

Abstract
The gene-mutation-cancer hypothesis holds that mutated cellular protooncogenes, such as point-mutated proto-ras, “play a dominant part in cancer,” because they are sufficient to transform transfected mouse cell lines in vitro [Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K. & Watson, J. D. (1994) Molecular Biology of the Cell (Garland, New York)]. However, in cells transformed in vitro mutated human ras genes are expressed more than 100-fold than in the cancers from which they are isolated. In view of the discrepancy between the very low levels of ras transcription in cancers and the very high levels in cells transformed in vitro, we have investigated the minimal level of human ras expression for transformation in vitro. Using point-mutated human ras genes recombined with different promoters from either human metallothionein-IIA or human fibronectin or from retroviruses we found dominant in vitro transformation of the mouse C3H cell line only with ras genes linked to viral promoters. These ras genes were expressed more than 120-fold higher than are native ras genes of C3H cells. The copy number of transfected ras genes ranged from 2–6 in our system. In addition, nondominant transformation was observed in a small percentage (2–7%) of C3H cells transfected with ras genes that are expressed less than 20 times higher than native C3H ras genes. Because over 90% of cells expressing ras at this moderately enhanced level were untransformed, transformation must follow either a nondominant ras mechanism or a non-ras mechanism. We conclude that the mutated, but normally expressed, ras genes found in human and animal cancers are not likely to “play a dominant part in cancer.” The conclusion that mutated ras genes are not sufficient or dominant for cancer is directly supported by recent discoveries of mutated ras in normal animals, and in benign human tissue, “which has little potential to progress” [Jen, J., Powell, S. M., Papadopoulos, N., Smith, K. J., Hamilton, S. R., Vogelstein, B. & Kinzler, K. W. (1994) Cancer Res. 54, 5523–5526]. Even the view that mutated ras is necessary for cancer is hard to reconcile with (i) otherwise indistinguishable cancers with and without ras mutations, (ii) metastases of the same human cancers with and without ras mutations, (iii) retroviral ras genes that are oncogenic without point mutations, and (iv) human tumor cells having spontaneously lost ras mutation but not tumorigencity.
All directly oncogenic retroviruses carry dominant cancer genes, termed oncogenes (1–5). These oncogenes are genetic hybrids that consist of strong retroviral promoters linked to coding sequences transduced from cellular genes, termed protooncogenes (2, 6). Retroviral promoters enhance transcription of protooncogene coding regions at least 100-fold compared with those of cellular protooncogenes (7–11). This 100-fold higher transcription of viral oncogenes compared with native cellular protooncogenes is the key to their oncogenic function (2, 7, 11).
In view of the coding sequence that cellular protooncogenes share with retroviral oncogenes, it has been proposed that these cellular genes are convertible to dominant cancer genes by mutation, e.g., a point mutation of human proto-ras is thought to convert this gene to an oncogene (1, 6, 12). According to this gene-mutation-cancer hypothesis, or oncogene hypothesis, the cell contains “about 60 protooncogenes … ; each of these can be converted into an oncogene that plays a dominant part in cancer of some sort or another” (1). In the light of this hypothesis, numerous mutations of protooncogenes have been identified in cancer, above all in ras genes (1). But practically none of these mutations ever elevates the low native levels of protooncogene transcription including that of proto-ras (2, 11, 13–15).
The protooncogene-mutation-cancer hypothesis derives support from two kinds of observations: (i) Mutations of proto-ras ond other protooncogenes are more frequent in cancer than predicted from the spontaneous incidence of mutation (1, 16). (ii) Point-mutated proto-Harvey- and proto-Kirsten-ras DNA from various nonviral human and animal cancer cells is able to dominantly transform the mouse 3T3 cell line—just like the ras-containing Harvey and Kirsten sarcoma viruses do (1, 16). This appeared to be functional support for the oncogene hypothesis (17–19).
But other facts challenge the gene-mutation hypothesis.
Several experimental observations indicate that mutated ras genes are not necessary for cancer: (i) Typically only a minority of a given cancer contains a specific protooncogene mutation, as, for example, mutated proto-ras. The majority of histologically and clinically indistinguishable cancers lack ras mutations (2, 14–16, 20–23). (ii) Some cancer patients carry metastases with and without ras mutations (24, 25). (iii) Human cancer cells that have lost mutated ras remain tumorigenic (26). (iv) Retroviruses from which ras (7, 27–29), src (9, 10), and myc-oncogene mutations (8, 30) have been removed remain oncogenic. Thus point mutation of proto-ras is not necessary for carcinogenesis.
According to the literature the level of ras transcription in mouse 3T3 cells and rodent embryo cells transformed in vitro with mutated ras genes is about 100-fold higher than in untransfected cells (2, 14, 31–43). Likewise the level of ras transcription in animal tumors formed with synthetic retroviruses, carrying human proto-ras coding regions (28, 29, 44), is as high as that in tumors caused by Harvey sarcoma virus (HaSV) (7), the virus in which ras was originally identified (45). Thus dominant transforming function of mutated human ras genes in vitro appears to depend on 100-fold elevated transcription, which is not seen in the cancers from which these genes are isolated.
The elevated transcription of human proto-ras in cells transformed in vitro appears to derive from two sources: (i) Heterologous promoters acquired by recombination with either plasmid DNA, cellular DNA used as carrier in transfection, or retroviral or DNA tumor viral helper genes that must be added to ras genes to transform rodent embryo cells (2, 31, 37, 38, 40–43); and (ii) the introduction of multiple ras copies into the same cell (2).
Both concatenation and recombination of DNAs are known artifacts of transfection (46–49).
Thus enhanced ras transcription in cells transformed in vitro by transfection with mutated cellular ras genes is an artifact of the transfection assay (2, 11). It follows that the in vitro assays may create, via artificial overexpression, dominant transforming genes from point-mutated proto-ras genes that have no transforming function at their native levels of expression.
In view of the discrepancy between the low, normal levels of transcription of mutated ras genes in natural cancers (2, 11, 13–15) and the very high levels in cells transformed in vitro, we have investigated here which level of transcription is necessary for dominant transformation by point-mutated cellular ras genes. For this purpose we have synthetically recombined mutated human proto-ras coding regions with heterologous promoters from human metallothionein IIA (MN) and fibronectin (FN) (50, 51) and from retroviruses. And we have analyzed the ability of these recombinant human ras genes to dominantly transform the mouse C3H cell line upon transfection. It was found that efficient, dominant transformation depends on at least 100-fold enhanced ras transcription compared with the expression in the natural tumors from which the ras DNAs were isolated. This result calls into question the hypothesis that mutated, but normally expressed, human ras genes of natural cancers play a dominant role in carcinogenesis.
MATERIALS AND METHODS
Recombinant ras Plasmid Constructs.
pHa#8 (pLTR/v-ras). pHa#8 is a pBR322-derived plasmid carrying an infectious HaSV flanked by redundant viral sequences extending from a proviral MstII (4317) site 5′ of the 5′-long terminal repeat (LTR) and to a PvuI (253) site 3′ of the 3′-LTR (6). This circularly permutated provirus was cloned into the EcoRI (4361) and PvuII (2064) sites of pBR322 (52) (Fig. 1B, part 1).
Figure 1.

(A) Genetic structures of plasmid vectors carrying the human FN promoter pFN-6 and the human MN promoter pMN-10. pFN-6 (part 1) and pMN-10 (part 2) are composed of DNA fragments from various sources: the origin (ori) of replication and the β-lactamase gene (AmpR) of pBR322; the promoters of human FN or human MN genes; the neomycin-resistance gene; the origin of DNA replication of SV40 (SV40 ori) containing the SV40 early promoter/enhancer region, which is in the opposite transcriptional orientation to the FN or MN promoter; and the SV40 poly(A)-addition sequences. (B) Genetic structures of human proto-Ha-ras and v-Ha-ras constructs. Part 1, pLTR/v-ras (pHa#8), part 2, pLTR/proto-rasm, part 3, pFN-SV40/proto-rasm, and part 4, pFN/proto-rasm. Construction of and origin of the complete plasmids are described in Materials and Methods. Three letter symbols identify restriction enzyme sites. Numbers following some v-ras restriction sites refer to the sequence position of HaSV (19). LTR is the retroviral promoter, and ras is the coding region of HaSV. E2, E3, E4, and E5 are the coding exons of human proto-Ha-ras.
pLTR/proto-rasm.
The coding region of the mutated human proto-Ha-ras-1 gene was derived from plasmid construct proto-rasm (ATCC, no. 41000), which contains a 6.6-kb BamHI–BamHI ras fragment from the human T24 bladder carcinoma cell line mutated at ras codon 12 (Gly-12 to Val-12). A 2.35-kb CelII–SacI fragment containing the full-length T24 proto-Ha-rasm coding sequence (exons 2 to 5) and the poly(A)-addition signal, but without the native cellular promoter of proto-Ha-ras, was cut from the proto-rasm plasmid, blunt-ended, and ligated with a blunt-ended SacII (940)–NcoI (3739) vector fragment from pHa#8. The resulting plasmid is termed pLTR/proto-rasm (Fig. 1B, part 2).
pFN-SV40/proto-rasm and pMN-SV40/proto-rasm.
The vectors carrying human FN and MN promoters (pFN-6 and pMN-10) were constructed by Shelley Blam (Lawrence Berkeley Laboratory, Berkeley, CA), by replacing the cytomegalo- virus promoter of the pSV2CMV plasmid with either the human FN or human MN-IIA promoter (50, 51, 53) (Fig. 1A). These two vectors also contain, in the opposite transcriptional orientation, the neomycin-resistance gene (NeoR) driven by the simian virus 40 (SV40) early promoter/enhancer region. The 2.35-kb CelII–SacI mutated proto-Ha-ras coding region was blunted and joined with EcoRI linkers, and inserted into the EcoRI site of pFN-6 and pMN-10 vectors. The resulting two plasmids were termed pFN-SV40/proto-rasm (Fig. 1B, part 3) and pMN-SV40/proto-rasm (not shown in Fig. 1) (53).
pFN/proto-rasm and pMN/proto-rasm.
The SV40 promoter/enhancer region and neomycin coding region were deleted from plasmid vectors pFN-6 and pMN-10 with PvuII digestion, resulting in plasmid vectors pFN-6/dSV40 and pMN-10/dSV40. The 2.35-kb CelII–SacI mutated proto-Ha-ras coding region joined with EcoRI linkers (see above) was inserted into the EcoRI site of pFN-6/dSV40 and pMN-10/dSV40 vectors to generate pFN/proto-rasm (Fig. 1B, part 4) and pMN/proto-rasm (not shown in Fig. 1) (53).
pFN/v-ras and pMN/v-ras.
These two plasmids were made by the following strategy: the v-Ha-ras coding sequence between the BamHI (409) and NaeI (5720) sites of pHa#8 was first cloned into the BamHI (375) and NaeI (1283) sites of pBR322, followed by XbaI digestion to partially delete the LTR downstream of the v-Ha-ras coding region (corresponding to the XbaI sites of 2023 and 5359 in pHa#8). The resulting plasmid, called pBR322/v-ras/dx, then was cut in the pBR322 sequence with EcoRI and NaeI to generate a 2,352-bp ras-containing fragment that was inserted into the pFN-6/dSV40 and pMN-10/dSV40 vectors. The vectors were prepared by cutting pFN-6/dSV40 and pMN-10/dSV40 with NotI, end-filling with Klenow enzyme, followed by digestion with EcoRI. The larger EcoRI–NotI vector fragments containing the FN or MN promoter were purified and ligated to the 2,352-bp EcoRI–NaeI v-Ha-ras insert, resulting in two new plasmids termed pFN/v-ras and pMN/v-ras (not shown in Fig. 1) (53).
RESULTS
Dominant Transformation with Mutated ras Coding Regions.
To determine the role of expression on dominant, in vitro transformation by mutated proto-Ha-ras genes from human cancers, ras coding regions artificially recombined with promoters of varying strength were examined. The constructs tested included the mutated human proto-ras coding region linked with the promoters of HaSV (pLTR/proto-rasm), of human FN with or without an added SV40 enhancer (pFN-SV40/proto-rasm and pFN/proto-rasm), or of human MN with or without an added SV40 enhancer (pMN-SV40/proto-rasm and pMN/proto-rasm). In addition constructs were tested in which the ras coding region of HaSV, naturally linked with promoters of HaSV (pLTR/v-ras), was linked with promoters of human FN (pFN/v-ras) and MN (pMN/v-ras) (Fig. 1, Table 1). About 10 μg of the plasmids carrying these ras constructs were transfected with 20 μg of salmon sperm carrier DNA and 1 μg of pSV2neo if the plasmids did not contain a neomycin resistance gene (47, 54).
Table 1.
Survey of the structure, transforming efficiency, and transcriptional activity of various human, mutated proto-Ha-ras, and Harvey sarcoma viral-ras constructs under heterologous promoters

| Construct | % of transfected cells transformed | ras copy number | ras overexpression factor | ||
|---|---|---|---|---|---|
Constructs: 1, pLTR/v-ras (pHA#8); 2, pLTR/proto-rasm; 3, pFN/v-ras; 4, pMN/v-ras; 5, pFN-SV40/proto-rasm; 6, pMN-SV40/proto-rasm; 7, pFN/proto-rasm; 8, pMN/proto-rasm; and 9, proto-rasm.
Transforming function of these ras genes was tested in mouse C3H cells instead of 3T3 cells. Based on simultaneous working experience with both cell lines for more than 12 years (7, 27, 47), we have determined that spontaneous transformation of C3H cells is much lower than that of 3T3 cells. Transformed and untransformed C3H colonies (Fig. 2) appeared in the presence of 600 μg/ml geneticin G418 (a neomycin derivative) about 1–2 weeks after transfection as described previously (28, 53). The efficiency of morphological transformation by ras genes compared to the efficiency of conferring neomycin resistance by the neomycin-resistance plasmid varied from 80% for LTR-ras to <1% for FN-promoted proto-ras (Table 1).
Figure 2.
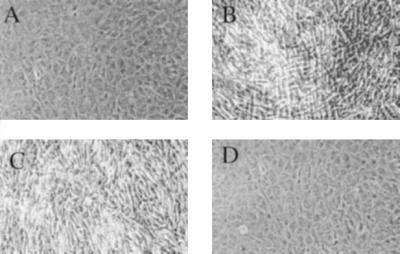
Morphology of mouse C3H cells transfected with mutated human proto-ras and viral ras constructs. (A) Untransfected normal C3H cells. (B) pLTR/proto-rasm-transformed C3H clone. (C) pFN-SV40/proto-rasm-transformed C3H clone. (D) Proto-rasm-transfected, untransformed C3H clone. (Cell pictures were taken at 100× magnification.)
Based on quantitative hybridization of total cellular RNA bound to nitrocellulose with 32P-labeled 564-bp HindIII (1088)–DraIII (1650) v-Ha-ras probe, derived from HaSV-provirus pHa#8 (Materials and Methods), ras was expressed in transformants generated by LTR-promoted ras genes 120- to 200-fold higher than in untransfected cells (Table 1 and Fig. 3) (53, 55). By contrast, transformants generated by ras genes with cellular FN and MN promoters, even with additional SV40 viral enhancers, expressed ras only 10- to 20-fold higher than untransformed cells (Table 1).
Figure 3.

Expression level of ras RNA in mouse C3H cells transfected with mutated human proto-ras- and HaSV-derived constructs. Total cellular RNA (A) or poly(A)-selected mRNA (B) was isolated with the guanidinium isothiocyanate method (55) from transformed (T) and nontransformed (NT) clonal cultures of G418-resistant C3H cells transfected with either proto-ras or viral ras constructs or from untransfected C3H control cells. The RNA was quantitated by spectrophotometry, equal amounts of RNA were dotted onto nitrocellulose membranes for each dilution and hybridized with the 32P-labeled ras-DNA probe described in the text. The blot in the lower left of A shows no detectable ras expression in untransfected C3H control cells and in proto-rasm transfected C3H cells after 2 days of exposure. After exposure for 9 days, the same blot showed barely detectable levels of ras expression in proto-rasm-transfected C3H cells, but not in untransfected C3H control cells (A, Lower Right).
The level of ras expression of untransfected C3H cells was too low to be detected by this method with total cell RNA (Fig. 3A). To estimate normal proto-ras expression, poly(A)-selected mRNA from untransfected C3H cells was compared with poly(A)-selected mRNA from cells transfected with mutated proto-ras (Fig. 3B). On this basis it was estimated that expression in untransfected cells is two times lower than in cells transfected with native, mutated ras genes from human cancers and 120–200 times lower than in cells transfected and transformed with ras genes with viral promoters (compare Fig. 3 A and B).
Nondominant in Vitro Transformation with Mutated ras Genes.
To determine whether the untransformed, neomycin-resistant colonies that had been transfected with ras genes linked to cellular promoters had in fact not received ras DNAs, the genomic DNAs of several untransformed neomycin-resistant clones were analyzed. The presence of transfected ras DNA with FN and MN promoters, plus or minus SV40 viral enhancers, in one group of five neomycin-resistant cultures, of which three were transformed (T) and two were untransformed (NT), is shown in Fig. 4A, Left. The presence of transfected ras DNA in another group of six clonal cultures, all of which were untransformed, is shown in Fig. 4A, Right. In each untransformed (NT) culture either the predicted 3.8-kb BamHI-resistant fragment from the proto-ras plasmids containing the FN promoter or the predicted 4.07-kb BamHI-resistant fragment from the proto-ras plasmids containing the MN promoter was detected. The predicted ras fragments are made up from the central 2.35-kb human proto-ras region flanked by CelII and SacI, and about 1.5 kb of pFN or pMN Bam-flanked vector elements (Fig. 1A). Based on the radiographic intensities there were about 4–6 copies of transfected DNA per each C3H genomic proto-ras copy of 3.4 kb, regardless of whether the cell was transformed or untransformed (Fig. 4A, Right). However, one transformed colony carried about 20 copies of human proto-ras under a FN promoter, pFN/proto-rasm (Fig. 4A, lane 5). Barring this one exception, there was no difference between transformed and untransformed cells with regard to the presence and even copy number of ras genes with cellular promoters.
Figure 4.

Identification of transfected and endogenous ras DNAs in G418-resistant C3H cells transfected with proto-Ha-ras or v-Ha-ras under human FN or MN promoters. About 20 μg cellular DNAs were digested with BamHI (A) or XbaI (B), resolved on a 0.8% agarose gel, transferred to positively charged nylon membrane by the alkaline transfer method (55), and hybridized with 32P-labeled ras DNA from HaSV (see text). The positions of HindIII-resistant fragments of lambda DNA were used as molecular weight standards and are indicated between the two filters in A and on each side of the two filters in B. (A, Left) Lane 1, untransfected C3H; lane 2, pFN-SV40/proto-rasm-transformed C3H clone; lane 3, pMN-SV40/proto-rasm-transformed C3H clone; lane 4, pFN/proto-rasm-transfected, untransformed C3H clone; lane 5, rare pFN/proto-rasm-transformed C3H clone; and lane 6, pMN/proto-rasm-transfected, untransformed normal C3H clone. (A, Right) Lane 1, untransfected C3H; lanes 2–4, pFN-SV40/proto-rasm-transfected, untransformed C3H clones 1–3; and lanes 5–7, pMN-SV40/proto-rasm-transfected, untransformed C3H clones 1–3. The 3.4-kb BamHI-resistant ras fragment represents the genomic proto-Ha-ras-1 gene of C3H mice. The 4.07-kb (A, Left, lanes 3 and and A, Right, lanes 4–7) and 3.81-kb (A, Left, lanes 2, 4 and 5 and A, Right, lanes 2–4) BamHI-resistant ras fragments are diagnostic of the plasmids pFN-SV40/proto-rasm and pMN-SV40/proto-rasm, respectively (see Fig. 1). (B) Lane 1, untransfected C3H; lane 2,: pLTR/v-ras (pHa#8)-transformed C3H; lane 3, untransfected C3H; and lane 4, pLTR/proto-rasm-transformed C3H. The ≈12-kb XbaI fragment represents the genomic mouse proto-Ha-ras-1 gene cut with XbaI. The 2.2-kb XbaI-resistant ras fragment (lane 2) is diagnostic of the pLTR/v-ras plasmid; and the 3.66-kb and 1.4-kb XbaI-resistant ras fragments (lane 4) are diagnostic of the pLTR/proto-rasm plasmid.
Thus differential expression was theoretically the only variable left for ras to exert a dominant role in transformation. But because the copy numbers of transfected ras genes in transformed and untransformed cells were the same, and because the input promoters were also the same, the chances for significant differences in expression were small. Nevertheless, ras expression was tested in four transformed and 12 untransformed clonal cultures transfected with proto-ras or viral ras under human cellular promoters, e.g., those of MN, FN, or proto-ras itself.
Indeed, Fig. 5 shows that ras expression was the same both in transformed and untransformed cells transfected by the same ras construct. Although expression levels in cultures transfected with ras genes linked to cellular promoters exceeded normal C3H ras expression levels up to 20-fold (Fig. 5), they were 5–10 times lower than in cells transfected with dominantly transforming viral LTR-linked ras genes (Fig. 3). Fig. 5 also shows that ras expression of nontransformed control cells, transfected with ras-free plasmid vectors, was not elevated.
Figure 5.

Expression level of ras RNA in transformed and untransformed, G418-resistant clonal cultures of mouse C3H cells that were transfected with ras constructs in which human proto-Ha-ras- and viral ras- coding regions are linked to human MN and FN promoters. Equal amounts of RNA were dotted onto nitrocellulose membranes for each dilution and hybridized with the 32P-labeled ras-DNA probe as described in Fig. 3.
Because only a small percentage of cells expressing transfected ras at relatively low levels are transformed (Table 1), the corresponding ras constructs are not sufficient, and thus not dominant transforming genes.
Role of ras Copy Number on Transformation of C3H Cells.
The copy number of transfected ras genes in transformed cells was 2- to 6-fold higher than that of the endogenous proto-Ha-ras standard of C3H cells (Fig. 4 and Table 1). For example, cells transformed by mutated human proto-ras linked to the promoter of HaSV (pLTR/proto-rasm) contained about four copies of the 3.66-kb Xba-resistant fragment of transfected pLTR/proto-rasm per genomic ras (Fig. 4B, lane 4). Likewise cells transfected with cloned HaSV DNA (pLTR/v-ras) contained about four copies of a 2.2-kb Xba-resistant fragment of that plasmid (Fig. 4B, lane 2; other data not shown). Cells transfected by mutated human proto-ras linked to FN or MN promoters (pFN and MN/proto-rasm) also contained 2–6 ras copies regardless whether they were transformed or untransformed (Fig. 4A and Table 1). It follows that the level of ras expression in in vitro transfection is increased not only by artificial promoters, but also by the copy number of transfected ras genes.
DISCUSSION
Dominant Transformation in Vitro.
Our results show that dominant transformation of C3H mouse cells by point-mutated human proto-ras genes requires at least 100-fold overexpression, compared with the levels observed in untransfected C3H cells. This overexpression is achieved in our system by two factors: artificial retrovirus-derived promoters and artificially increased copy numbers of transfected ras genes. Surprisingly, not even the strong cellular promoters of human FN and MN were sufficient by themselves to confer dominant transforming function to mutated human proto-ras genes (Table 1).
Our evidence for the essential role of overexpression in dominant transformation with mutated proto-ras genes confirms and extends the results obtained by transfection of mouse 3T3 cells (2, 14, 32–36), and rodent embryo cells (2, 31, 37, 38, 40–43), and by infection of animals with ras-containing retroviruses (28, 29, 44). Cells previously transformed by transfection in vitro also contained multiple transfected ras genes (34, 35, 38, 39, 56). Thus earlier studies confirm overexpression and overdosage of ras genes in cells transformed by transfection, but considered point mutation as the critical requirement for dominant transformation, rather than enhanced expression. However, experiments by ourselves and others with proto-ras genes whose natural mutations had been reverted in vitro have shown since 1986 that point mutations are not necessary for transforming function of ras genes (7, 27–29).
Nondominant Transformation in Vitro.
Only 2–7% of C3H cells transfected with recombinant ras genes driven by cellular FN or MN promoters were transformed. Unexpectedly, these cells expressed ras at the same level as untransformed cells transfected with the same ras constructs. These ras constructs were expressed less than 20-fold over the level of untransfected C3H cells. Because 93–98% of cells expressing this level of ras were untransformed, transformation of a few cells must have followed one of two nondominant mechanisms: cooperation of ras with another mutated gene or ras-independent transformation. However, this nondominant transformation is not necessarily relevant to the hypothetical role that point-mutated proto-ras genes play in cancer, because transfection with recombinant ras genes, particularly those that include an SV40 virus enhancer, may artificially activate unknown cellular genes via downstream promotion.
Relevance of Point-Mutated ras to Natural Carcinogenesis.
Because mutated ras genes are not overexpressed and are not amplified in most human and animal cancers (see above), and because dominant transformation by mutated ras genes depends on at least 100-fold overexpression, we conclude that point-mutated ras genes do not “play a dominant part in cancer” (1).
Several experimental and theoretical arguments support the conclusion that mutated proto-ras is not dominant in, or sufficient for, natural carcinogenesis.
Point-mutated proto-ras genes have been identified in normal animals (57, 58) and in benign human tissue “which has little potential to progress” (59).
As demonstrated here, point-mutated human proto-ras with native human and heterologous human FN and MN promoters do not transform C3H mouse cells.
All humans can be calculated to contain at least 105 cells with mutated proto-ras genes. This calculation is based on the facts that the spontaneous mutation rate of human and nonhuman cells is 1 in 109 nucleotides per cell division (1, 2, 11, 16), and that humans and most mammals contain about 109 nucleotides per cell (1, 2, 11). Thus 1 in 109 cells contains a mutation in any specific nucleotide of the human genome. Because humans contain about 1014 cells, each person contains at least 105 cells with point-mutated proto-ras. Because several point mutations convert ras genes to 3T3 cell-transforming genes, and because there are several families of ras genes (1, 6), even more than 105 cells should be cancer cells at any given time if point-mutated ras genes were dominant cancer genes.
In view of this it has been argued that point-mutated proto-ras genes are not sufficient, but necessary for carcinogenesis, and that carcinogenesis depends on the cooperation of multiple mutated genes (20, 31). However, according to several studies by ourselves and others point mutation of proto-ras is not even necessary for carcinogenesis.
We have shown that oncogenic retroviruses from which ras (7, 27–29), src (9, 10), and myc-oncogene mutations (8, 30) have been removed remain oncogenic.
Others have shown that typically only a minority of a given cancer contains a specific protooncogene mutation, as, for example, mutated proto-ras. The majority of histologically and clinically indistinguishable cancers lack ras mutations (2, 14–16, 20–23).
Some cancer patients have metastases with and without ras mutations (24, 25).
Human cancer cells that have lost mutated ras remain tumorigenic (26).
Moreover it is unlikely that the same ras protein that plays a dominant, or sufficient, role in transformation by transfection and by retrovirus infection, also would function as a cofactor of other gene(s) in a completely different mechanism of carcinogenesis. Thus mutated proto-ras under its native promoter, as it is in most human and animal cancers, appears neither sufficient nor necessary for carcinogenesis.
Finally, it appears that the dominantly transforming host range even of highly expressed retroviral oncogenes, including ras genes, does not include human cells (62).
This leaves open the possibility that point-mutated, normally expressed, ras genes play an indirect role in cancer. For example, mutated proto-ras could stimulate cell proliferation, and enhanced proliferation is a known cancer risk (60, 61). This hypothesis would provide a plausible explanation for the unexpectedly high, but not consistent, presence of ras mutations in certain cancers.
Perhaps aneuploidy, which according to Hollstein et al. (63) is “almost always found in human cancers,” including those with mutated ras genes, is the cause of these cancers (11).
Acknowledgments
We gratefully acknowledge critical reviews by Harry Rubin (Berkeley, CA) and Alwin Kraemer (Mannheim, Germany), and thank Shelley Blam (Berkeley, CA) for the plasmids carrying the human FN and MN genes. P.D. thanks Ruediger Hehlmann, director of the III Medizinische Klinik of the University of Heidelberg at Mannheim, for offering his lab and facilities for a sabbatical semester in Mannheim, Germany. This investigation was supported in part by the Council for Tobacco Research, and private donations from Robert Leppo, San Francisco; Carol Wilhelmy, San Mateo (CA); Tom Boulger, Los Angeles; the Anthony Robbins Research International (San Diego), and a foundation that prefers to remain anonymous.
ABBREVIATIONS
- HaSV
Harvey sarcoma virus
- LTR
long terminal repeat
- SV40
simian virus 40
- FN
fibronectin
- MN
metallothionein
References
- 1.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. New York: Garland; 1994. [Google Scholar]
- 2.Duesberg P H, Schwartz J R. Prog Nucleic Acid Res Mol Biol. 1992;43:135–204. doi: 10.1016/s0079-6603(08)61047-8. [DOI] [PubMed] [Google Scholar]
- 3.Lai M M C, Duesberg P H, Horst J, Vogt P K. Proc Natl Acad Sci USA. 1973;70:2266–2270. doi: 10.1073/pnas.70.8.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duesberg P H, Vogt P K. Proc Natl Acad Sci USA. 1970;67:1673–1680. doi: 10.1073/pnas.67.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duesberg P H. Cancer Res. 1987;47:1199–1220. [PubMed] [Google Scholar]
- 6.Weiss R, Teich N, Varmus H, Coffin J. Molecular Biology of RNA Tumor Viruses. Plainview, NY: Cold Spring Harbor Lab. Press; 1985. [Google Scholar]
- 7.Chakraborty A K, Cichutek K, Duesberg P H. Proc Natl Acad Sci USA. 1991;88:2217–2221. doi: 10.1073/pnas.88.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou R-P, Duesberg P H. Proc Natl Acad Sci USA. 1988;85:2924–2928. doi: 10.1073/pnas.85.9.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H, Duesberg P H. Proc Natl Acad Sci USA. 1990;87:9128–9132. doi: 10.1073/pnas.87.23.9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Zhou H, Duesberg P. Proc Natl Acad Sci USA. 1992;89:6393–6397. doi: 10.1073/pnas.89.14.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duesberg P. Science. 1995;267:1407–1408. doi: 10.1126/science.7794335. [DOI] [PubMed] [Google Scholar]
- 12.Lodish H, Baltimore D, Berk A, Zipursky S L, Matsudaira P, Darnell J. Molecular Cell Biology. New York: Freeman; 1995. [Google Scholar]
- 13.Nagai M A, Habr-Gama A, Oshima C T F, Brentani N M. Dis Colon Rectum. 1992;35:444–451. doi: 10.1007/BF02049400. [DOI] [PubMed] [Google Scholar]
- 14.Barbacid M. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 15.Forrester K, Almoguera C, Han K, Grizzle W, Perucho M. Nature (London) 1987;327:298–303. doi: 10.1038/327298a0. [DOI] [PubMed] [Google Scholar]
- 16.Strauss B S. Cancer Res. 1992;52:249–253. [PubMed] [Google Scholar]
- 17.Tabin C J, Bradley S M, Bargmann C I, Weinberg R A, Papageorge A G, Scolnick E M, Dhar R, Lowy D R, Chang E H. Nature (London) 1982;300:143–149. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 18.Reddy E P, Reynolds R K, Santos E, Barbacid M. Nature (London) 1982;300:149–152. doi: 10.1038/300149a0. [DOI] [PubMed] [Google Scholar]
- 19.Logan J, Cairns J. Nature (London) 1982;300:104–105. doi: 10.1038/300104a0. [DOI] [PubMed] [Google Scholar]
- 20.Vogelstein B, Fearon E R, Stanley B A, Hamilton R, Kern S E, Preisinger A C, Leppert M, Nakamura Y, White R, Smits A M M, Bos J L. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 21.Cooper G M. Oncogenes. Boston: Jones and Bartlett; 1990. [Google Scholar]
- 22.Bos J L, Fearon E R, Hamilton S R, Verlaan-de Vries M, van Boom J H, van der Eb A J, Vogelstein B. Nature (London) 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 23.Feinberg A P, Vogelstein B, Droller M J, Baylin S B, Nelkin B D. Science. 1983;220:1175–1177. doi: 10.1126/science.6304875. [DOI] [PubMed] [Google Scholar]
- 24.Albino A P, Le Strange R, Oliff A I, Furth M E, Old L J. Nature (London) 1984;308:69–72. doi: 10.1038/308069a0. [DOI] [PubMed] [Google Scholar]
- 25.Shibata D, Schaeffer C, Li Z H, Capella G, Perucho M X. J Natl Cancer Inst. 1993;85:1058–1063. doi: 10.1093/jnci/85.13.1058. [DOI] [PubMed] [Google Scholar]
- 26.Plattner R, Anderson M J, Sato K Y, Fasching C L, Der C J, Stanbridge E J. Proc Natl Acad Sci USA. 1996;93:6665–6670. doi: 10.1073/pnas.93.13.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cichutek K, Duesberg P H. Proc Natl Acad Sci USA. 1986;83:2340–2344. doi: 10.1073/pnas.83.8.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cichutek K, Duesberg P H. J Virol. 1989;63:1377–1383. doi: 10.1128/jvi.63.3.1377-1383.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velu T J, Vass W C, Lowy D R, Tambourin P E. J Virol. 1989;63:1384–1392. doi: 10.1128/jvi.63.3.1384-1392.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou R-P, Duesberg P H. Proc Natl Acad Sci USA. 1989;86:7721–7725. doi: 10.1073/pnas.86.20.7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Land H, Parada L F, Weinberg R A. Nature (London) 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 32.Chang E H, Furth M E, Lowy D R. Nature (London) 1982;297:479–483. doi: 10.1038/297479a0. [DOI] [PubMed] [Google Scholar]
- 33.Goldfarb M, Shimizu K, Perucho M, Wigler M. Nature (London) 1982;296:404–409. doi: 10.1038/296404a0. [DOI] [PubMed] [Google Scholar]
- 34.Parada L F, Tabin C J, Shih C, Weinberg R A. Nature (London) 1982;297:474–478. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 35.Pulciani S, Santos E, Lauver A V, Long L K, Robins K C, Barbacid M. Proc Natl Acad Sci USA. 1982;79:2845–2849. doi: 10.1073/pnas.79.9.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Der C J, Finkel T, Cooper G M. Proc Natl Acad Sci USA. 1982;79:3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruley H E. Nature (London) 1983;304:602–606. doi: 10.1038/304602a0. [DOI] [PubMed] [Google Scholar]
- 38.Schwab M, Varmus H E, Bishop J M. Nature (London) 1985;316:160–162. doi: 10.1038/316160a0. [DOI] [PubMed] [Google Scholar]
- 39.Bizub D, Wood A W, Skalka A M. Proc Natl Acad Sci USA. 1986;83:6048–6052. doi: 10.1073/pnas.83.16.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spandidos D A, Wilkie N M. Nature (London) 1984;310:469–475. doi: 10.1038/310469a0. [DOI] [PubMed] [Google Scholar]
- 41.Lee W F, Schwab M, Westaway D, Varmus H E. Mol Cell Biol. 1985;5:3345–3356. doi: 10.1128/mcb.5.12.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pozzatti R, Muschel R, Williams J, Padmanabhan R, Howard B, Liotta L, Khoury G. Science. 1986;232:223–227. doi: 10.1126/science.3456644. [DOI] [PubMed] [Google Scholar]
- 43.Brown P O, Bowerman B, Varmus H E, Bishop J M. Cell. 1987;49:347–356. doi: 10.1016/0092-8674(87)90287-x. [DOI] [PubMed] [Google Scholar]
- 44.Mangues R, Seidman I, Gordon J W, Pellicer A. Oncogene. 1992;7:2073–2076. [PubMed] [Google Scholar]
- 45.Maisel J, Klement V, Lai M M-C, Ostertag W, Duesberg P. Proc Natl Acad Sci USA. 1973;70:3536–3540. doi: 10.1073/pnas.70.12.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbins D, Ripley S, Henderson A, Axel R. Cell. 1981;23:29–39. doi: 10.1016/0092-8674(81)90267-1. [DOI] [PubMed] [Google Scholar]
- 47.Goodrich D W, Duesberg P H. Proc Natl Acad Sci USA. 1988;85:3733–3737. doi: 10.1073/pnas.85.11.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goff S P, Tabin C J, Wang J Y-J, Weinberg R, Baltimore D. J Virol. 1982;41:271–285. doi: 10.1128/jvi.41.1.271-285.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perucho M, Hanahan D, Wigler M. Cell. 1980;22:309–317. doi: 10.1016/0092-8674(80)90178-6. [DOI] [PubMed] [Google Scholar]
- 50.Karin M, Haslinger A, Holtgreve H, Richards R I, Krauter P, Wesphal H M, Beato M. Nature (London) 1984;308:513–519. doi: 10.1038/308513a0. [DOI] [PubMed] [Google Scholar]
- 51.Dean D C, Bowlus C, Bourgeois S. Proc Natl Acad Sci USA. 1987;84:1876–1880. doi: 10.1073/pnas.84.7.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R, Zhou R-P, Duesberg P. Proc Natl Acad Sci USA. 1996;93:7522–7527. doi: 10.1073/pnas.93.15.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hua V Y. Ph.D. thesis. Berkeley: Univ. of California; 1996. [Google Scholar]
- 54.Southern P J, Berg P. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- 55.Ausubel F M, Brent R, Kingston R E, Moore D E, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1988. [Google Scholar]
- 56.Pulciani S, Santos E, Long L K, Sorrentino V, Barbacid M. Mol Cell Biol. 1985;5:2836–2841. doi: 10.1128/mcb.5.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finney R E, Bishop J M. Science. 1993;260:1524–1527. doi: 10.1126/science.8502998. [DOI] [PubMed] [Google Scholar]
- 58.Cha R S, Thilly W G, Zarbl H. Proc Natl Acad Sci USA. 1994;91:3749–3753. doi: 10.1073/pnas.91.9.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jen J, Powell S M, Papadopoulos N, Smith K J, Hamilton S R, Vogelstein B, Kinzler K W. Cancer Res. 1994;54:5523–5526. [PubMed] [Google Scholar]
- 60.Rous P. Science. 1967;157:24–28. doi: 10.1126/science.157.3784.24. [DOI] [PubMed] [Google Scholar]
- 61.Parsons R, Li G-M, Longley M, Modrich P, Liu B, Berk T, Hamilton S R, Kinzler K W, Vogelstein B. Science. 1995;268:738–740. doi: 10.1126/science.7632227. [DOI] [PubMed] [Google Scholar]
- 62.Li R, Zhou R-P, Duesberg P. Proc Natl Acad Sci USA. 1996;93:7522–7527. doi: 10.1073/pnas.93.15.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hollstein M, Sidransky D, Vogelstein B, Harris C C. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]