

Abstract
Age-related skeletal muscle sarcopenia has been extensively studied and smooth muscle sarcopenia has been recently described, but age-related cardiac sarcopenia has not been previously examined. Therefore, we evaluated adult (7.5±0.5 months; n=27) and senescent (31.8±0.4 months; n=26) C57BL/6J mice for cardiac sarcopenia using physiological, histological, and biochemical assessments. Mice do not develop hypertension, even into senescence, which allowed us to decouple vascular effects and monitor cardiac-dependent variables. We then developed a mathematical model to describe the relationship between age-related changes in cardiac muscle structure and function. Our results showed that, compared to adult mice, senescent mice demonstrated increased left ventricular (LV) end diastolic dimension, decreased wall thickness, and decreased ejection fraction, indicating dilation and reduced contractile performance. Myocyte numbers decreased, and interstitial fibrosis was punctate but doubled in the senescent mice, indicating reparative fibrosis. Electrocardiogram analysis showed that PR interval and QRS interval increased and R amplitude decreased in the senescent mice, indicating prolonged conduction times consistent with increased fibrosis. Intracellular lipid accumulation was accompanied by a decrease in glycogen stores in the senescent mice. Mathematical simulation indicated that changes in LV dimension, collagen deposition, wall stress, and wall stiffness precede LV dysfunction. We conclude that age-related cardiac sarcopenia occurs in mice and that LV remodeling due to increased end diastolic pressure could be an underlying mechanism for age-related LV dysfunction.
Keywords: aging, sarcopenia, cardiac, hypertrophy, fibrosis
1. Introduction
Sarcopenia is defined as age-related muscle atrophy characterized by the involuntary decline in fatty-free muscle mass, strength, and function (Evans 1995; Morley et al. 2001; Roubenoff and Castaneda 2001). Sarcopenia is due to both muscle disuse and the inherent biological deterioration associated with age, which decreases quality of life in the elderly and contributes significantly to morbidity and mortality. Sarcopenia has been well documented in skeletal muscle (Lindle et al. 1997; Muller et al. 2006; Melov et al. 2007) and has also been demonstrated in aortic smooth muscle (Kunieda et al. 2006). Sarcopenia in skeletal muscle is associated with a decline in protein synthesis, resulting in a decrease in type II fibers and a decrease in myosin heavy chains (Morley et al. 2001; Hameed et al. 2002). It has not been determined, however, whether sarcopenia occurs in cardiac muscle. Based on what has been shown in skeletal muscle, our working definition of cardiac sarcopenia is the age-related involuntary decline in cardiac myocyte numbers and myocardial function.
Age-related cardiac changes are well described in humans and rats (Khan et al. 2002; Lakatta 2003). Quantitative studies have shown that a continuous loss of myocytes accompanied by reactive hypertrophy of the remaining cells in aging heart (Anversa et al. 1986; Anversa et al. 1990; Olivetti 1991; Khan et al. 2002). However, it is not clear whether age-related cardiac sarcopenia is an outcome of these observed changes. Since mice are widely used in cardiovascular studies, it is important to study cardiac aging in mice. Mice do not develop hypertension with age, even into senescence (Batkai et al. 2007), which allowed us to evaluate the cardiac sarcopenia in aging mice independent of vascular effects.
In addition, while various changes in ventricular structure and function have been observed in aging heart (Lim et al. 1999; Kawaguchi et al. 2003; Li et al. 2005; Batkai et al. 2007), the relationship between these changes and the underlying mechanism that explains them are poorly understood.
Mechanical stress induces tissue growth and remodeling (Fung 1993; Taber 1998; Berenji et al. 2005). Therefore, changes in LV end diastolic pressure can lead to changes in LV wall stress, structure and function. In addition, LV function and structural changes can also affect mechanical load and wall stress. Age-related changes in mechanical properties of myocardial and vascular tissue have been characterized experimentally (Nguyen et al. 2001; Cheitlin 2003), and collagen turnover in response to changes in wall stress has been demonstrated in adult myocardium (Miller and Tyagi 2002). End-diastolic wall strain is normalized in hypertrophic remodeling (Emery and Omens 1997). Mathematical models have been used to describe the dynamics of remodeling in bone, skeletal muscle, and arteries (Fung 1993; Rachev et al. 1998; Taber 1998), but a model for cardiac aging is lacking. Establishing a cardiac aging model would provide a tool for us to understand underlying mechanisms and to predict outcomes.
In order to test the hypothesis that age-related sarcopenia occurs in cardiac muscle, we evaluated left ventricular muscle mass and function in senescent mice for indications of cardiac sarcopenia and then developed a mathematical model to characterize the dynamic process of LV remodeling in aging.
2. Materials and Methods
2.1 Animal procedures
All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington, DC, 1996) and were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio.
Male and female C57BL/6J wild type adult (7.5±0.5 months, n=27, 13 male and 14 female) and senescent (31.8±0.4 months, n=26, 14 male and 12 female) mice were used. Equal numbers of males and females were used for each measurement and the results of both male and female data were combined and analyzed together.
2.2 Noninvasive blood pressure measurements
After 3 training sessions over 1 week, arterial blood pressure was measured in awake mice (6 female and 6 male adult and 6 female and 6 male senescent mice) using a tail-cuff recording device (Model MC 4000MSP, Hatteras Instruments). Each training session and the recording session consisted of 15 min equilibration followed by 5 preliminary cycles and 10 measurement cycles.
2.3 Echocardiography
Transthoracic echocardiography was performed in all mice in this study under light anesthesia (0.5% isoflurane) with spontaneous respiration. The mice were maintained at ambient body temperature using a temperature controlled surgical board underneath the mice in a supine position. Heart rate was determined from a surface electrocardiogram and maintained at a minimum of 400 beats per min. Two-dimensional targeted M-mode echocardiographic recordings were obtained with an optimized 15.7 MHz transducer (ATL, HDI 5000). Left ventricular volumes and wall thickness were measured from the long axis and short axis views, respectively. Fractional shortening and ejection fraction were then calculated using the formulas: Fractional shortening = [(End diastolic dimension − End systolic dimension) / End diastolic dimension] * 100; and Ejection fraction = [(End diastolic volume − End systolic volume) / End diastolic volume] * 100.
2.4 Electrocardiography (ECG)
Limb-lead electrocardiograms were acquired for all mice at the time of sacrifice. The ECGs were acquired at 4000HZ using the THM100 electrocardiogram monitoring system for mice (Indus Instruments), with the mice under light (0.5%) isoflurane anesthesia. An average of 29±3 cycles were analyzed for each mouse, using the Chart 5.5 electrocardiogram module (ADInstruments).
2.5 Necropsy measurements
Following echocardiogram and electrocardiogram acquisition, mice were sacrificed under anesthesia. All mice were initially anesthetized by placing in a flow-through system containing 3-4% isoflurane in a 100% oxygen mix. Following loss of consciousness, the mice were placed on a modified mask assembly that allows a continuous flow of 2-3% isoflurane in an oxygen mix. An endotracheal tube was inserted and connected to a rodent ventilator. The ventilator was set at a volume of 0.20-0.25 cc and a rate of 200-250 cycles/ min maintaining a flow rate of 2-3% isoflurane. The inhalation anesthetic flow rate was adjusted based upon heart rate, pedal reflex, and pulse oximetry. Arterial oxygen saturation, pulse rate, breath rate, and pulse distension were all monitored using the MouseOx™ (STARR Life Science) to confirm comparable sedation between groups.
Body and lung weights were measured at the time of sacrifice. Wet and dry lung weights were taken to determine water percentage. Hearts were harvested by two different procedures: perfusion fixation for histology and snap frozen for biochemical analysis.
Perfusion fixation
Thirteen adult mice (7 female and 6 male) and thirteen senescent mice (6 female and 7 male) were used for this protocol. Retrograde perfusion with glutaraldehyde was achieved by hanging the hearts and perfusing the coronary vasculature by aortic cannulation at a pressure of 80 mmHg. The hearts were first perfused with cardioplegia for 1 min to arrest them in diastole, and the coronary bed was then perfused for 2 min with 2.5% glutaraldehyde. Left ventricle (LV) and right ventricle (RV) were then separated and weighed. The LV was cut in half along the circumference and postfixed in 2.5% glutaradehyde for 24 hours. To determine whether paraffin-embedding affected myocyte cross sectional area measurements, half of the samples were dehydrated in ethanol and embedded in paraffin, while the other half of the samples were frozen in OCT media.
Biochemical analysis
Fourteen adult mice (7 female and 7 male) and thirteen senescent mice (6 female and 7 male) were used for this protocol. Hearts were arrested in diastole by intraventricular injection of cardioplegia. The LV and RV were separated and weighed. The LV was then sliced into 3 sections and photographed for gross examination. A biopsy from the LV apex was used to determine water content by weighing the sample before and after drying in a 50°C incubator overnight. The mid section was homogenized in soluble and insoluble buffers for protein extraction and collagen evaluation, and the base of LV was frozen in OCT media for lipid and glycogen analyses.
2.6 Histology
Myocyte cross sectional area
Glutaraldehyde-fixed sections were stained with hematoxylin and eosin (H&E). Five regions from each slide were randomly scanned at 40x magnification, and 10 myocytes were measured from each section using Image-Pro Plus (Version 5.0.1, MediaCybernetics). Only myocytes with a round shape and central nuclei were measured, as described previously (Lindsey et al. 2003). Because sections paraffin-embedded or frozen in OCT media showed similar results, the data for the two techniques were combined.
Myocyte number and inter-myocyte space
Myocyte numbers were quantified by determining nuclei number (as a percentage of total tissue area) with hematoxylin and eosin stained sections. Intermyocyte space was calculated as the percent of white space within the section boundary. Six images were scanned at 100x magnification for each section and then analyzed with Image-Pro Plus.
Collagen content
Picrosirius red staining was used to stain interstitial collagen on paraffin sections (Dolber and Spach 1987). Five regions from each slide were randomly scanned at 40x magnification. The percentage of collagen was measured with Image-Pro Plus.
Glycogen Content
Periodic acid schiff staining was used to detect carbohydrate content (glycogen) (Vargas et al. 1999). Serial sections digested with amylase were used to evaluate a same blockpositive control for each sample. Images were scanned at 40x magnification.
Lipid content
Fresh cryosections (5μm) were fixed with formalin and washed with water and 60% isopropanol after warming up the slides for 30 min at room temperature. Slides were then stained with freshly prepared oil red o stain for 15 min (Klett et al. 1995). After rinsing with 60% isopropanol, nuclei were lightly stained by hematoxylin. Images were scanned at 100x magnification.
2.7 Biochemistry
Total protein concentration
Fresh myocardial samples from the mid section of the LV were homogenized in soluble protein extraction buffer (250 mM sucrose, 1 mM EDTA and 10 mM Tris-HCL, pH 7.4) supplemented with 1x protein inhibitor cocktail (Complete Mini, Roche). Following centrifugation at 14,000 rpm for 40 min, the pellets were homogenized in insoluble protein extraction reagent (Sigma Reagent 4), which contained urea, thiourea, Trizma and the detergent C7BzO. Both soluble and insoluble homogenates were subsequently analyzed for protein concentration using the Bradford assay. The insoluble homogenates were diluted 1:40 to avoid interference from urea. Total protein (5 μg) from each sample (soluble and insoluble fractions) were separated on 4-12% polyacrylamide gels to confirm concentration accuracy.
Total collagen concentration
A microplate reader–based quantitation of collagens was performed as described previously (Ducharme et al. 2000). Briefly, collagen standards and protein lysates (10 μg total protein) were plated on microtiter wells in triplicate, and dried onto the plates at 37°C. The wells were stained with 0.1% Sirius red F3BA in saturated picric acid for 1 hour, and washed with 10 mM HCl and 0.1 mM NaOH. Absorbance was read at 540 nm. Collagen levels were determined from the collagen standard curve.
2.8 Statistics
All data are expressed as mean±SEM. The unpaired Student's t test was used to compare values between the two groups. A value of p<0.05 was considered statistically significant.
2.9 Mathematical modeling
To illustrate possible links between changes in LV dimensions, wall properties, mechanical stress, and LV function, we employed a linear-elastic cylindrical LV model to simulate the changes in the LV wall during aging. This model is based on the growth-remodeling theory in biomechanics (Fung 1990; Humphrey 2002; Gleason and Humphrey 2004).
Our underlying hypothesis was that changes in LV dimensions, wall properties, and function were driven by LV adaptation to mechanical stress. Our model focused on the end-diastolic properties, because end-diastolic properties have been shown to most influence remodeling (Emery and Omens 1997). In addition, end-diastolic strain is normalized in hypertrophic remodeling (Emery and Omens 1997). We assumed that collagen turnover responded to changes in wall stress, based on previous experimental results of adult myocardium and arteries (Miller and Tyagi 2002; Gleason and Humphrey 2004). In addition, LV wall volume was assumed to be the same between the two age groups, since our experimental data showed that LV mass and total protein concentrations isolated from the adult and senescent groups were similar.
Let the LV be cylindrical and the free and end diastolic (deformed) radius be denoted by R and r respectively. The circumferential deformation (stretch ratio λ) of LV was given by
| (1) |
The wall stress was estimated by Laplace's law
| (2) |
where p is the LV end diastolic pressure and h is the wall thickness. The linear elastic stress-strain relationship was approximated by
| (3) |
where E is the Young's modulus of the LV wall.
We used the following remodeling rate equation as a first order approximation to account for age-related changes:
| (4), |
| (5) |
where subscript 0 represents the initial values, is the initial circumferential deformation ratio, represents the growth ratio of the LV radius, vc represents the volume ratio of the collagen in the LV wall, τc and τR are time constants. Accordingly, the wall thickness can be determined using the constant LV volume assumption and the incompressible conditions (Fung 1993).
Furthermore, using the mixture theory, the Young's modulus E of the myocardium was determined by the Young's modulii of muscle and collagen (Fung 1993; Gleason and Humphrey 2004):
| (6) |
where Ec, Em and vc, vm are the modulus and volume ratios of myocardial collagen and muscle, respectively.
These equations were implemented on a PC using Matlab to simulate the cardiac aging process. Experimental data from the adult mice were used as the initial input.
3. Results
3.1 Changes in left ventricular function
Left ventricular (LV) function in adult and senescent mice was assessed by echocardiography and electrocardiography, and the results are summarized in Table 1. End diastolic and systolic dimensions and volume all increased in senescent mice compared to adult mice (all p<0.05), indicating dilation. LV wall thickness in both the posterior wall and interventricular septum decreased in senescent mice compared with the adult mice, also indicating dilation. Despite the decrease in LV function, as evidenced by a 9% decline in ejection fraction, stroke volume was maintained in the senescent mice. Systolic and diastolic arterial blood pressures were not different between adult and senescent groups.
Table 1.
Echocardiographic and blood pressure measurements.
| Adult | Senescent | p Value | |
|---|---|---|---|
| Age (months) | 7.5±0.5 | 31.8±0.4 | <0.0001 |
| Heart Rate (beats per min) | 484±6 | 463±7 | 0.03 |
| End Systolic Dimension (mm) | 2.45±0.06 | 2.86±0.09 | <0.001 |
| End Diastolic Dimension (mm) | 3.79±0.06 | 4.15±0.08 | <0.001 |
| Fractional Shortening (%) | 35±1 | 31±1 | <0.01 |
| Interventricular Septal Wall Thickness (mm; diastolic) | 0.81±0.01 | 0.71±0.02 | <0.001 |
| Posterior Wall Thickness (mm; diastolic) | 0.80±0.02 | 0.72±0.02 | <0.001 |
| End Systolic Volume (μl) | 12.1±0.7 | 18.6±1.4 | <0.001 |
| End Diastolic Volume (μl) | 38.4±1.5 | 45.8±2.4 | 0.02 |
| Ejection Fraction (%) | 69±1 | 60±2 | <0.001 |
| Stroke Volume (μl) | 26±1 | 27±1 | 0.15 |
| Systolic Blood Pressure (mmHg) | 114±3 | 111±3 | 0.48 |
| Diastolic Blood Pressure (mmHg) | 102±3 | 102±4 | 0.96 |
Data presented are AVG±SEM. Sample sizes for the echocardiographic analysis are 27 adult (14 female and 13 male) and 26 senescent (12 female and 14 male). Sample sizes for the blood pressure measurements are 12 adult (6 female and 6 male) and 12 senescent (6 female and 6 male).
Electrocardiogram analysis demonstrated that the PR interval and QRS interval increased and R amplitude decreased in the senescent mice (Figure 1), indicative of prolonged conduction times in senescence consistent with increased fibrosis.
Figure 1.
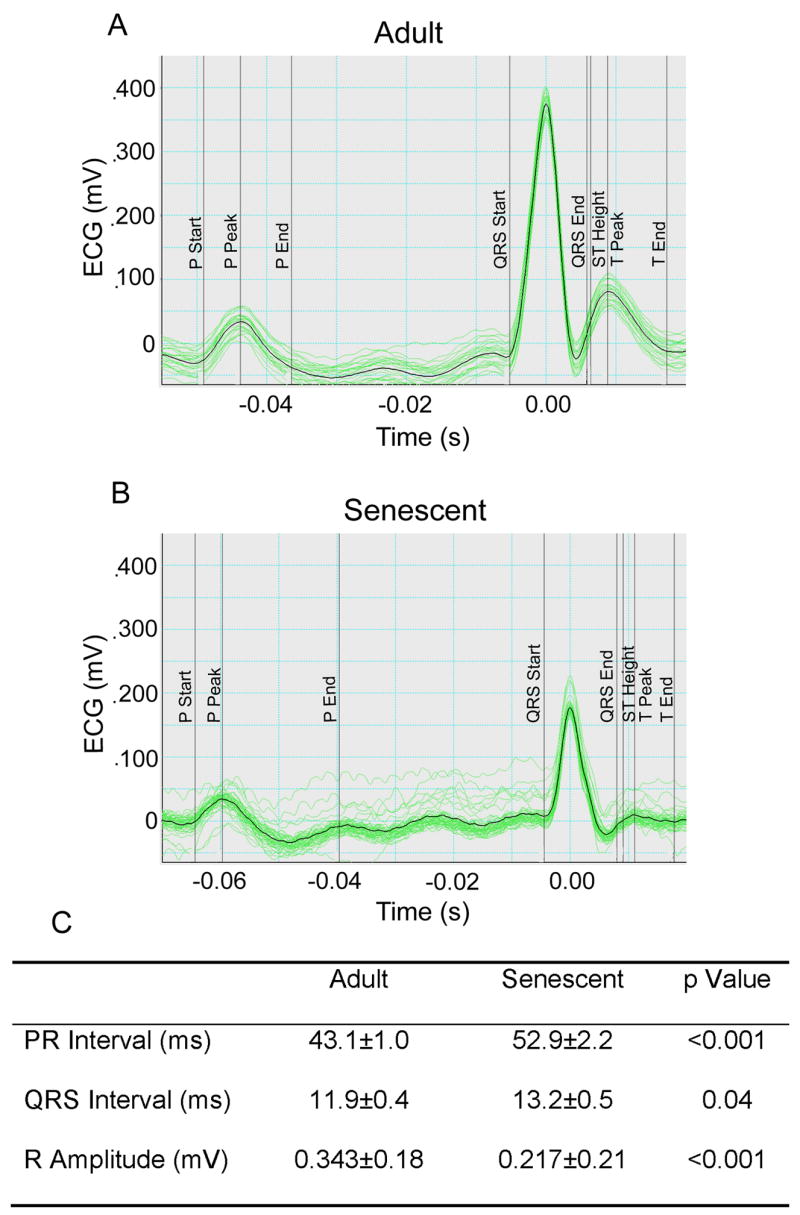
Representative electrocardiograms (ECGs) in lead II recorded from lightly anesthetized adult (A; n=27) and senescent (B; n=26) mice. Senescence increased the PR and QRS intervals and decreased R amplitude (C).
3.2 Necropsy results
Body weight, LV mass and right ventricular (RV) mass were similar between the two groups (Table 2). Compared to adult mice, lung weight significantly increased 17% in the senescent mice (p<0.05). To determine whether the increase in lung weight indicated pulmonary edema, wet to dry ratios were calculated. The increase in lung weight was not due to pulmonary edema, as both groups showed similar percentages of water.
Table 2.
Necropsy results.
| Adult | Senescent | p Value | |
|---|---|---|---|
| Body Weight (g) | 26.4±0.8 | 25.2±0.9 | 0.29 |
| Left Ventricle Mass (mg) | 92.6±3.1 | 93.2±3.7 | 0.68 |
| Right Ventricle Mass (mg) | 25.1±1.0 | 26.3±1.0 | 0.92 |
| Lung Weight (mg) | 141±3 | 165±16 | <0.01 |
| Lung water composition (%) | 78±1 | 79±1 | 0.20 |
Data presented are AVG±SEM. Sample sizes for body weight, left ventricle mass, and right ventricle mass measurements are 27 adult (14 female and 13 male) and 26 senescent (12 female and 14 male). Sample sizes for the lung weight measurements are 12 adult (6 female and 6 male) and 12 senescent (6 female and 6 male).
3.3 Histology and biochemistry results
Myocyte cell numbers, measured by percentage of nuclei, decreased from 6.3±0.4% in adult mice to 4.9±0.3% in senescent mice (p<0.05) (Figure 2C). Compared with the adult group, myocyte cross sectional areas increased 30% (p<0.05) (Figure 2D) and interstitial (inter-myocyte) space increased 62% (p<0.05) (Figure 2E) in the senescent mice. Interstitial fibrosis was punctate but significantly increased in the senescent mice, indicating reparative fibrosis (Figure 3). In the senescent mice, oil red o staining revealed increased intracellular lipid accumulation (Figure 4A and B), while periodic acid schiff staining showed a concomitant decrease in glycogen stores (Figure 4C and D).
Figure 2.

H&E staining images in paraffin sections of adult (n=15) and senescent mice (n=14) were scanned at a magnifications of 40x and 100x. Myocyte cross sectional areas were calculated from circumstance of myocyte with round and central nuclear measured with the software of Echo-Station. Compared with adult group, myocyte cross sectional areas increased 40% in senescent group (p<0.05) (A). Quantitation of the percentage of white space to the total stained area and blue color staining (hematoxylin) to the pink (Eosin) were performed. Intermyocyte space increased 62% in senescent group compared with adult group (p<0.05) (B). Myocyte cell numbers decreased in senescent mice (C).
Figure 3.

Picrosirius red staining was performed for detection of interstitial collagen on the slides both in adult (A) and senescent mice (B). Slides were scanned at 40x magnification. The percentages of red staining to the background (yellow) were quantified with the software of Image-Pro (C). Interstitial fibrosis was punctate but significantly increased in the senescent mice, indicative of reparative fibrosis.
Figure 4.

Intercellular lipids and extracellular lipid were detected by Oil Red O staining with the cryosections from adult mice (A) and senescent mice (B). Arrows show the positive staining (red). The images were photographed at 100x magnification. Periodic acid Schiff (PAS) staining on paraffin embedded slides from adult mice (C) and senescent mice (D) were used for detection of carbohydrate content (Glycogen). Images were photographed at 40x magnification. In the senescent mice, oil red o staining revealed increased intracellular lipid accumulation while periodic acid schiff staining showed a concomitant decrease in glycogen stores.
Water composition in the myocardial tissue was not significantly different between groups (71±1% water for adult vs 69±2% water for senescent; p=0.22). There were also no changes in total protein, soluble protein, and insoluble protein in the myocardium of the adult and senescent mice.
3.4 Model simulation results
The model described by equations 1 to 6 indicated that an increase in pressure-induced stress will stimulate both cell growth and ECM deposition, and thus change the LV dimension and collagen volume. Collagen deposition increases wall stiffness. The cell growth and ECM deposition form feedback loops to change the LV end diastolic dimension -- the former is a positive feedback while the latter is a negative feedback as shown in Figure 5. Furthermore, the adaptative changes in LV end diastolic dimension feed back to adjust changes in wall stress/strain.
Figure 5.
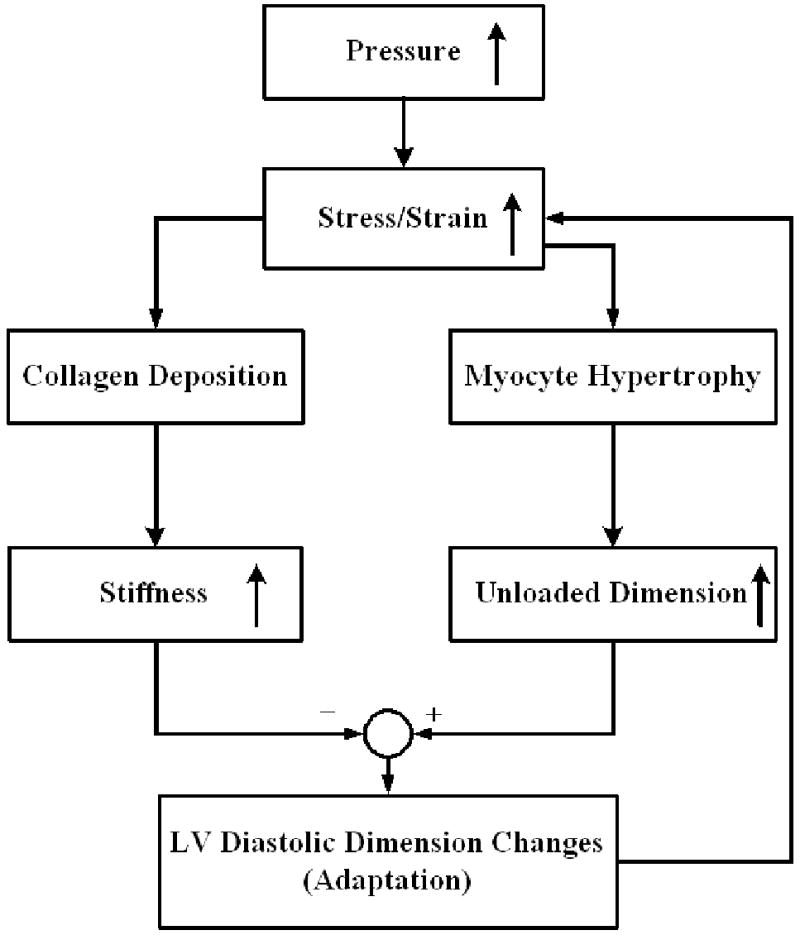
A flow chart illustrating the positive and negative feedback loops that lead to the age-related LV remodeling. Our working model is that changes in LV structure and function are driven by LV adaptation to mechanical stress and the model focuses on end diastolic changes (see text for details). The increase stress and strain activates both negative (collagen deposition and increased stiffness) and positive (myocyte hypertrophy and increased dimensions) feedback loops. The end diastolic dimension changes in turn feed back to change the wall stress.
To simulate age-related remodeling, we used data for adult mice, either from our experimental measurements or from the literature, as the initial input values (see Table 3). The simulation time spanned 24 months, corresponding to the difference between the 7.5 month old and 31.5 month old data points. The simulation results (Figure 6) predict changes in wall dimensions (R) and the corresponding changes in wall thickness (Hd), and collagen volume (vc) with the same tendency as the experimental results given in Table 1 and Figure 3. By fitting the dimension and collagen content changes obtained from our experimental measurements, the time constants τR and τc were determined to be 0.25 and 1.125, respectively. Additionally, the model simulations predict age-related increases in wall stress σ and elastic modulus E. These results indicate that the LV dimension, collagen deposition, and wall stress are inter-related. These results also support the hypothesis that cardiac sarcopenia is associated with stress-driven remodeling that leads to further LV structural changes in aging mice.
Table 3.
Input parameter values used in model simulation.
| Initial Values
(7.5 months) |
Final Values
(31.5 months) |
Source | |
|---|---|---|---|
| LV End Diastolic radius (rd0; mm) | 1.895 | 2.075 | Table 1 |
| LV End Diastolic wall thickness (h0; mm) | 0.80 | 0.72 | Table 1 |
| Collagen volume ratio (vc0; %) | 2.26 | 4.77 | Figure 3 |
| LV End Diastolic Pressure (mm Hg) | 4.5 | 7.0 | (Batkai et al. 2007) |
| Collagen modulus (Ec0; kPa) | 500 | (Fung 1993) | |
| Muscle modulus (Em0; kPa) | 370 | (Fung 1993) |
Figure 6.
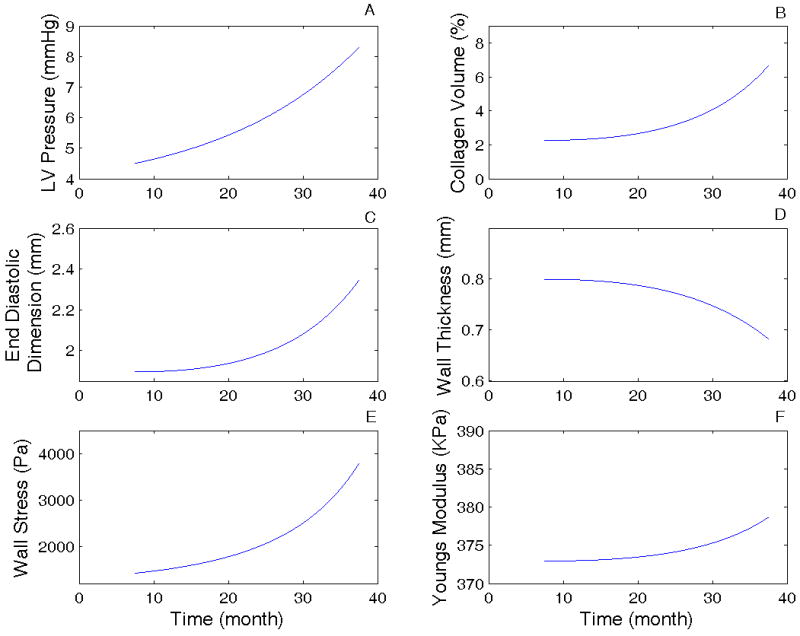
Model simulation results of the LV remodeling process with aging based on the model equations and the flow chart (see equations 1-6 and Figure 5). (A) LV end-diastolic pressure changes with age. The change in pressure was assumed to be the driving force for the remodeling (see text for details); (B) Collagen volume ratio changes with aging; (C) End diastolic LV dimension (radius) changes with age; (D) LV wall mechanical stress changes with age; and (E) Young's modulus changes with age. See Table 3 for initial input values used in the simulation.
4. Discussion
This study is the first integrated investigation of age-related cardiac sarcopenia in mice using physiology, histology, and biochemistry assessments in combination with mathematical modeling. The most important findings of this study are that: (1) in senescent mice, the number of myocytes decreases while the remaining individual myocytes hypertrophy; intracellular lipids accumulate with a concomitant decrease in glycogen stores; and interstitial fibrosis increases, indicative of increased reparative fibrosis. (2) Changes in LV structure are accompanied by changes in LV function, with decreased LV wall thickness and ejection fraction indicating dilation and reduced contractile performance in the senescent mice. Together, these changes are indicative of cardiac sarcopenia. (3) Mathematical modeling indicates an inter-relationship between these events, suggesting that strain driven remodeling might be a mechanism for the age-related structural and functional changes. Our results suggest that age-related cardiac sarcopenia likely contributes to depressed LV function in the absence of overt cardiovascular disease, which may contribute to the poor response of the elderly to cardiac injury.
We and others have previously reported that from middle-age and old-age (but not senescent), mice showed increases in wall thickness and LV mass, with no difference in ejection fraction (Burgess et al. 2001; Slama et al. 2004; Lindsey et al. 2005; Rosa et al. 2005). Age-related cardiac hypertrophy has also been demonstrated in middle-aged and old rats (Yin et al. 1980; Besse et al. 1993). Anversa and colleagues demonstrated that age-related hypertrophy is a compensatory mechanism in response to a loss in the total number of cardiomyocytes, primarily those located in the endocardium (Anversa et al. 1990). Our current results showed that LV mass was not different between the adult and senescent groups, while myocytes mass decreased in the senescent group. These results indicate that myocardial compensatory growth that occurs primarily during the transition from young to middle-age groups, and is maintained into old age, is decompensated in senescence for a net effect loss of myocyte mass. Myocyte loss with aging has been identified in the rat, consistent with our results (Katzberg et al. 1977; Korecky and Rakusan 1978; Anversa et al. 1990). Increased myocyte size, indicated by increased myocyte cross sectional area, may be an important compensatory mechanism to maintain normalized cardiac output in unstressed conditions in senescent mice, while LV contractile performance is depressed. Jan Vijg's laboratory has demonstrated that cell to cell variation in cardiac myocyte gene expression increases with age, suggesting a stochastic deregulation of gene expression (Bahar et al. 2006). Whether this deregulation contributes to myocyte loss remains to be determined.
The electrocardiogram results were consistent with the changes in dimensions and increased fibrosis. A significant senescent prolongation of the PR interval reflected local fibrotic changes in the transitional cell zone, compact portion of the AV node, or His-Purkinje system. The expected prolongation of the QRS complex was also noted in senescence, consistent with ventricular fibrotic and geometric changes affecting the Purkinje fibers.
Earlier pathological studies in human and in rat have reported increased myocardial fibrosis in aged hearts (Tomanek et al. 1972; Eghbali et al. 1989; Besse et al. 1993). Similarly our results showed that the total protein levels were maintained in the senescent group while the collagen levels were increased. These results indicated that other individual proteins (e.g., contractile proteins, membrane proteins and additional extracellular proteins) were likely decreased. Based on previous measurements on collagen RNA and protein turnover rates during aging (Meerson et al. 1978; Crie et al. 1981), the increase in total collagen is likely a reflection of increased synthesis and/or decreased turnover rather than increased mRNA levels.
Furthermore, a metabolic shift from glucose to fat is also indicative of sarcopenia (Rosa et al. 2005; Marzetti and Leeuwenburgh 2006). Increases in both myocardial intracellular and extracellular lipids and decreases in myocardial glycogen stores in senescent mice suggest that the transformation rate from lipid to glycogen utilization occurs with aging, so that glycogen storage in the myocardium is insufficient. In the young healthy myocardium, glycogen is preferentially oxidized over exogenous glucose and undergoes significant turnover (Goodwin et al. 1995; Goodwin et al. 1996; Henning et al. 1996). Our results suggest that the decrease of glycogen storage may contribute to LV dysfunction in senescent mice. Melov and colleagues recently reported that the skeletal muscle strength of older adults was 59% weaker than younger adults, indicative of sarcopenia (Melov et al. 2007). Examination of the skeletal muscle transcriptome revealed enrichment in genes associated with mitochondrial function, suggesting an age-related mitochondrial impairment. Future studies associating cardiac sarcopenia with mitochondrial dysfunction would be warranted.
This study included equal numbers of both male and female mice, in order to determine whether cardiac sarcopenia occurred in both genders. This study was not designed, however, to evaluate differences between genders. The prevalence of cardiac disease increases significantly with age, independent of gender, and we wanted to evaluate whether sarcopenia occurred in both male and female mice. We did observe some obvious gender differences, including higher body weights and LV masses in males compared to females at both age groups. Several studies have reported differences between men and women in skeletal muscle sarcopenia, including different rates of concentric strength decline and degree of grip strength loss (Hurley 1995; Castillo et al. 2003). Future studies are needed to determine whether the rate of cardiac sarcopenia development differs with gender.
There were several limitations in this study. Blood pressure was measured using the tail-cuff method, which is noninvasive and widely used in mice (Krege et al. 1995). However, we did not evaluate LV pressures. Therefore, values of LV diastolic blood pressure used in our model simulation were taken from the literature (Batkai et al. 2007). More studies examining the roles of other cell types, particularly cardiac fibroblasts, will help to elucidate additional mechanisms associated with age. Future directions also include evaluating whether therapies such as exercise training alter the kinetics of age-related cardiac sarcopenia (Kwak et al. 2006).
Nonetheless, using the cylindrical LV model and adaptation equations, we were able to simulate the temporal changes in LV dimensions structure, and wall stiffness. First, the model predicted an increase in LV dimension, which agreed with the changes observed in our experiments and previous studies (Lim et al. 1999; Batkai et al. 2007). Secondly, our model also predicted an increase in stress and Young's modulus. Though we did not measure the wall stiffness directly, the predicted results showed the same trends in literatures (Brooks and Conrad 2000; Lieber et al. 2004). Thirdly, our model results also suggest that these changes are inter-related and that strain-driven remodeling could be a possible mechanism to link the observed structural and functional changes in aging mice. While the model constructed in this project does not include every conceivable parameter, it does demonstrate that the experimentally observed changes are most likely connected. The model suggests that changes in the aging heart are part of an adaptation/growth process and involve interactions between the biological remodeling and mechanical stress. The current model provides a foundation for further studies to develop a comprehensive model to better describe the LV aging process. Though limitations exist, modeling provides us a useful tool to simulate LV remodeling process to illustrate the quantitative relationships between the structural and functional changes and predict possible outcomes in the aging heart at normal conditions and following pharmacologic or genetic interventions. Furthermore, the model could also be useful for human aging since human LV end diastolic pressure shows similar trend of increased LV pressure and end diastolic dimension with aging (Kawaguchi et al. 2003).
In conclusion, this is the first report to document and mathematically model age-related cardiac sarcopenia in mice. The decrease in myocyte cell numbers, the increase in individual myocyte hypertrophy, and the increased reparative fibrosis likely combine to depress left ventricular function. Cardiac sarcopenia may explain, in part, why elderly patients respond poorly to cardiovascular events.
Acknowledgments
The authors acknowledge support from the NIH (R01 HL-75360- MLL and P01 AG-20591- HVR) and the NSF (0602834-HCH, CAREER award 0644646- HCH and 0649172-YJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anversa P, Hiler B, Ricci R, Guideri G, Olivetti G. Myocyte cell loss and myocyte hypertrophy in the aging rat heart. J Am Coll Cardiol. 1986;8:1441–1448. doi: 10.1016/s0735-1097(86)80321-7. [DOI] [PubMed] [Google Scholar]
- Anversa P, Palackal T, Sonnenblick EH, Olivetti G, Meggs LG, Capasso JM. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res. 1990;67:871–885. doi: 10.1161/01.res.67.4.871. [DOI] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle MET, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Batkai S, Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Cravatt BF, Csiszar A, Ungvari ZI, Pacher P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression and apoptosis in mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol. 2007;293:H909–918. doi: 10.1152/ajpheart.00373.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:H8–16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- Besse S, Assayag P, Delcayre C, Carre F, Cheav S, Lecarpentier Y, Swynghedauw B. Normal and hypertrophied senescent rat heart: mechanical and molecular characteristics. American Journal of Physiology: Legacy Content. 1993;265:H183–190. doi: 10.1152/ajpheart.1993.265.1.H183. [DOI] [PubMed] [Google Scholar]
- Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000;32:187–195. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- Burgess ML, McCrea JC, Hedrick HL. Age-associated changes in cardiac matrix and integrins. Mechanisms of Ageing and Development. 2001;122:1739–1756. doi: 10.1016/s0047-6374(01)00296-2. [DOI] [PubMed] [Google Scholar]
- Castillo EM, Goodman-Gruen D, Kritz-Silverstein D, Morton DJ, Wingard DL, Barrett-Connor E. Sarcopenia in elderly men and women: the Rancho Bernardo study. Am J Prev Med. 2003;25:226–231. doi: 10.1016/s0749-3797(03)00197-1. [DOI] [PubMed] [Google Scholar]
- Cheitlin MD. Cardiovascular physiology-changes with aging. Am J Geriatr Cardiol. 2003;12:9–13. doi: 10.1111/j.1076-7460.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- Crie J, Millward DJ, Bates PC, Griffin EE, K W. Age-related alterations in cardiac protein turnover. Journal of Molecular and Cellular Cardiology. 1981;13:589–598. doi: 10.1016/0022-2828(81)90329-1. [DOI] [PubMed] [Google Scholar]
- Dolber PC, Spach MS. Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain Technol. 1987;62:23–26. doi: 10.3109/10520298709107961. [DOI] [PubMed] [Google Scholar]
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M, Eghbali M, Robinson TF, Seifter S, Blumenfeld OO. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc Res. 1989;23:723–729. doi: 10.1093/cvr/23.8.723. [DOI] [PubMed] [Google Scholar]
- Emery JL, Omens JH. Mechanical regulation of myocardial growth during volume-overload hypertrophy in the rat. Am J Physiol. 1997;273:H1198–1204. doi: 10.1152/ajpheart.1997.273.3.H1198. [DOI] [PubMed] [Google Scholar]
- Evans WJ. What is sarcopenia? J Gerontol A Biol Sci Med Sci. 1995;50 doi: 10.1093/gerona/50a.special_issue.5. Spec No: 5-8. [DOI] [PubMed] [Google Scholar]
- Fung YC. Biomechanics: Motion, Flow, Stress, and Growth. Springer; New York: 1990. [Google Scholar]
- Fung YC. Biomechanics: Mechanical Properties of Living Tissues. Springer Verlag; New York: 1993. [Google Scholar]
- Gleason RL, Humphrey JD. A mixture model of arterial growth and remodeling in hypertension: altered muscle tone and tissue turnover. J Vasc Res. 2004;41:352–363. doi: 10.1159/000080699. [DOI] [PubMed] [Google Scholar]
- Goodwin G, Ahmad F, H T. Preferential oxidation of glycogen in isolated working rat heart. Journal of Clinical Investigation. 1996;97:1409–1416. doi: 10.1172/JCI118561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin G, Arteaga JR, H T. Glycogen turnover in the isolated working rat heart. Journal of Biological Chemistry. 1995;270:9234–9240. doi: 10.1074/jbc.270.16.9234. [DOI] [PubMed] [Google Scholar]
- Hameed M, Harridge SD, Goldspink G. Sarcopenia and hypertrophy: a role for insulin-like growth factor-1 in aged muscle? Exerc Sport Sci Rev. 2002;30:15–19. doi: 10.1097/00003677-200201000-00004. [DOI] [PubMed] [Google Scholar]
- Henning S, Wambolt RB, Schonekess BO, Lopaschuk GD, MF A. Contribution of glycogen to aerobic myocardial glucose utilization. Circulation. 1996;93:1549–1555. doi: 10.1161/01.cir.93.8.1549. [DOI] [PubMed] [Google Scholar]
- Humphrey JD. Cardiovascular Solid Mechanics: Cells, Tissues, and Organs. Springer; New York: 2002. [Google Scholar]
- Hurley BF. Age, gender, and muscular strength. J Gerontol A Biol Sci Med Sci. 1995;50 doi: 10.1093/gerona/50a.special_issue.41. Spec No: 41-44. [DOI] [PubMed] [Google Scholar]
- Katzberg A, Farmer B, Harris R. The predominance of binucleation in isolated rat heart myocytes. American Journal of Anatomy. 1977;149:489–499. doi: 10.1002/aja.1001490406. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Hay I, Fetics B, Kass DA. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation. 2003;107:714–720. doi: 10.1161/01.cir.0000048123.22359.a0. [DOI] [PubMed] [Google Scholar]
- Khan AS, Sane DC, Wannenburg T, Sonntag W. Growth hormone, insulin-like growth factor-1 and the aging cardiovascular system. Cardiovascular Research. 2002;54:25–35. doi: 10.1016/s0008-6363(01)00533-8. [DOI] [PubMed] [Google Scholar]
- Klett CP, Palmer AA, Dirig DM, Gallagher AM, Riosecco-Camacho N, Printz MP. Evidence for differences in cultured left ventricular fibroblast populations isolated from spontaneously hypertensive and Wistar-Kyoto rats. J Hypertens. 1995;13:1421–1431. [PubMed] [Google Scholar]
- Korecky B, Rakusan K. Normal and hypertrophic growth of the rat heart: changes in cell dimensions and number. American Journal of Physiology: Legacy Content. 1978;234:H123–128. doi: 10.1152/ajpheart.1978.234.2.H123. [DOI] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Hagaman JR, Smithies O. A Noninvasive Computerized Tail-Cuff System for Measuring Blood Pressure in Mice. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, Nagai T, Kaneko S, Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Song W, Lawler JM. Exercise training attenuates age-induced elevation in Bax/Bcl-2 ratio, apoptosis, and remodeling in the rat heart. Faseb J. 2006;20:791–793. doi: 10.1096/fj.05-5116fje. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, LaCour KH, Yang X, Wilbert CJ, Sreejayan N, Ren J. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell. 2005;4:57–64. doi: 10.1111/j.1474-9728.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- Lieber SC, Aubry N, Pain J, Diaz G, Kim SJ, Vatner SF. Aging increases stiffness of cardiac myocytes measured by atomic force microscopy nanoindentation. Am J Physiol Heart Circ Physiol. 2004;287:H645–651. doi: 10.1152/ajpheart.00564.2003. [DOI] [PubMed] [Google Scholar]
- Lim CC, Liao R, Varma N, Apstein CS. Impaired lusitropy-frequency in the aging mouse: role of Ca(2+)-handling proteins and effects of isoproterenol. Am J Physiol. 1999;277:H2083–2090. doi: 10.1152/ajpheart.1999.277.5.H2083. [DOI] [PubMed] [Google Scholar]
- Lindle RS, Metter EJ, Lynch NA, Fleg JL, Fozard JL, Tobin J, Roy TA, Hurley BF. Age and gender comparisons of muscle strength in 654 women and men aged 20-93 yr. J Appl Physiol. 1997;83:1581–1587. doi: 10.1152/jappl.1997.83.5.1581. [DOI] [PubMed] [Google Scholar]
- Lindsey ML, Goshorn DK, Squires CE, Escobar GP, Hendrick JW, Mingoia JT, Sweterlitsch SE, Spinale FG. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovascular Research. 2005;66:410–419. doi: 10.1016/j.cardiores.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Lindsey ML, Yoshioka J, MacGillivray C, Muangman S, Gannon J, Verghese A, Aikawa M, Libby P, Krane SM, Lee RT. Effect of a Cleavage-Resistant Collagen Mutation on Left Ventricular Remodeling. Circ Res. 2003;93:238–245. doi: 10.1161/01.RES.0000085580.45279.60. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp Gerontol. 2006;41:1234–1238. doi: 10.1016/j.exger.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Meerson F, Javich M, Lerman M. Decrease in the rate of RNA and protein synthesis and degradation in the myocardium under long-term compensatory hyperfunction and on aging. Journal of Molecular and Cellular Cardiology. 1978;10:145–159. doi: 10.1016/0022-2828(78)90039-1. [DOI] [PubMed] [Google Scholar]
- Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS ONE. 2007;2:e465. doi: 10.1371/journal.pone.0000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Tyagi SC. Mutation in collagen gene induces cardiomyopathy in transgenic mice. J Cell Biochem. 2002;85:259–267. doi: 10.1002/jcb.10130. [DOI] [PubMed] [Google Scholar]
- Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med. 2001;137:231–243. doi: 10.1067/mlc.2001.113504. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ, 2nd, Csete M, Faulkner JA, Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Nguyen CT, Hall CS, Scott MJ, Zhu Q, Marsh J, Wickline SA. Age-related alterations of cardiac tissue microstructure and material properties in Fischer 344 rats. Ultrasound Med Biol. 2001;27:611–619. doi: 10.1016/s0301-5629(01)00343-x. [DOI] [PubMed] [Google Scholar]
- Olivetti G. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circulation Research. 1991;68:1560–1568. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- Rachev A, Stergiopulos N, Meister JJ. A model for geometric and mechanical adaptation of arteries to sustained hypertension. J Biomech Eng. 1998;120:9–17. doi: 10.1115/1.2834313. [DOI] [PubMed] [Google Scholar]
- Rosa EF, Silva AC, Ihara SSM, Mora OA, Aboulafia J, Nouailhetas VLA. Habitual exercise program protects murine intestinal, skeletal, and cardiac muscles against aging. J Appl Physiol. 2005;99:1569–1575. doi: 10.1152/japplphysiol.00417.2005. [DOI] [PubMed] [Google Scholar]
- Roubenoff R, Castaneda C. Sarcopenia-understanding the dynamics of aging muscle. Jama. 2001;286:1230–1231. doi: 10.1001/jama.286.10.1230. [DOI] [PubMed] [Google Scholar]
- Slama M, Ahn J, Varagic J, Susic D, Frohlich ED. Long-term left ventricular echocardiographic follow-up of SHR and WKY rats: effects of hypertension and age. American Journal Of Physiology. Heart And Circulatory Physiology. 2004;286:H181–H185. doi: 10.1152/ajpheart.00642.2003. [DOI] [PubMed] [Google Scholar]
- Taber LA. Biomechanical growth laws for muscle tissue. J Theor Biol. 1998;193:201–213. doi: 10.1006/jtbi.1997.0618. [DOI] [PubMed] [Google Scholar]
- Tomanek R, Taunton C, Liskop K. Relationship between age, chronic exercise, and connective tissue of the heart. Journal of Gerontology. 1972;27:33–38. doi: 10.1093/geronj/27.1.33. [DOI] [PubMed] [Google Scholar]
- Vargas SO, Sampson BA, Schoen FJ. Pathologic detection of early myocardial infarction: a critical review of the evolution and usefulness of modern techniques. Mod Pathol. 1999;12:635–645. [PubMed] [Google Scholar]
- Yin F, Spurgeon H, Weisfeldt M, Lakatta E. Mechanical properties of myocardium from hypertrophied rat hearts. A comparison between hypertrophy induced by senescence and by aortic banding. Circulation Research. 1980;46:292–300. doi: 10.1161/01.res.46.2.292. [DOI] [PubMed] [Google Scholar]