Bim is a particularly attractive candidate target of miR-17~92 given its role in controlling lymphocyte apoptosis and in suppressing Myc-induced B cell lymphomagenesis (Bouillet et al., 1999; Bouillet et al., 2002; Egle et al., 2004; Hemann et al., 2005; O'Connor et al., 1998).
The 3’-UTR of Bim contains four predicted binding sites for members of the miR-17~92 cluster and its paralogs (Figure 6A). Two binding sites for miR-19 and miR-92 that are located 42 nucleotides apart in the distal portion of the Bim 3’UTR (Figure 6A) are of particular interest, as it has been shown that closely spaced sites often act synergistically (Grimson et al., 2007). To determine whether Bim expression is modulated by miR-17~92, we examined its expression in miR-17~92-null animals. A significant increase of Bim protein levels compared to controls was observed in miR-17~92-deficient pro- and pre-B cells from fetal livers (Figure 6B). Similarly, we detected higher Bim levels in adult pre-B cells following acute deletion of miR-17~92 in hematopoietic cells in vivo (Figure 6C). Bim up-regulation was also observed in the lungs of miR-17~92-null late gestation embryos (Figure 6D). Next we cloned a fragment of the Bim 3’UTR spanning the closely spaced miR-19/miR-92 binding sites downstream of the Renilla luciferase coding sequence, and compared its activity in HeLa cells to an equivalent construct where the miR-19/miR-92 seed sites were mutated. These experiments demonstrated that the presence of the two binding sites in the 3’UTR confers significant and reproducible repression (Figure 6E). Together these results indicate that Bim is a direct target of miR-17~92, and suggest a possible mechanism through which deletion or overexpression of miR-17~92 affects B cell development and lymphomagenesis.
Figure 6. Bim regulation by miR-17~92.
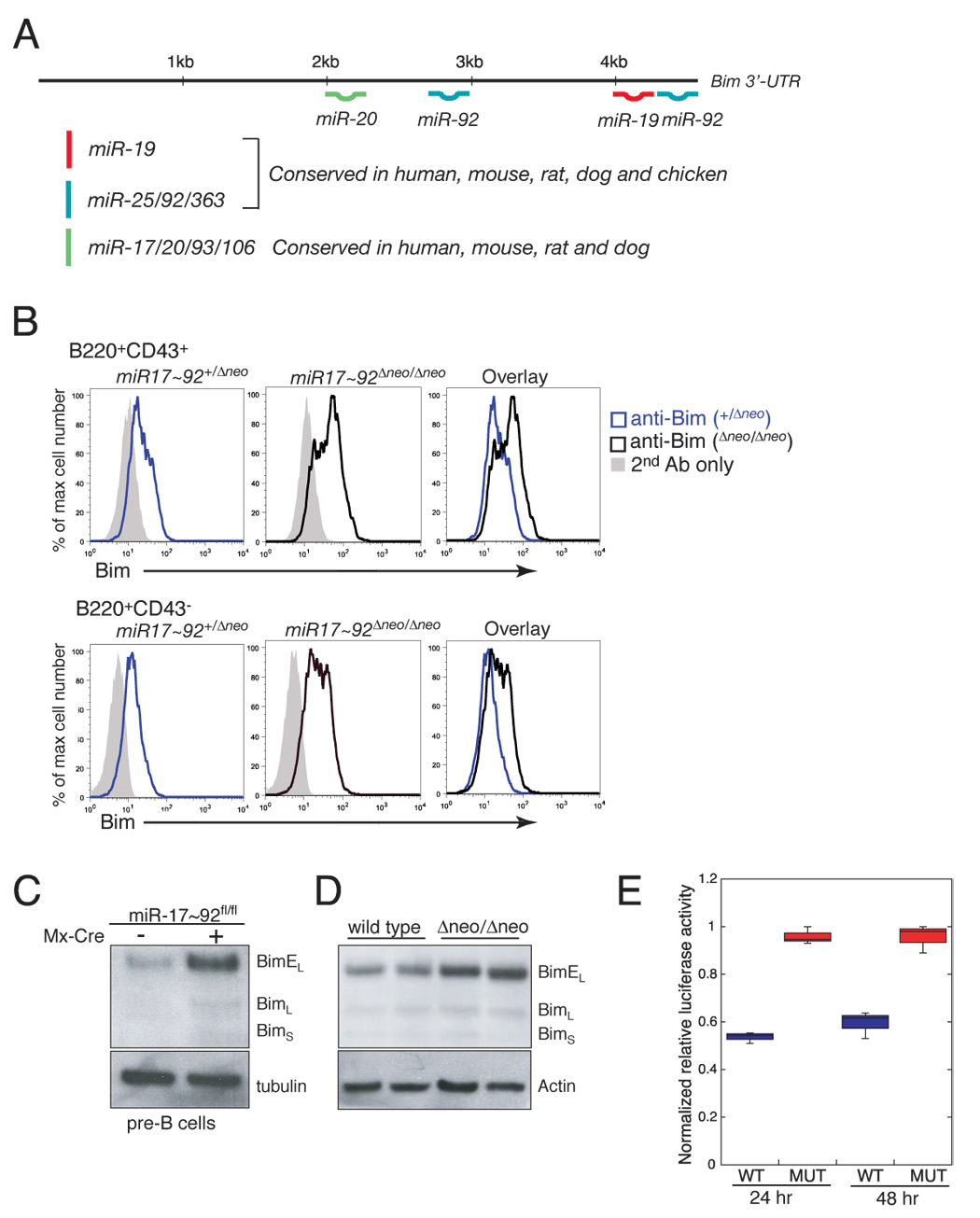
(A) Schematic representation of the 3’UTR of Bim and the predicted binding sites for members of the miR-17~92 cluster and its paralogs. (B) Detection of Bim expression by flow cytometry in miR-17~92+/Δneo and miR-17~92Δneo/Δneo pro-B (upper panels) and pre-B cells (lower panel) from E18.5 fetal livers. (C) Detection of Bim expression by western blotting in pre-B cells from adult mice following conditional deletion of miR-17~92. miR-17~92fl/fl (left lane) and miR-17~92fl/fl;Mx-Cre mice (right lane) were treated with pI:pC to induce expression of the Cre transgene. Pre-B cells were sorted and analyzed by Western blotting four weeks after treatment. (D) Detection of Bim expression by western blotting in E18.5 lungs from wild-type and miR-17~92Δneo/Δneo embryos. (E) Box plot of normalized relative luciferase activity of wildtype and mutant Bim UTR constructs 24 and 48 hours after transfection in HeLa cells. Data represent three independent experiments performed in triplicate.
Functional interaction between miR-17~92 and miR-106b~25
The occurrence of multiple, highly similar miRNAs is common in animal genomes and raises the question of functional redundancy and compensation. Given the similarity in sequence and expression pattern among miRNAs expressed from miR-17~92 and miR-106b~25 (Figure 1B, Figure S1 and Figure 5E), we decided to investigate the consequences of simultaneous deletion of these miRNA clusters in the mouse. Intercrosses between double heterozygous animals (miR-17~92+/Δ;miR-106b~25+/Δ) led to the generation of viable miR-17~92Δ/+;miR-106b~25Δ/Δ mice that could not be distinguished from wild-type controls. In similar experiments including the miR-106a~363Δ allele, we also obtained viable and fertile miR-17~92Δ/+;miR-106b~25Δ/Δ;miR-106a~363Δ/y mice, thus demonstrating that a single wild-type miR-17~92 allele is compatible with normal mouse development even in the absence of the two paralog clusters. In contrast, neither double knockout (miR-17~92Δ/Δ;miR-106b~25Δ/Δ, hereafter indicated as DKO) nor triple knockout (miR-17~92Δ/Δ;miR-106b~25Δ/Δ;miR-106a~363Δ/y, hereafter referred to as TKO) animals were recovered at birth. Timed-mating experiments revealed that DKO and TKO embryos die before embryonic day 15 and exhibit a much more severe phenotype compared to embryos lacking miR-17~92 alone (Figure 7A). Macroscopic and microscopic examination of E13.5 and E14.5 DKO embryos revealed edema and vascular congestion (n=3). This was associated with severe cardiac developmental abnormalities, consisting of defective ventricular and atrial septation and thinner ventricles (Figure 7B). In addition, we observed areas of intense staining for cleaved caspase 3, a marker of apoptosis, in the fetal liver, the ventral horns of the spinal cord, and the lateral ganglionic eminences of DKO and TKO embryos, but not in controls or in miR-17~92Δneo/Δneo embryos (Figure 7C and data not shown). These results provide genetic evidence that the miR-17~92 and miR-106b~25 clusters functionally cooperate in regulating embryonic development and apoptosis in the mouse.
Figure 7. Functional cooperation between miR-17~92 and miR-106b~25.
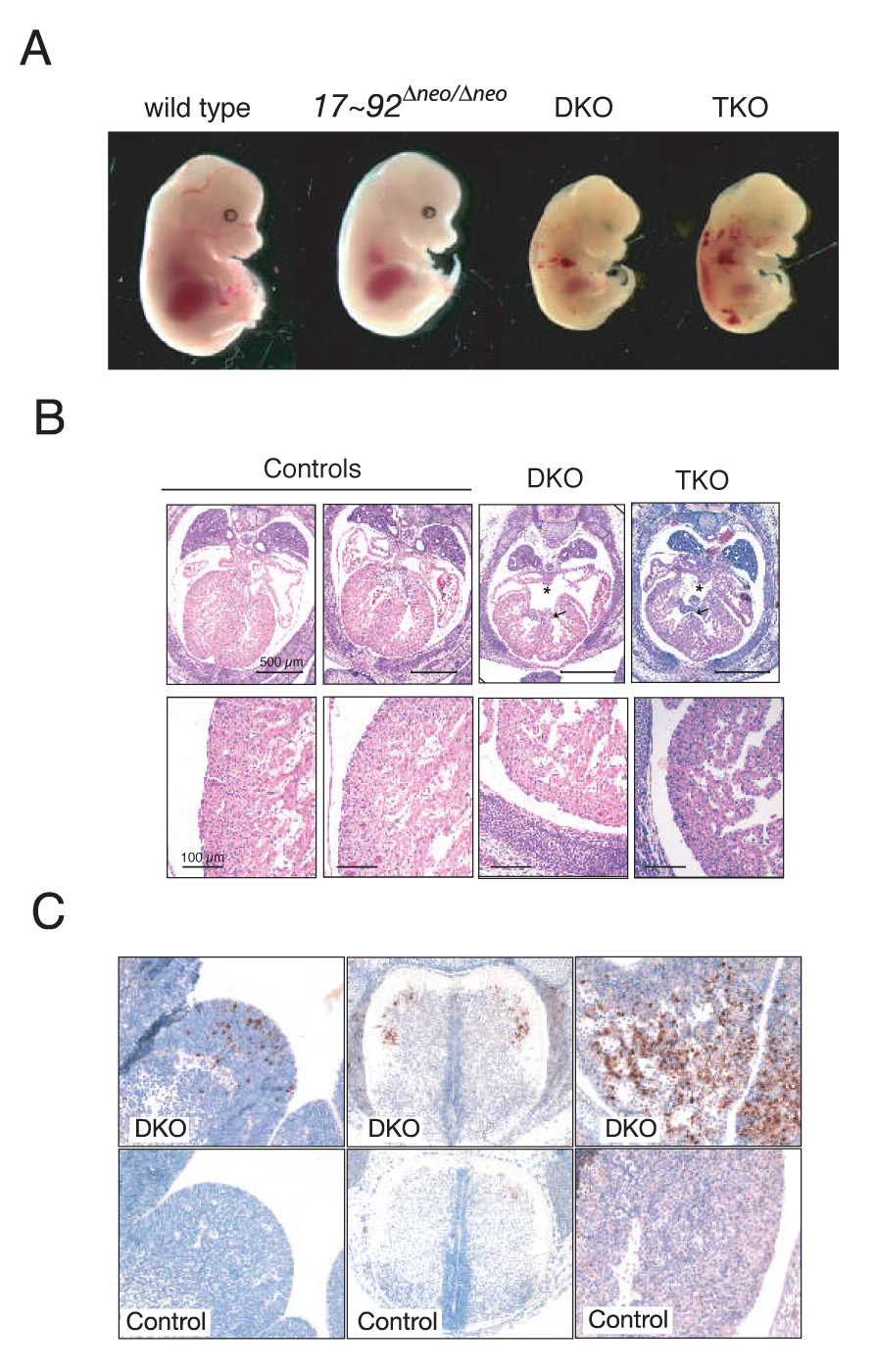
(A) Effects of compound mutations of miR-17~92, miR-106b~25 and miR-106a~363 on embryonic development. E14.5 embryos with the indicated genotypes are shown. DKO = miR-17~92Δ/Δ;miR-106b~25Δ/Δ; TKO = miR-17~92Δ/Δ;miR-106b~25Δ/Δ;miR-106A~363Δ/y (B) Representative transverse sections of E14.5 mice showing the presence of ventricular (arrow) and atrial (asterisk) septal defects in DKO and TKO embryos (upper panels). Lower panels show a higher magnification of the lateral wall of the left ventricle. Notice the thinner wall in double and triple mutant embryos. The black bars correspond to 500 µm (upper panels) and 100 µm (lower panels). The controls were a miR-17~92+/+;miR-106b~25+/Δ;miR-106a~363Δ/y (left) and a miR-17~92+/Δ;miR-106b~25+/Δ;miR-106a~363wt (right) mouse. (C) Cleaved caspase 3 immunostaining of sections from an E14.5 DKO miR-17~92Δ/Δ;miR-106b~25Δ/Δ (DKO) embryo, and a miR-17~92+/+; miR-106b~25Δ/Δ (control) embryo showing the presence of areas of apoptosis in the liver (right panels), the ventral horns of the spinal cord (middle), and the lateral ganglionic eminence (left) in the DKO.
DISCUSSION
In the present study we have investigated the biological functions of the miR-17~92 cluster and its two paralogs: miR-106b~25 and miR-106a~363. We show that deletion of miR-17~92 leads to neonatal lethality and very specific defects in the development of heart, lungs and B cells. We also provide genetic evidence of functional cooperation between related miRNA clusters in the mouse by showing that concomitant deletion of miR-106b~25 and miR-17~92 results in severe cardiac defects, increased apoptosis and embryonic death by mid-gestation.
Role of miR-17~92 in heart and lung development
Cardiac development is a carefully regulated process that is extremely susceptible to even small perturbations, as attested by the high frequency of congenital cardiac malformations in humans and in genetically-modified mice (Chen and Fishman, 2000; Hoffman et al., 2004). In mice, closure of the ventricular septum is normally completed by gestation day 14.5 (Webb et al., 1998) and several mutations are known to interfere with this process (Chen and Fishman, 2000), including deletion of miR-1-2 (Zhao et al., 2007). A defective ventricular septum can be sufficient to cause postnatal lethality, although mice can sometimes reach adulthood (Sakata et al., 2002). However, in the case of miR-17~92 deletion, it is likely that the associated severe lung hypoplasia significantly contributes to the complete penetrance of neonatal lethality.
The mechanism leading to lung hypoplasia in response to miR-17~92 deletion is presently unclear. Over-expression and amplification of miR-17~92 has been reported in lung cancers (Hayashita et al., 2005), and antisense inhibition of miR-17 and miR-20 leads to increased apoptosis in lung cancer cell lines (Matsubara et al., 2007). It is tempting to speculate that this cluster plays an important role in the proliferation or survival of normal lung cells. Consistent with this hypothesis is the recent finding that forced expression of miR-17~92 under the control of a lung-specific promoter leads to hyper-proliferation and blocks differentiation of the lung epithelium in vivo (Lu et al., 2007).
Role of miR-17~92 in normal B cell development and lymphomagenesis
Pro-B cells are the committed B cell progenitors in the bone marrow and fetal liver. Productive V-DJ rearrangement of the immunoglobulin heavy chain leads to the assembly of the pre-B cell receptor (pre-BCR) and signaling via the pre-BCR allows progression from the pro-B to the pre-B cell stage (Martensson et al., 2007; Hardy and Hayakawa, 2001). After rearrangement of the immunoglobulin light chains, the pre-BCR is replaced by the B cell receptor (BCR), an event that marks the transition to immature B cells and eventually to mature B cells. Signaling through the pre-BCR first, and the BCR later, appears to be required for the survival of B cell precursors and for their progression through the various stages.
Our results suggest that miR-17~92 promotes, in a cell-autonomous manner, survival of early B cell progenitors. A specific role for this miRNA cluster in cell survival at the pre-B cell checkpoint is also suggested by our finding that its expression is tightly regulated during B cell development and it is induced in pre-B cells. Of note, Koralov and colleagues have demonstrated that deletion of the miRNA processing enzyme Dicer in lymphocyte progenitors leads to an arrest at the pro-B to pre-B transition that is highly reminiscent of what we observe in miR-17~92-deficient mice (Koralov et al., accompanying manuscript).
Although additional work is required to elucidate the detailed molecular mechanisms, our results suggest a model for miR-17~92 function in B cell development and in lymphomagenesis. We propose that miR-17~92 expression is induced at the pro-B to pre-B transition where it acts to antagonize the expression of Bim, and possibly additional pro-apoptotic genes. We also propose that in the event of over-expression or amplification of the miR-17~92 locus, as it occurs in human B cell lymphomas, inappropriate suppression of the same pro-apoptotic gene(s) facilitates transformation by concomitant oncogenic events. This model is consistent with the previously published observation that forced over-expression of miR-17~92 cooperates with the c-myc oncogene to induce B cell lymphomas by reducing the degree of apoptosis of lymphoma cells (He et al., 2005). One prediction of this model with potential therapeutic implications is that inhibition of miR-17~92 expression in human diffuse large B cell lymphomas, and possibly in other tumor types, will lead to cell death.
Functional cooperation between miR-17~92 and miR-106b~25
Given their sequence similarity, these three miRNA clusters are predicted to have a highly overlapping set of targets (Lewis et al., 2005; Lewis et al., 2003; Sethupathy et al., 2006). However, the results presented in this work demonstrate that only miR-17~92 is required for normal mouse development, while miR-106b~25 and miR-106a~363 are dispensable. A function for miR-106b~25 is revealed only in the context of miR-17~92 loss, as shown by our analysis of DKO and TKO embryos. Furthermore, our preliminary results using the conditional miR-17~92 allele, show that deletion of a single allele of miR-106b~25 synergizes with miR-17~92 deletion in causing a more severe defect in B cell development (data not shown). Strikingly, the DKO and TKO embryos not only exhibit increased severity of the defects observed in miR-17~92-null embryos, but also display additional defects, in particular apoptosis in specific regions of the central nervous system and in the liver.
There are several possible explanations for these observations. For example, expression of these three clusters could be spatially and temporally segregated. This is probably the case for miR-106a~363, that appears to be expressed at much lower levels compared to the other two clusters. However, miR-106b~25 and miR-17~92 are very similar in terms of both tissue distribution and expression levels. One potentially relevant difference between these two clusters is that miR-17~92, but not miR-106b~25, expresses members of the miR-19 and miR-18 families (Figure 1A, B). It is tempting to speculate that loss of miR-19a, miR-19b and miR-18 is largely responsible for the phenotype caused by deletion of miR-17~92.
Bim and regulation of cell death by miR-17~92 and its paralogs
It is of great interest to determine the identity of the genes regulated by miR-17~92 and its paralogs that are responsible for the specific defects observed in single and compound mutant animals. In principle, the complex phenotypes observed in the mutant neonates and embryos could be explained by the effects of a limited number of target mRNAs. Given the increased cell death observed in the B cell lineage in single miR-17~92 mutants and the more widespread apoptosis observed in miR-17~92;miR-106b~25 compound mutants, obvious candidates are proapoptotic genes that have predicted binding sites for microRNAs belonging to these clusters. Our observation that expression of the pro-apoptotic gene Bim is modulated by miR-17~92 is of particular interest in this context. Bim belongs to the BH3-only family of pro-apoptotic genes and its overexpression leads to apoptosis (Bouillet et al., 2002; O'Connor et al., 1998). Importantly, Bim is a crucial regulator of leukocyte homeostasis, as its deletion in mice leads to leukocytosis, with increased number of circulating B and T cells due to reduced apoptosis (Bouillet et al., 1999; Bouillet et al., 2002). Bim is also an important suppressor of Myc-induced B cell lymphomagenesis (Egle et al., 2004; Hemann et al., 2005), and its tumor suppressive activity is dosage-sensitive. Even deletion of a single Bim allele leads to accelerated Myc-induced lymphomagenesis without loss of the wild-type allele (Egle et al., 2004). Moreover, Koralov and colleagues have shown that that Bim deletion partially rescues the defective B cell development caused by Dicer (Koralov et al., accompanying manuscript). Future work will determine the extent to which Bim deregulation is responsible for the phenotypes caused by miR-17~92 deletion.
In conclusion, we have demonstrated both essential and overlapping functions for the miR-17~92 family of miRNA clusters. These results provide new insights into the regulation of critical developmental processes by miRNAs and indicate an additional level of control by this class of regulatory molecules in the form of functional overlap.
EXPERIMENTAL PROCEDURES
ES cell manipulation, generation of chimeras and tetraploid blastocyst complementation
V6.5 ES cells were cultivated on irradiated MEFs in DMEM containing 15% FBS, leukemia inhibiting factor, penicillin/streptomycin, L-glutamine and non-essential amino acids.
Targeting constructs were generated by either conventional subcloning using appropriate restriction enzymes (miR-17~92 and miR-106a~363) or by “recombineering” (Liu et al., 2003) (miR-106b~25). The constructs were electroporated in V6.5 embryonic stem cells and cells plated under selective conditions (2 µg/ml puromycin) 48 hours later. Individual clones were expanded and tested by Southern blot using probes external to the targeting region. Clones that had undergone successful homologous recombination were injected into diploid (miR-17~92 and miR-106b~25 targeting) or tetraploid blastocysts (miR-106a~363 targeting) as previously described (Eggan et al., 2001). Deletion of the floxed miR-17~92 allele was obtained by crossing mice heterozygous for the targeted allele to a Cre-deletor strain (Actin-Cre) (Lewandoski et al., 1997). Deletion of the Neo cassette was obtained by crossing to a flpe-deletor mouse (Actin-Flpe) (Rodriguez et al., 2000). Successful recombination was verified by PCR. Primers and protocols for genotyping PCR are available upon request.
Fetal liver reconstitution experiments
For fetal liver reconstitution experiments, 6–8 weeks old recipient mice (B6.SJL, Jackson Laboratories) congenic for the CD45.1 allele were irradiated with 9 Gy and injected retro-orbitally with 4×105− 1×106 fetal liver cells from either wild-type or miR-17~92 Δneo/Δneo E14.5 embryos. Mice were analyzed 8–10 weeks after transplantation. Peripheral blood was obtained by tail bleeding.
RNAse protection assays and qPCR
RNA samples were isolated by homogenizing tissue or cells in Trizol (Invitrogen). RNAse Protection assays were performed using the Ambion miRvana miRNA detection kit. All probes were purchased from Dharmacon (Supplemental Experimental Procedures) and end-labeled with NEB Polynucleotide Kinase and γ32P-ATP. qPCR reactions were performed using the Applied Biosystems miRNA-detection kit. Sno-142 was used for normalization.
Bim 3’UTR reporter assays
A 165 nucleotide fragment of the Bim 3’ UTR, including the distal miR-19 and miR-92 binding sites, was amplified from mouse genomic DNA by PCR using primer BimUTRfwd and primer BimUTRrev (Supplemental Experimental Procedures) and cloned into a modified pRL-TK vector. The mutant version was generated from the wild type construct by Quikchange multi site-directed mutagenesis kit (Promega) according to manufacturers protocol using primers QCmiR92 and QCmiR19. All constructs were sequenced. For luciferase assays, 24 hours before transfection 10^5 Hela cells were seeded per well in 24 well plates. Cells were transfected with 100ng of pGL3 (Promega) and 140ng of pRL-TK Bim UTR with Lipofectamine 2000 (Invitrogen). Cell were lysed at 24 or 48 hrs and luciferase activity was measured using Dual luciferase reporter assay system (Promega) on the Glomax 20/20 luminometer (Promega). Data are expressed as ratio between Renilla and firefly luciferase activity. Wild type Bim luciferase activity was normalized to mutant Bim luciferase activity.
Antibodies, Immunohistochemistry and flow cytometry analysis
Four-color and seven-color flow cytometry was performed using FACSCalibur and LSRII cytometers (BD Biosciences). Cells were sorted on a FacsAria cell sorter (BD Biosciences). Antibodies were from Pharmingen (TCRβ, IgD, Gr-1, CD11b, CD19, CD21, CD23, B220) and eBioscience (c-kit, Sca-1, IL7R, IgM, Ter119, BP-1, CD5, CD24, CD45.1, CD45.2, CD43, CD71). Permeabilization for DAPI staining of DNA content was performed with cytofix/cytoperm buffers (BD Pharmingen). Annexin V staining was performed with Annexin V (BD) in the recommended staining buffer. Flow cytometry data were analyzed with FlowJo software (TreeStar). The Mcm-7 antibody was from Santa Cruz (sc-300). The Bim antibody was a rabbit polyclonal from Stressgen (AAP-330) and was used at a dilution of 1:1000 for flow cytometry and 1:2000 for western blotting experiments.
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded 4µm sections using the ABC Vectastain kit. Sections were developed with DAB and counterstained with hematoxylin. The Cleaved Caspase 3 antibody (1:200) was from Cell Signaling.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grant 2-PO1-CA42063-21 from the National Institutes of Health and in part by Cancer Center Support (core) grant P30-CA14051 from the National Cancer Institute, by United States Public Health Service RO1-GM34277 from the National Institutes of Health and by an RNA Interference as a Weapon Against Terrorism Grant U19 AI056900 from the National Institutes of Health to PAS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. TJ is a Howard Hughes Investigator and a Daniel K. Ludwig Scholar. AV was supported by a fellowship from the American Italian Cancer Research Foundation. AGY is recipient of a David Koch Graduate Fellowship. MMW is a Merck Fellow of the Damon Runyon Cancer Research Foundation. SE was supported by Dutch Cancer Society, KWF kankerbestrijding. We thank Alice Shaw for help and discussion, and Sergei Koralov, Stefan Muljo and Klaus Rajewsky for sharing their results prior to publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, Ambros V. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegant. Dev Cell. 2005;9:403–414. doi: 10.1016/j.devcel.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- Chen JN, Fishman MC. Genetics of heart development. Trends Genet. 2000;16:383–388. doi: 10.1016/s0168-9525(00)02075-8. [DOI] [PubMed] [Google Scholar]
- Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122:6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggan K, Akutsu H, Loring J, Jackson-Grusby L, Klemm M, Rideout WM, 3rd, Yanagimachi R, Jaenisch R. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A. 2001;98:6209–6214. doi: 10.1073/pnas.101118898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM. MicroRNAs as oncogenes. Curr Opin Genet Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, Lowe SW. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am Heart J. 2004;147:425–439. doi: 10.1016/j.ahj.2003.05.003. [DOI] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38(Suppl):S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–168. [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra EA, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005 doi: 10.1038/nature03702. in press. [DOI] [PubMed] [Google Scholar]
- Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–453. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson IL, Keenan RA, Licence S. The pre-B-cell receptor. Curr Opin Immunol. 2007;19:137–142. doi: 10.1016/j.coi.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M, Nimura Y, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007 doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny GW, Sonne SB, Nielsen JE, Jonstrup SP, Hansen MA, Skakkebaek NE, Meyts ER, Kjems J, Leffers H. Translational repression of E2F1 mRNA in carcinoma in situ and normal testis correlates with expression of the miR-17-92 cluster. Cell Death Differ. 2007 doi: 10.1038/sj.cdd.4402090. [DOI] [PubMed] [Google Scholar]
- O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sakata Y, Kamei CN, Nakagami H, Bronson R, Liao JK, Chin MT. Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci U S A. 2002;99:16197–16202. doi: 10.1073/pnas.252648999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3:881–886. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P. An E2F/miR-20a auto-regulatory feed-back loop. J Biol Chem. 2006 doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Webb S, Brown NA, Anderson RH. Formation of the atrioventricular septal structures in the normal mouse. Circ Res. 1998;82:645–656. doi: 10.1161/01.res.82.6.645. [DOI] [PubMed] [Google Scholar]
- Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic microRNA cluster by E2F transcription factors. J Biol Chem. 2006 doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.