

SUMMARY
The means by which Ca2+ store depletion evokes the opening of store-operated Ca2+ channels (SOCs) in the plasma membrane of excitable and non-excitable cells has been a longstanding mystery. Indirect evidence has supported local interactions between the ER and SOCs as well as long-range interactions mediated through a diffusible activator. The recent molecular identification of the ER Ca2+ sensor (STIM1) and a subunit of the CRAC channel (Orai1), a prototypic SOC, has now made it possible to visualize directly the sequence of events that links store depletion to CRAC channel opening. Following store depletion, STIM1 moves from locations throughout the ER to accumulate in ER subregions positioned within 10-25 nm of the plasma membrane. Simultaneously, Orai1 gathers at discrete sites in the plasma membrane directly opposite STIM1, resulting in local CRAC channel activation. These new studies define the elementary units of store-operated Ca2+ entry, and reveal an unprecedented mechanism for channel activation in which the stimulus brings a channel and its activator/sensor together for interaction across apposed membrane compartments. We discuss the implications of this choreographic mechanism with regard to Ca2+ dynamics, specificity of Ca2+ signaling, and the existence of a specialized ER subset dedicated to the control of the CRAC channel.
1. Introduction
Store-operated Ca2+ entry (SOCE) is a ubiquitous signaling mechanism in non-excitable and excitable cells that is triggered by a reduction in the level of Ca2+ in intracellular stores [1]. Physiologically, SOCE most commonly results from stimulation of cell surface receptors that evoke Ca2+ release through IP3 receptors or ryanodine receptors in the ER, which then activates store-operated Ca2+ channels (SOCs) in the plasma membrane (PM). By supplying a source of Ca2+ for refilling stores, SOCE enables the ER to release Ca2+ over extended periods and thereby evoke sustained Ca2+ oscillations, exocytosis, and other Ca2+-dependent events in response to prolonged stimulation [1]. In addition, SOCE can contribute directly to the elevation of cytosolic Ca2+ levels ([Ca2+]i), as in T lymphocytes where SOCs generate the sustained Ca2+ signals needed to drive gene expression underlying T cell activation by antigen [2].
In the years following the original proposal of the “capacitative Ca2+ entry” hypothesis by Putney [3], [Ca2+]i measurements in a variety of cells treated with thapsigargin (TG) and other SERCA inhibitors to deplete stores revealed that SOCE was widespread [4]. However, because [Ca2+]i is influenced by many factors in addition to channels (e.g., pumps, transporters, membrane potential, and organelles), an essential step in defining the entry pathway for SOCE was the identification of a store-operated Ca2+ current (the Ca2+ release-activated Ca2+ current, or ICRAC) in mast cells and T cells using patch-clamp techniques. The discovery of the CRAC current progressed in stages, beginning with the description of an extremely small Ca2+ current stimulated by IP3 in mast cells [5] and by mitogens in Jurkat T cells [6], and proceeding to the demonstration that it was store-dependent, based on activation by intracellular Ca2+ buffers and ionomycin [7] and by thapsigargin [8].
The CRAC channel emerged as a prototypic SOC for several reasons. First, it has fulfilled the most stringent criteria for being store-operated, in that it is activated by a variety of procedures that reduce free Ca2+ in the ER under conditions of constant cytosolic [Ca2+] [1, 9]. Second, its properties have been extensively characterized, establishing a distinctive “fingerprint” of the channel that later proved to be essential in identifying the CRAC channel gene [9]. Finally, the CRAC channel was found to have a critical physiological function, in that a spontaneous loss of CRAC channel activity in T cells causes a severe combined immunodeficiency (SCID) syndrome in human patients [10-12].
Despite many efforts over the past 20 years to unravel the mechanism underlying SOCE, relatively little progress was made largely due to a complete lack of identified SOCE proteins [9]. However, the recent identification of genes encoding the CRAC channel and the Ca2+ sensor in the ER has made possible for the first time direct experiments that reveal the activation process and the structure of the fundamental units of SOCE. In this review we present a historical overview of attempts to understand coupling between the ER and SOCs, followed by a discussion of recent work describing the elementary units of SOCE and their assembly in response to store depletion.
2. Communication between Ca2+ stores and the CRAC channel: local or global?
The fundamental question underlying SOCE is how does a reduction of free [Ca2+] in the ER lumen cause channels in the PM to open? At its most basic, the mechanism must include a Ca2+ sensor in the ER and a SOC (e.g., the CRAC channel) in the PM, but how and where they communicate with each other has long been a mystery. The notion that the ER might signal locally to SOCs in the PM dates back to the origins of the capacitative Ca2+ entry model, which was based in part on observations that extracellular Ca2+ could reload intracellular stores without an apparent increase in [Ca2+]i ([13, 14], see also [15, 16]). The ability of intracellular BAPTA to slow reloading, and of Ba2+ and Mn2+, which are not transported by Ca2+ pumps, to enter the cytosolic pool following store depletion, was taken as evidence that Ca2+ entering the cell traverses a narrow gap of cytosol before being actively pumped into the ER [17]. Later studies of Xenopus oocytes, endothelial cells and astrocytes showed that local receptor stimulation evoked Ca2+ entry (as detected by Cl− channel activation or Ca2+-dependent dye signals) that was confined to within 10's to 100's of μm from where Ca2+ release was thought to occur [18-21].
Further indirect evidence for a local interaction came from studies showing that calyculin A and jasplakinolide inhibit SOCE in smooth muscle cells [22]. This effect was attributed to the ability of these agents to promote polymerization of cortical actin and thereby prevent the ER from forming close contacts with the PM, and led to the proposal of a “secretion-like induced coupling model” in which store depletion induces the migration of the ER towards the PM to activate SOCE. However, these effects have not been seen in other cells [23], and in some cases actin breakdown rather than polymerization inhibits SOCE [24]. A membrane fusion model based on store depletion-stimulated insertion of CRAC channels in the PM followed from the inhibitory effects of SNARE inhibitors [25], but again, these results are not generalizable to all cells [26, 27]. As a whole, these studies led to the notion that the stores are close to the PM, and that some sort of local communication activates SOCs. The most explicitly defined model along these lines was the conformational coupling model, inspired by analogy to the Ca2+ release units of skeletal muscle, in which SOCE was envisioned to result from direct physical interactions of the IP3 receptor and the CRAC channel [28-30]. Although it was later shown that CRAC channel activation is independent of IP3 receptors [31, 32], the possibility of physical coupling between the CRAC channel and other ER proteins remains.
Other studies have argued against a strictly local mode of activation, proposing instead that SOCE is driven by a diffusible factor released from the ER. Early evidence for this idea included the activation of mixed-ion currents in membrane patches that were crammed into agonist-treated Xenopus oocytes [33]. Ca2+ influx factor (CIF), a cytosolic extract of store-depleted cells, was shown to activate Ca2+ entry when applied to cells with full stores [34], but this effect was later attributed to nonspecific activities [35]. In subsequent work a modified CIF preparation was proposed to activate ICRAC through stimulation of iPLA2 and generation of lysolipids [36, 37], but definitive tests of this model await the isolation of the active molecular component of the CIF extract. Experiments on RBL cells, in which various conditions believed to distance the ER from the PM (actin polymerization, microtubule depolymerization, and “cell ballooning”) failed to affect ICRAC, were cited as evidence against local coupling models [23]. However, the ER was not actually imaged in these experiments, and therefore disruption of local contacts could not be verified.
A major difficulty in distinguishing local from global models stemmed from the lack of molecular markers with which to probe SOCE. Without identified proteins it was impossible to directly observe where the CRAC channel and its sensor are located relative to each other.
3. Molecular identification of the ER Ca2+ sensor and the CRAC channel
In 2005, STIM1 was identified through RNAi screens and soon thereafter shown to be the ER Ca2+ sensor of SOCE [38-40]. STIM1 is a 77-kD, type I membrane protein residing primarily in the ER [39-41], but to a limited extent also in the PM [40, 42, 43]. STIM1 has a variety of predicted protein-interaction or signaling domains, including an unpaired EF-hand, sterile-α motif (SAM), coiled-coil, ERM, serine/proline-rich, and lysine-rich domains. Knockdown of STIM1 suppresses SOCE, and EF-hand mutants of STIM1, designed to have low Ca2+ affinity and thereby mimic the store-depleted state, activate ICRAC even when stores are full [39, 40, 44, 45]. The isolated EF-SAM domain has a Ca2+ binding affinity of 200-600 μM [46], overlapping the range of [Ca2+]i reported for the ER lumen by fluorescent protein measurements [47].
Orai1 (also called CRACM1) was identified in 2006 as a second essential component of SOCE in mammalian cells by a combination of linkage analysis of human CRAC-deficient SCID T cells and an RNAi screen for suppression of NFAT translocation in Drosophila S2 cells [48], as well as by RNAi screens for direct suppression of SOCE in S2 cells [49, 50]. Orai1 is a 33-kD, 4-transmembrane protein present in the PM. A single mutation in Orai1 (R91W) was shown to be responsible for the loss of CRAC channel function in human T cells, leading to the defect in T cell activation and immunodeficiency [48]. Overexpression of Orai1 and STIM1 (or their Drosophila homologs) gave rise to extremely large CRAC-like currents [45, 50-52], suggesting that either Orai1 was the CRAC channel, or that it encodes a non-channel component that is present in limiting amounts. Definitive evidence that Orai1 is a pore subunit of the CRAC channel was provided by several studies showing that mutagenesis of acidic residues in Orai1 changes the divalent and monovalent selectivity of ICRAC. The isolation of STIM1 and Orai1 are reviewed in greater detail elsewhere in this volume and in other recent reviews [53-56].
4. Redistribution of STIM1 in response to store depletion
A remarkable finding appearing soon after the identification of STIM1 was that store depletion causes it to relocate from a dispersed distribution throughout the ER into discrete puncta near the PM (Fig. 1) [39, 40]. This finding has been confirmed by many groups in a variety of mammalian cells expressing STIM1 tagged at its N-terminus with fluorescent proteins [41, 43, 45, 57-59]. Redistribution to peripheral sites has also been shown in one study by immunostaining of endogenous STIM1 in human T cells and Jurkat cells, RBL cells, and PC12 cells [40]. This is an important result, as it validates the use of exogenous labeled STIM1 to follow its trafficking and localization.
Figure 1.

Redistribution of STIM1 following store depletion. Two Jurkat cells expressing GFP-STIM1 were imaged by wide-field epifluorescence microscopy (A) or TIRF microscopy (B) at rest and after treatment with 8 μM cyclopiazonic acid (a SERCA inhibitor) for > 2 min (store depleted). After store depletion, STIM1 moves from being diffusely distributed throughout the ER to accumulate in junctional ER located close to the PM. Scale bars, 5 μm.
Several lines of evidence suggest that the redistribution of STIM1 into puncta is a necessary step in CRAC channel activation. First, the timing of STIM1 puncta formation at a cell population level roughly parallels the time course of ICRAC in separate patch-clamp experiments [39, 40]. Because cell-to-cell variability can obscure the precise temporal order of puncta formation and channel opening in population measurements, the time courses have also been compared simultaneously in single cells. In Jurkat cells stimulated by intracellular EGTA dialysis in whole-cell recordings, GFP-STIM1 translocation preceded ICRAC activation by 0-20 sec, with an average of 6-10 s [41]. Thus, the time course of redistribution is rapid enough to be involved in activating CRAC channels. The source of the variable delay between translocation and ICRAC activation is unknown, but one possibility is that additional events must occur before CRAC channels can open, such as the accumulation of Orai1 opposite STIM1 puncta (see below). A similar lag was observed whether stores were depleted passively in whole-cell recording by intracellular EGTA or actively in a more physiological manner, using the perforated-patch configuration with anti-CD3 mAb to crosslink the T cell receptor complex (generating IP3 as an activating signal) [41].
Further evidence that STIM1 redistribution is required for CRAC channel activation comes from observations that STIM1 puncta colocalize with open CRAC channels [59] (see next section). In addition, EF-hand mutants of STIM1 which constitutively activate CRAC channels are prelocalized in puncta [39, 40], and deletion of several domains of STIM1 (SAM, coiled-coil/ERM, and other C-terminal regions) prevent the formation of puncta and activation of SOCE in parallel [57]. These findings together argue that understanding the spatial organization of the STIM1 puncta, in particular their location relative to the CRAC channel, the ER and the PM, is a prerequisite for elucidating the mechanism of SOCE.
5. Where does STIM1 accumulate after store depletion and how does it get there?
From confocal and total internal reflection fluorescence (TIRF) microscopy studies it is clear that puncta form close to the PM, but there has been debate over whether they reside in the ER or some other organelle near the PM, or whether they result from insertion of STIM1 into the PM. The strongest evidence in support of PM insertion comes from one study in which STIM1 reactivity over the plasma membrane was shown by immunoelectron microscopy, and streptavidin was able to pull down STIM1 following biotinylation of cell surface proteins [40]. The amount of STIM1 recovered in this way increased after store depletion, consistent with a depletion-stimulated insertion of STIM1 into the PM. This conclusion carries the important implication that STIM1 in the PM could interact in cis with CRAC channels to cause activation. However, the dimensions of antibodies used for immunolabeling are comparable to the distance between the ER and the PM at sites of STIM1 accumulation (see below), making it difficult to distinguish ER from PM localization in immunoelectron microscopy [41]. Also, another group found no increase in STIM1 pulled down by streptavidin after depletion [43], and biotinylation of STIM1 itself was not verified in either study, raising the possibility that STIM1 in the ER may have been recovered by binding to biotinylated PM proteins. This seems plausible given the close interaction of the ER and PM at sites of CRAC channel activation [41, 59] and the fact that STIM1 and Orai1 can be co-immunoprecipitated [60, 61] (see below).
On the other hand, a large number of studies using labeled STIM1 proteins have shown that it does not enter the PM acutely after store depletion. In these studies, STIM1 was tagged by inserting a label (a fluorescent protein or a FLAG or HA tag) at the N-terminus, just downstream of the signal sequence. Assuming a membrane fusion mechanism for PM insertion, the labels would be expected to change from an intraluminal to an extracellular location. However, store depletion failed to cause extracellular exposure of the tags, as shown by the inability of acidic extracellular pH to quench GFP fluorescence [41, 58], and a lack of staining with anti-GFP [39, 45], anti-HA, or anti-FLAG antibodies [41, 57]. These studies indicate that labeled STIM1 does not enter the PM on the time scale of CRAC channel activation. One caveat is that the tags (8 to >300 amino acids) added to the N-terminus of STIM1 could alter its trafficking, possibly preventing its insertion into the PM. Nevertheless, although these exogenously supplied STIM1 proteins are confined to the ER, they are able to activate CRAC channels after knockdown [39, 45, 62] or even knockout [57] of endogenous STIM1. Thus, it appears that STIM1 relays the depletion signal to the CRAC channel from its location in the ER.
Although a small fraction of endogenous STIM1 can be labeled by antibodies to the N-terminus in intact cells [42, 43], the function of STIM1 in the PM is unclear at this point. Antibodies to STIM1 in the PM were reported to inhibit ICRAC [44] but this was not confirmed in a second study [62]. STIM1 can form homodimers via its cytoplasmic coiled-coil domains [57], raising the notion that STIM1 in the PM may interact with ER-localized STIM1 at ER-PM junctions [40, 44]; however, the function of such a complex is uncertain given that puncta formation and ICRAC activation is apparently normal in STIM1-knockout DT40 cells after expression of FLAG-tagged STIM1 that is confined to the ER [57].
The most precise description of STIM1 puncta thus far comes from electron microscopy of Jurkat cells expressing an HRP-labeled STIM1 [41]. In resting cells, HRP reaction product is found in tubular structures throughout the cytoplasm that closely resemble ER tubules labeled with an ER-targeted HRP. Following TG treatment, however, dense HRP reaction product is seen in tubules juxtaposed to the PM, with reduced labeling of the cytoplasmic tubules. Importantly, these structures, found within 10-25 nm of the PM, resemble the junctional ER seen in Jurkat cells expressing ER-targeted HRP [41] and described previously in a variety of cells from oocytes to yeast [63-66]. Thus, these results suggest that upon store depletion, STIM1 migrates within the ER membrane to a subset of ER tubules juxtaposed to the PM. Interestingly, even though bulk movement of the ER towards the PM was undetectable by TIRF microscopy of Jurkat cells expressing an ER-targeted DsRed, a quantitative analysis of the EM data showed that junctional tubules in Jurkat cells increase in number by ∼50% after store depletion [41]. These results imply that the increase in contacts involves small local movements of the ER; our current hypothesis is that the ER is constantly probing the cell surface, and by increasing the affinity of the ER for the PM, store depletion lengthens the lifetime of these transient contacts. STIM1 itself is a candidate for mediating this effect, as it is able to sense intralumenal [Ca2+] and STIM1 overexpression increases the lengths of ER-PM contacts in a store depletion-dependent way [41].
These EM studies have several important implications. First, they show for the first time that the N-terminus of STIM1 actually resides within the ER, based on the accumulation of the HRP reaction product in that location. This places the EF hand of STIM1 also within the lumen where it can “sense” ER Ca2+. Second, they show that STIM1 puncta represent the accumulation of STIM1 in ER subregions that are separated from the PM by a gap small enough to permit proteins in the two membranes to interact directly. Finally, the increase in ER-PM contacts after store depletion provides the first direct evidence for the “induced-coupling” model of SOCE, in which store depletion causes the ER to approach the PM to activate SOCs. The model was originally based on the ability of cortical actin polymerization to inhibit SOCE in transformed smooth muscle cells [22]. The fact that cortical actin does not inhibit SOCE in other cells [23] might be explained if junctions in some cells are more labile than in others.
An important question yet to be resolved is how does STIM1 travel to peripheral ER sites to form puncta? One simple mechanism would be a diffusion trap, in which STIM1 diffuses randomly within the ER and is trapped at peripheral sites by binding to an anchoring protein. This idea is consistent with measurements showing that the ER does not as a whole move towards the PM after store depletion [41]. An alternative mechanism has been proposed by Baba et al. [57], who suggest that STIM1 is transported actively to puncta in an ER subcompartment that moves along microtubules. This idea is based in part on observations that a fraction of overexpressed GFP-STIM1 moves as a “tubulovesicular shape” along linear tracks in resting DT40 cells, and that these structures diminish after store depletion as puncta form. We have observed similar rapidly moving aggregates of labeled STIM1 after overexpression in Jurkat T cells (Supplemental Movie 1) and HEK 293 cells (M. Wu, E. Covington, R. Luik, and R. Lewis, unpublished data). However, the nature of these moving aggregates and their possible physiological role in SOCE is not yet clear. The prominence of the aggregates increases with the level of STIM1 overexpression, and they persist after store depletion (Supplemental Movie 1). In cells expressing very low levels of GFP-STIM1, TG induces puncta to form in the absence of rapidly moving tubular aggregates (Supplemental Movie 2). These observations raise the possibility that the aggregates are the result of high level overexpression of STIM1. An important unresolved question is therefore whether STIM1 transport occurs by an active, motor-driven, or passive, diffusion-based mechanism, and what causes it to accumulate at ER-PM junctions.
6. The dynamic dyad: defining the elementary unit of store-operated Ca2+ entry
To begin to understand how STIM1 at ER-PM junctions activates CRAC channels, it is essential to know first where CRAC channels open. The only available marker for active CRAC channels is the elevated [Ca2+]i microdomains that form near open channels. Several prior studies using Ca2+-sensitive dyes have shown that SOCE occurs within 10-100 μM of the ER [19-21], but the applied imaging techniques lacked sufficient resolution to pinpoint the relationship between individual influx sites and junctional ER structures. Much higher resolution is possible with TIRF microscopy, which has been used to image Ca2+ microdomains near voltage-gated Ca2+ (CaV) channels on a submicron scale [67, 68]. Luik et al. [59] adapted this technique to image [Ca2+]i microdomains following store depletion in Jurkat cells. This measurement was particularly challenging because the CRAC channel has a unitary conductance <1/100 of that of most CaV channels [8, 69]. To restrict the Ca2+ signal to within 0.5 μm from its source, cells were loaded through the patch pipette with a low-affinity, fast on-rate fluorescent Ca2+ indicator (fluo-5F) and a 50-fold excess of a high-affinity, slow on-rate Ca2+ chelator (EGTA) [67, 70]; TIRF microscopy provided an additional increase in signal-to-noise ratio by rejecting fluorescence signals arising more than 100-200 nm from the PM.
Using this approach, Luik et al. [59] determined that STIM1 puncta overlap generally with regions of Ca2+ influx, but that puncta are normally too close together to be able to resolve individual influx sites given the spatial spread of the fluo-5F signal by diffusion. Interestingly, the space between puncta is increased by treatment with cytochalasin D, an agent that promotes actin filament depolymerization and causes the STIM1 puncta to coalesce. Importantly, when applied during or after store depletion, cytochalasin D reorganizes but does not disrupt the basic CRAC activation machinery: the amount of peripheral STIM1 remains constant, the sites of STIM1 accumulation at junctional ER look normal at the EM level (Fig. 2), and ICRAC is unchanged [59]. In cytochalasin-treated cells, individual puncta were spatially well resolved, and overlapped very closely with [Ca2+]i elevations, to within the resolution limit of the light microscope (Fig. 2). These results demonstrate that STIM1 delivers its activating signal locally to CRAC channels in the PM.
Figure 2.

The elementary unit of store-operated Ca2+ entry. Electron micrograph and TIRF images of individual Jurkat cells treated with TG to deplete stores and exposed to cytochalasin D to disperse puncta. (A) HRP-STIM1 accumulates in ER tubules (arrows) located 10-25 nm from the plasma membrane (pm). (B) TIRF images of Cherry-STIM1 fluorescence at the cell footprint (left) and at a single punctum within the boxed area. Ca2+ influx (middle) at the same punctum is expressed as pseudocolored ΔF/F0 ratios from minimum (blue; −0.01) to maximum (red; 1.05) levels. Pseudocolored contour lines of Ca2+ influx density overlaid on the Cherry-STIM1 image (right) show a close association between the Ca2+ influx site and the STIM1 punctum. (C) TIRF images of Cherry-STIM1 fluorescence at the cell footprint (left) and at a single punctum within the boxed area. Cherry-STIM1 (red) and GFP-myc-Orai1 (green) colocalize (merged image). Reproduced from [59]. Copyright 2006 Rockefeller University Press.
Local activation could in principle stem from the local activation of a subset of widely dispersed channels or from activation of a tightly packed cluster of channels opposite STIM1. By coexpressing labeled STIM1 and Orai1 in Jurkat cells, Luik et al. found that while Orai1 is widely dispersed through the cell in the resting state, store depletion causes it to accumulate at PM sites directly opposite the STIM1 puncta, with the same approximate time course as STIM1 redistribution (Fig. 2) [59]. Similar results were also reported in HEK 293 cells overexpressing labeled STIM1 and Orai1 [58] (P. Hoover and R. Lewis, unpublished observations). These findings reveal an unprecedented mechanism for channel activation in which the stimulus for channel activation (in this case store depletion) causes a channel and its sensor, residing in separate membranes, to collect at sites of close apposition where interactions leading to channel opening can occur.
Just how STIM1 communicates with the CRAC channel at these ER-PM junctions is not clear, although several kinds of evidence suggest that the two proteins may be physically connected. First, the distance separating the ER and PM at the junctions (10-25 nm) is small enough to permit protein-protein interactions [41]. Second, Orai1 remains closely associated with STIM1 even after reorganization of STIM1 puncta by cytochalasin D [59]. Third, STIM1 and Orai1 (or their Drosophila homologs) can be co-immunoprecipitated [60, 61] to an extent that may increase following store depletion [61], as might be expected from the accumulation of STIM1 and Orai1 at junctional sites. It is not known whether the interaction of the two proteins is direct or is mediated through intermediary partners or a diffusible messenger.
An important recent finding is that the cytoplasmic C-terminus of STIM1, when expressed in Jurkat cells, is able to activate ICRAC in cells with presumably full stores [71]. This result suggests that the C-terminus of STIM1 is constitutively “active.” If this is the case, the lack of ICRAC in resting cells could be explained by the fact that only a small proportion of STIM1 is present at ER-PM junctions when stores are full; accordingly, store depletion would elicit activation by concentrating STIM1 at these sites. Oligomerization of STIM1 is likely to play an important role in this process. Thus far, two parts of the molecule have been implicated in oligomer formation: the coiled-coil region appears to dimerize in a constitutive fashion [57], and the isolated EF-SAM region in vitro has been shown to form dimers and higher order oligomers upon Ca2+ removal [46]. The accumulation of STIM1 oligomers at the junctions may serve to stabilize interactions with Orai1 leading to channel activation.
It is important to note that although much of the evidence thus far is consistent with a physical interaction model, this does not rule out a role for a diffusible activator such as Ca2+ influx factor (CIF) [37]. In fact, both classes of mechanism are known to underlie excitation-contraction coupling in striated muscle, where CaV channels in the PM stimulate Ca2+ release through ryanodine receptors in the SR across a similarly narrow cleft (see next section). In skeletal muscle, communication occurs through conformational coupling of the two channels, whereas in cardiac muscle it occurs by a diffusible messenger, Ca2+ [72]. One approach to reconciling the two types of models for SOCE will be to test whether the C-terminus of STIM1 activates ICRAC independently of the ER, CIF, and its associated iPLA2 pathway.
7. Implications of CRAC-channel choreography
The studies on STIM1/Orai1 movements and CRAC channel opening described above establish directly and for the first time the elementary structural and functional units underlying SOCE (Fig. 3). These units assemble through the accumulation of STIM1 and Orai1 at closely apposed sites in the ER and PM, respectively, to trigger Ca2+ entry at discrete sites. This mechanism has several interesting implications.
Figure 3.
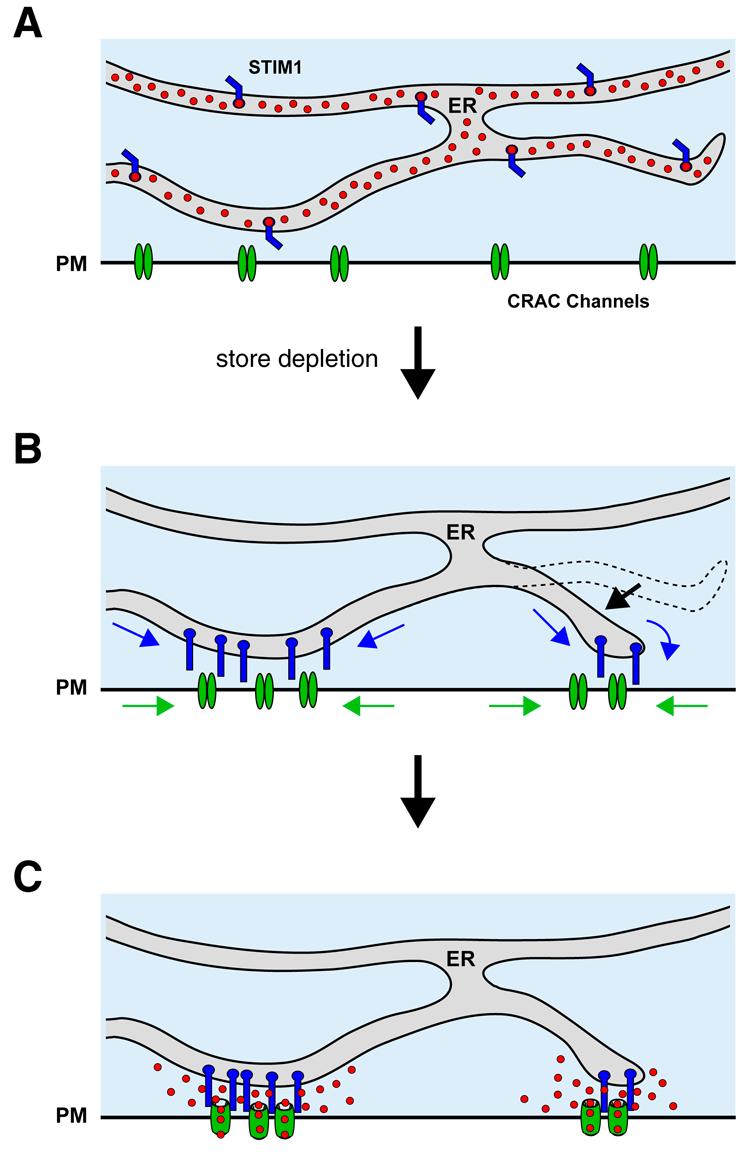
A current model for store-operated Ca2+ entry. (A) In cells with full ER Ca2+ stores, Ca2+-bound STIM1 and Orai1 are distributed throughout the endoplasmic reticulum and the PM, respectively. (B) Store depletion promotes functional coupling of Ca2+-free STIM1 and Orai1 by driving the coordinated redistribution of both proteins to closely apposed sites in the ER and PM, and by increasing the number of these sites. (C) The parallel accumulation of both proteins allows STIM1 and Orai1 to interact, causing the local activation of CRAC channels at individual junctions. Reproduced from [53].
The “dynamic dyad.”
The ER-PM junctions in SOCE resemble in some ways the specialized SR-PM couplings that underlie e-c coupling in striated muscle, where the SR comes to within 10 nm of the PM [73]. At these sites, excitation of CaV in the PM activates Ca2+ release through ryanodine receptors in the SR to initiate muscle contraction. Despite the similarities in structure, there are interesting differences as well. First, the flow of information in muscle is outside-in, with the CaV channel in the PM acting on the SR ( a “channel-operated store”) while SOCE signaling is inside-out, with the ER controlling the activation of a store-operated channel in the PM. More importantly, however, the muscle unit is fully organized and ready to respond well before the stimulus arrives, a means of ensuring reliable and rapid muscle contraction; in contrast, the SOCE unit is assembled on demand, forming only in response to the stimulus (store depletion), and requiring seconds for the sensor and the channel to reach their sites of interaction. The time needed for the migration of STIM1 and Orai1 to these locations settles an old argument against the physical coupling model, namely that activation by physical coupling must be extremely fast because it is so direct. The slow time course of STIM1/Orai1 redistribution creates lags between changes in store content and CRAC channel activity, possibly explaining findings that in T cells, a variety of agents that partially deplete Ca2+ stores (e.g., low doses of TG, ionomycin and TCR crosslinking) generate [Ca2+]i oscillations through the periodic activation of ICRAC[6, 74].
The specificity of Ca2+ signaling
The restriction of SOCE to discrete junctional sites that comprise only a few percent of the cell surface creates opportunities for selective signaling. By concentrating Ca2+ influx near to SERCA pumps, it optimizes the efficiency of store refilling, perhaps explaining why in certain systems stores can reload with extracellular Ca2+ without a detectable rise in [Ca2+]i [15, 16, 75]. In general, the specificity of Ca2+ signaling through SOCE could be enhanced by targeting downstream effectors to the ER-PM junctions. Several enzymes have been reported to respond selectively or preferentially to SOCE, including adenylate cyclase, nitric oxide synthase, Cl− channels, and PM Ca2+-ATPase [1]. Thus, an important goal will be to dissect the protein contingent of the ER-PM junctions.
The “CRAC” store
Under some conditions, Ca2+ can be released from the ER without triggering ICRAC, and this uncoupling phenomenon has prompted the idea that a specialized subset of ER (the “CRAC store”) is responsible for controlling SOCE (reviewed in [1]). Given that store depletion triggers the formation of STIM1/Orai1 clusters at junctional sites, we expect that local Ca2+ depletion at these sites would be required to maintain those clusters. Thus, if Ca2+ is released distally it may not reduce ER Ca2+ at the junctions and therefore may fail to activate ICRAC. Conversely, local depletion at junctional sites may play the determining role under physiological conditions, and give the appearance of a separate Ca2+ store whose content is more directly connected to CRAC channel activation.
8. Perspectives
The discoveries of STIM1 and Orai1 have unleashed a barrage of new studies that have taken our understanding of SOCE to the next level. Yet, major puzzles remain. A primary question is still how does STIM1 activate the CRAC channel on a molecular level? How is the formation and maintenance of junctional ER sites controlled, and what signals direct the migration and accumulation of STIM1 and Orai1 at these sites? The complexity of the CRAC channel activation mechanism portends a multitude of potential sites for modulation. Ultimately, the dependence of SOCE on the architecture of the ER and its precise apposition to PM sites may explain why so many different experimental interventions over the years were able to modulate SOCE without illuminating the underlying mechanism.
Supplementary Material
Rapidly moving GFP-STIM1 aggregates under conditions of high-level overexpression. The TIRF movie shows a Jurkat cell overexpressing GFPSTIM1 at a high level before and after treatment with TG (1 μM). In such cells, GFPSTIM1 was often seen in rapidly moving aggregates in addition to the normal ER localization. The behavior of GFP-STIM1 aggregates was not significantly affected by store depletion. Frame interval, 20 s; scale bar, 5 μm.
STIM1 puncta form without “tubulovesicular shapes” in cells expressing a low level of GFP-STIM1. The TIRF movie shows a Jurkat cell expressing GFP-STIM1 before and after treatment with TG (1 μM). At this low level of overexpression, GFP-STIM1 exhibits a characteristic ER localization in the resting cell without tubular aggregates. Store depletion causes GFP-STIM1 to redistribute into puncta near the plasma membrane. Frame interval, 20 s; scale bar, 5 μm. Reproduced from [41]. Copyright 2006 Rockefeller University Press.
Acknowledgements
The authors gratefully acknowledge support from the NIH and a gift from the Mathers Charitable Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parekh AB, Putney JW., Jr. Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 2.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 3.Putney JW., Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 4.Putney JW, Jr., Bird GS. The inositol phosphate-calcium signaling system in nonexcitable cells. Endocr. Rev. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. [DOI] [PubMed] [Google Scholar]
- 5.Penner R, Matthews G, Neher E. Regulation of calcium influx by second messengers in rat mast cells. Nature. 1988;334:499–504. doi: 10.1038/334499a0. [DOI] [PubMed] [Google Scholar]
- 6.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regulation. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 8.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. U S A. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prakriya M, Lewis RS. Store-operated calcium channels: properties, functions and the search for a molecular mechanism. Adv. Mol. Cell Biol. 2004;32:121–140. [Google Scholar]
- 10.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- 11.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 12.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 2005;202:651–662. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aub DL, McKinney JS, Putney JW., Jr. Nature of the receptor-regulated calcium pool in the rat parotid gland. J. Physiol. 1982;331:557–565. doi: 10.1113/jphysiol.1982.sp014391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muallem S, Fimmel CJ, Pandol SJ, Sachs G. Regulation of free cytosolic Ca2+ in the peptic and parietal cells of the rabbit gastric gland. J. Biol. Chem. 1986;261:2660–2667. [PubMed] [Google Scholar]
- 15.Negulescu PA, Machen TE. Release and reloading of intracellular Ca stores after cholinergic stimulation of the parietal cell. Am. J. Physiol. 1988;254:C498–504. doi: 10.1152/ajpcell.1988.254.4.C498. [DOI] [PubMed] [Google Scholar]
- 16.Mogami H, Nakano K, Tepikin AV, Petersen OH. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- 17.Putney JW., Jr. Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 18.Petersen CC, Berridge MJ. Capacitative calcium entry is colocalised with calcium release in Xenopus oocytes: evidence against a highly diffusible calcium influx factor. Pflügers Arch. 1996;432:286–292. doi: 10.1007/s004240050135. [DOI] [PubMed] [Google Scholar]
- 19.Hüser J, Holda JR, Kockskamper J, Blatter LA. Focal agonist stimulation results in spatially restricted Ca2+ release and capacitative Ca2+ entry in bovine vascular endothelial cells. J. Physiol. 1999;514:101–109. doi: 10.1111/j.1469-7793.1999.101af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaconi M, Pyle J, Bortolon R, Ou J, Clapham D. Calcium release and influx colocalize to the endoplasmic reticulum. Curr. Biol. 1997;7:599–602. doi: 10.1016/s0960-9822(06)00259-4. [DOI] [PubMed] [Google Scholar]
- 21.Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J. Physiol. 2005;564:737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- 23.Bakowski D, Glitsch MD, Parekh AB. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBL-1 cells. J. Physiol. 2001;532:55–71. doi: 10.1111/j.1469-7793.2001.0055g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 25.Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- 26.Scott CC, Furuya W, Trimble WS, Grinstein S. Activation of store-operated calcium channels: assessment of the role of snare-mediated vesicular transport. J. Biol. Chem. 2003;278:30534–30539. doi: 10.1074/jbc.M304718200. [DOI] [PubMed] [Google Scholar]
- 27.Bakowski D, Burgoyne RD, Parekh AB. Activation of the store-operated calcium current ICRAC can be dissociated from regulated exocytosis in RBL-1 cells. J. Physiol. 2003;553:387–393. doi: 10.1113/jphysiol.2003.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates--a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 29.Berridge MJ. Capacitative calcium entry. Biochem. J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- 31.Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999;18:1303–1308. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parekh AB, Terlau H, Stühmer W. Depletion of InsP3 stores activates a Ca2+ and K+ current by means of a phosphatase and a diffusible messenger. Nature. 1993;364:814–818. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- 34.Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 35.Gilon P, Bird GJ, Bian X, Yakel JL, Putney JW., Jr. The Ca2+-mobilizing actions of a Jurkat cell extract on mammalian cells and Xenopus laevis oocytes. J. Biol. Chem. 1995;270:8050–8055. doi: 10.1074/jbc.270.14.8050. [DOI] [PubMed] [Google Scholar]
- 36.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 37.Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem. Sci. 2005;30:378–387. doi: 10.1016/j.tibs.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Roos J, Digregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manji SS, Parker NJ, Williams RT, van Stekelenburg L, Pearson RB, Dziadek M, Smith PJ. STIM1: a novel phosphoprotein located at the cell surface. Biochim. Biophys. Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 43.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr. Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 44.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc. Natl. Acad. Sci. U S A. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of STIM1 via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 47.Demaurex N, Frieden M. Measurements of the free luminal ER Ca2+ concentration with targeted “cameleon” fluorescent proteins. Cell Calcium. 2003;34:109–119. doi: 10.1016/s0143-4160(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 48.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 49.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. U S A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat. Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 53.Luik RM, Lewis RS. New insights into the molecular mechanisms of store-operated Ca2+ signaling in T cells. Trends Mol. Med. 2007;13:103–107. doi: 10.1016/j.molmed.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 55.Soboloff J, Spassova MA, Dziadek MA, Gill DL. Calcium signals mediated by STIM and Orai proteins--a new paradigm in inter-organelle communication. Biochim. Biophys. Acta. 2006;1763:1161–1168. doi: 10.1016/j.bbamcr.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 56.Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim. Biophys. Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 57.Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U S A. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem. Biophys. Res. Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 59.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store-depletion or translocation to the plasma membrane. J. Physiol. 2006 doi: 10.1113/jphysiol.2006.122432. DOI: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pichler H, Gaigg B, Hrastnik C, Achleitner G, Kohlwein SD, Zellnig G, Perktold A, Daum G. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur. J. Biochem. 2001;268:2351–2361. doi: 10.1046/j.1432-1327.2001.02116.x. [DOI] [PubMed] [Google Scholar]
- 64.Gardiner DM, Grey RD. Membrane junctions in Xenopus eggs: their distribution suggests a role in calcium regulation. J. Cell Biol. 1983;96:1159–1163. doi: 10.1083/jcb.96.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dadsetan S, Shishkin V, Fomina AF. Intracellular Ca2+ release triggers translocation of membrane marker FM1-43 from the extracellular leaflet of plasma membrane into endoplasmic reticulum in T lymphocytes. J. Biol. Chem. 2005;280:16377–16382. doi: 10.1074/jbc.M501202200. [DOI] [PubMed] [Google Scholar]
- 66.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 67.Zenisek D, Davila V, Wan L, Almers W. Imaging calcium entry sites and ribbon structures in two presynaptic cells. J. Neurosci. 2003;23:2538–2548. doi: 10.1523/JNEUROSCI.23-07-02538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaumont V, Llobet A, Lagnado L. Expansion of calcium microdomains regulates fast exocytosis at a ribbon synapse. Proc. Natl. Acad. Sci. U S A. 2005;102:10700–10705. doi: 10.1073/pnas.0501961102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prakriya M, Lewis RS. Regulation of CRAC Channel Activity by Recruitment of Silent Channels to a High Open-probability Gating Mode. J. Gen. Physiol. 2006;128:373–386. doi: 10.1085/jgp.200609588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J. Physiol. 1998;512:677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat. Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 72.Tanabe T, Mikami A, Numa S, Beam KG. Cardiac-type excitation-contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature. 1990;344:451–453. doi: 10.1038/344451a0. [DOI] [PubMed] [Google Scholar]
- 73.Franzini-Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann. N Y Acad. Sci. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- 74.Dolmetsch RE, Lewis RS. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol. 1994;103:365–388. doi: 10.1085/jgp.103.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jousset H, Frieden M, Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to SERCA pumps silently refill the endoplasmic reticulum. J. Biol. Chem. 2007 doi: 10.1074/jbc.M609551200. doi:10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rapidly moving GFP-STIM1 aggregates under conditions of high-level overexpression. The TIRF movie shows a Jurkat cell overexpressing GFPSTIM1 at a high level before and after treatment with TG (1 μM). In such cells, GFPSTIM1 was often seen in rapidly moving aggregates in addition to the normal ER localization. The behavior of GFP-STIM1 aggregates was not significantly affected by store depletion. Frame interval, 20 s; scale bar, 5 μm.
STIM1 puncta form without “tubulovesicular shapes” in cells expressing a low level of GFP-STIM1. The TIRF movie shows a Jurkat cell expressing GFP-STIM1 before and after treatment with TG (1 μM). At this low level of overexpression, GFP-STIM1 exhibits a characteristic ER localization in the resting cell without tubular aggregates. Store depletion causes GFP-STIM1 to redistribute into puncta near the plasma membrane. Frame interval, 20 s; scale bar, 5 μm. Reproduced from [41]. Copyright 2006 Rockefeller University Press.