

Summary
The first female meiotic division (MI) is uniquely prone to chromosome segregation errors through non-disjunction, resulting in trisomies and early pregnancy loss1. Here, we show a fundamental difference in the control of mammalian meiosis which may underlie such susceptibility. It involved a reversal in the well-established timing of activation of the Anaphase-Promoting Complex (APC)2, 3 by its co-activators cdc20 and cdh1. APCcdh1 was active first, during prometaphase I, and was needed in order to allow homologue congression, since loss of cdh1 speeded up MI, leading to premature chromosome segregation and a non-disjunction phenotype. APCcdh1 targeted cdc20 for degradation but not securin and cyclin B1. These were degraded later in MI through APCcdc20, making cdc20 re-synthesis essential for successful meiotic progression. The switch from APCcdh1 to APCcdc20 activity was controlled by increasing CDK1 and cdh1 loss. These findings demonstrate a fundamentally different mechanism of control for the first meiotic division in mammalian oocytes not observed in meioses of other species.
The E3 ligase activity of the Anaphase-Promoting Factor (APC) bound to its co-activator cdh1 (APCcdh1) is commonly associated with late M- and early G1-phases of the cell cycle, where it contributes to M-phase exit by degradation of mitotic proteins, while simultaneously preventing precocious DNA replication3-6. APCcdh1 activity is also observed in germinal vesicle stage (GV) mouse oocytes, equivalent to late G2, where it contributes to cyclin B1 degradation and as such is required for maintenance of GV arrest7, 8.
We wanted to establish if cdh1 had any role in meiosis I (MI) after GV breakdown (GVB), independent of its role in maintaining GV arrest. Therefore, we examined the ability of oocytes to progress through MI following microinjection with a cdh1 antisense morpholino (cdh1MO). Culture in milrinone-containing medium for 24 h following cdh1MO microinjection is sufficient to reduce cdh1 levels by >90% (hereafter ‘cdh1 knockdown oocytes’) but maintain GV arrest in the majority of oocytes, with longer term culture (48h) needed to promote GVB7. In cdh1 knockdown oocytes, which maintained arrest over 24 h, we found that progression through MI was accelerated following milrinone wash-out. Oocytes extruded a polar body (PB), which forms on completion of MI, 1.5 h earlier than non-injected oocytes (Fig 1a). This effect was attributed specifically to loss of cdh1, since it was not observed in mock cdh1 depleted oocytes through addition of a 5-base-mismatch cdh1 morpholino (5MM-cdh1MO). The acceleration of PB could have been due to a loss in cdh1 during either GV arrest or MI. However, it was attributed specifically to a function in MI because we could restore a normal PB timing by rescuing cdh1 expression after GVB. Thus, in cdh1 knockdown oocytes cdh1 cRNA injection, which was made on release from milrinone (hereafter ‘cdh1 rescue oocytes’), rescued a normal phenotype (Fig 1a).
Figure 1.
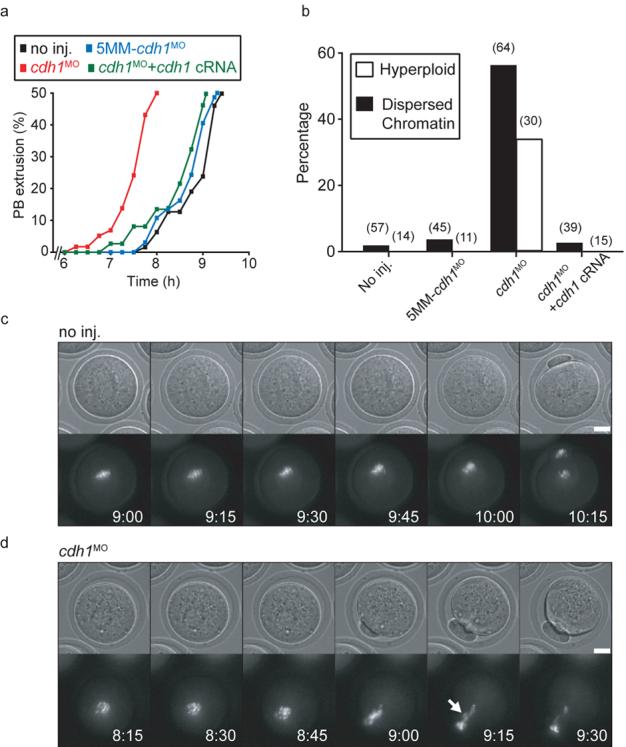
Cdh1 loss accelerated meiosis I leading to premature homologue segregation and non-disjunction. a, Timecourse of PB extrusion rates in oocytes injected with cdh1MO (n=58), 5MM-cdh1MO (n=37), cdh1MO and cdh1 cRNA (n=67); or no injections (n=64). Cdh1 knockdown accelerated PB extrusion. b, Percentages of dispersed chromatin and hyperploidy observed in matured non-injected control oocytes, or those injected with 5MM-cdh1MO, cdh1MO and cdh1 cRNA as stated. Number of oocytes examined in parentheses. c and d, Chromatin (Hoechst) and brightfield images of maturing non-injected oocytes (c) and those injected with cdh1MO (d). Cdh1 knockdown oocytes had no discernible congression of chromosomes on a metaphase I plate and had lagging chromosomes during anaphase (arrow). (c) and (d) time stamp is h:min, scale bar is 20μm. (c) and (d) are selected frames from Supplementary Movies 1 and 2. (a), (c) and (d) times are relative to GVB.
The MI acceleration in cdh1 knockdown oocytes led to premature segregation and non-disjunction of homologues. Following oocyte maturation, chromatin in cdh1 knockdown oocytes often failed to fully align on a metaphase II spindle and sometimes was very dispersed suggesting segregation of homologues had not occurred properly (Fig 1b; Supplementary Fig 1). To examine this further, chromosome spreads were performed in which the hyperploidy rate was calculated, to circumvent problems associated with chromosome loss during the fixing procedure9. About one-third of the cdh1 knockdown oocytes were hyperploid (Fig 1b), suggesting that non-disjunction had occurred in the majority of oocytes if chromosome segregation was stochastic between the PB and oocyte. These effects were attributed to loss of cdh1, since both 5MM-cdh1MO injected oocytes and cdh1 rescue oocytes formed metaphase II spindles and were not hyperploid (Fig 1b). Live-cell chromatin imaging during maturation of uninjected and cdh1 knockdown oocytes confirmed the immunofluorescence and chromosome spread results. In control oocytes, chromosomes underwent condensation after GVB, congressed on a metaphase I plate, migrated to the oocyte cortex and underwent anaphase (Fig 1c; Supplementary Movie 1). Condensed chromosomes in cdh1 knockdown oocytes however underwent anaphase without congression onto an obvious metaphase plate and so generated lagging chromosomes (Fig 1d; Supplementary Movie 2).
We wanted to establish if the acceleration of MI by cdh1 knockdown was associated with accelerated degradation of proteins known to be degraded at this time. At the mitotic metaphase-anaphase transition both cyclin B1, the regulatory subunit of CDK1, and securin, which prevents loss of cohesin from attached sisters by inhibiting separase, are degraded simultaneously10. Loss of endogenous and fluorescent protein tagged cyclin B1 and securin during MI have been reported previously but comparison of degradation in the same oocyte has not been performed11, 12. Therefore we visualised cyclin B1 and securin degradation together in an oocyte by coupling to separable fluorescent proteins, and found simultaneous degradation (Supplementary Fig S2a). When levels of cdh1 were knocked down, the initiation of both cyclin B1 (Fig 2 a,b) and securin (Supplementary Fig S2b and Fig S2c), were brought forward by about 1.5 h, a timing which is consistent with the accelerated PB extrusion observed above. This was effect was specific for cdh1, since it was not observed for the control 5MM-cdh1MO and normal timing of cyclin B1 degradation was restored in cdh1 rescue oocytes. Although cdh1 was involved in the timing of cyclin B1 and securin degradation, it appeared not to contribute to actual degradation, since their rates of loss were unaffected by cdh1 knockdown (Supplementary Fig S2d). This suggests that cyclin B1 and securin are degraded through the same mechanism in MI, possibly by APCcdc20 activity as they are in mitosis. Indeed, both cyclin B1 degradation (Fig 2c,d) and securin degradation (not shown) were blocked by addition of the spindle poison nocodazole or microinjection of cRNA to mad2, a component of the Spindle Assembly Checkpoint (SAC) pathway, which in mitosis is a well-established means of inhibiting APCcdc20 (Ref. 3). This was the case in control oocytes with unperturbed levels of cdh1 (Fig 2c), and in cdh1 knockdown oocytes (Fig 2d), suggesting that loss of cdh1 does not accelerate MI by overriding any SAC imposed arrest.
Figure 2.
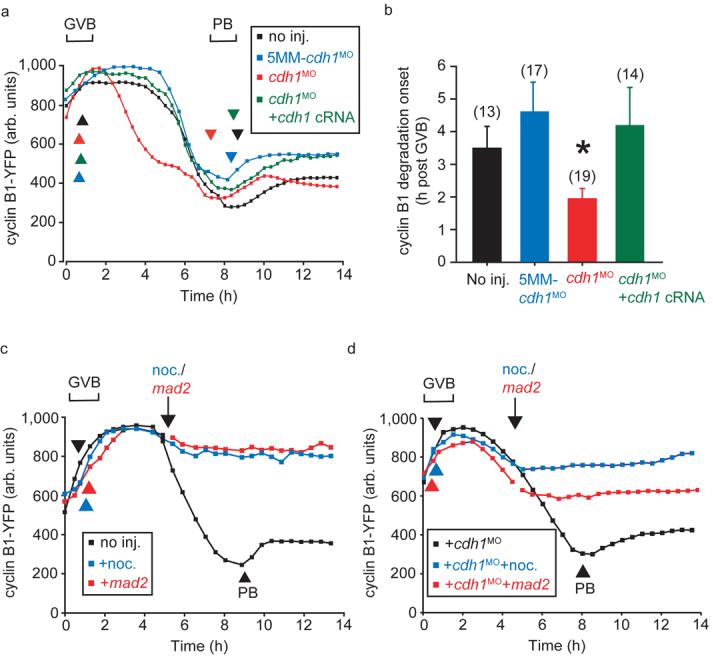
Premature cyclin B1 degradation following cdh1 knockdown. a, Cyclin B1-YFP fluorescence during maturation of oocytes injected with cdh1MO, 5MM-cdh1MO and cdh1 cRNA as stated. b, Calculated from (a), the timing of initiation of cyclin B1-YFP degradation after GVB (asterisk indicates P=0.034, significantly different from non-injected, ANOVA). The error bars represent s.d. Numbers of oocytes in parentheses, pooled for each condition from at least 2 independent experiments. c, Cyclin B1-YFP fluorescence in oocytes during maturation, at the arrow when indicated oocytes were injected with mad2 cRNA or nocodazole was added to the medium. d, As for (c), in oocytes following cdh1 knockdown. Mad2 and nocodazole both inhibited PB extrusion, which has been marked in control oocytes by the arrowheads. (c) and (d) are representative recordings of at least 12 oocytes.
The acceleration of MI and resulting non-disjunction observed following cdh1 knockdown suggested prometaphase APCcdh1 may delay homologue segregation to allow full congression on a metaphase plate. In order to demonstrate APCcdh1 activity directly during prometaphase I we examined for degradation of its substrate cdc20 (Ref 2, 3). Cdc20-GFP was used as a reporter for APCcdh1 activity but was itself biologically inactive since it lacked ability to rescue cdc20 knockdown (see Fig 4b), and we attribute this to occlusion of the C-terminal IR tail which is involved in APC binding13. However, cdc20-GFP proved to be a good reporter of APCcdh1 activity since it was degraded in oocytes shortly after GVB in a period lasting 2-4 h (Fig 3a; Supplementary Fig S3a, b). Degradation was due to cdh1, since it was blocked in cdh1 knockdown oocytes, not observed with control 5MM-cdh1MO, and was recovered in cdh1 rescue oocytes (Fig 3a). Interestingly, with respect to these fluorescent protein chimeras, the time of initiation for cdc20 degradation appeared to occur earlier than either cyclin B1 or securin, despite all being APCcdh1 substrates. Therefore, to determine if such timings were an artefact of overexpressed fluorescent protein chimeras, we examined for changes in endogenous proteins levels in uninjected maturing oocytes. Importantly we observed the same relative timing of endogenous cdc20, cyclin B1 and securin degradation as that for the chimeras (Fig 3b). Maximum cdc20 degradation was seen 3-4 h after GVB, whilst at this time both cyclin B1 and securin levels were still rising, their degradation began some 1-2 h later. Cdh1 knockdown resulted in a stabilisation of endogenous cdc20 and cdh1 rescue recovered this loss (Fig 3c). Therefore with both fluorescent protein chimeras and endogenous proteins we have observed APCcdh1 mediated cdc20 degradation ahead of any loss in cyclin B1 and securin.
Figure 4.

Essential role of cdc20 in meiosis I. a, Western blot of matured oocytes (50 per lane) following injection of cdc20MO or cdc20 cRNA as indicated at the GV stage. The morpholino prevented re-accumulation of cdc20 during MI. The uncropped Western is displayed in the Supplementary Information, Fig S4. b, PB extrusion rates following maturation in oocytes injected with cdc20MO, 5MM-cdc20MO, cdc20 cRNA, or cdc20-GFP cRNA as stated. Cdc20 knockdown blocked completion of MI which was rescued by cdc20 cRNA. c, Cyclin B1-Venus degradation rate, plotted with respect to time as a percentage of the maximum recorded fluorescence, in oocytes injected with cdc20MO (n=18); 5MM-cdc20MO (n=15); or without further microinjection (n=15), pooled for each condition from at least 2 independent experiments. d, Cyclin B1-Venus degradation rate plotted as for (c), in cdh1 knockdown oocytes injected cdc20-CeFP cRNA (n=38) or no further injections (n=34), pooled for each condition from at least 2 independent experiments. The pipette concentration of cdc20-CeFP was high, four-fold that used in any other experiment. In (b), (c) and (d), asterisks imply significantly different from non-injected group, (p<0.05; b, ANOVA; c and d, t-test). In (c) and (d) the range over which PB extrusion occurred is as marked.
Figure 3.
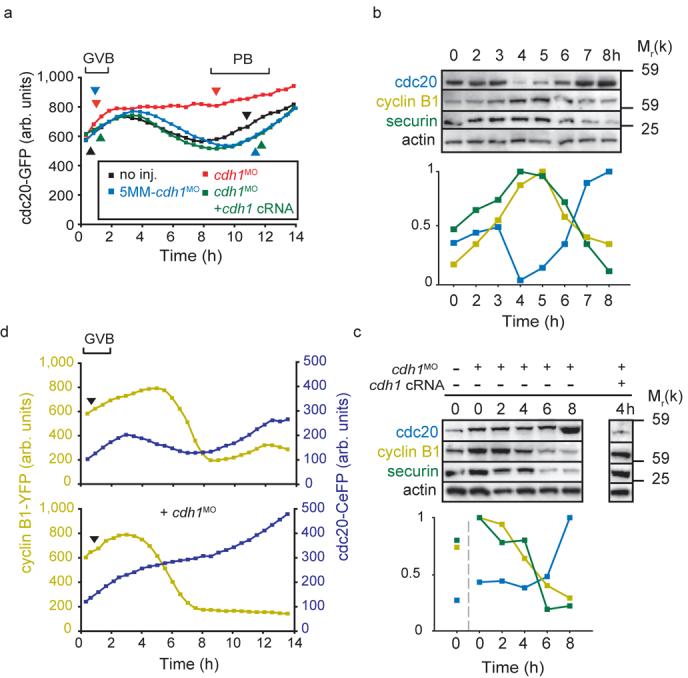
Cdc20 degradation precedes that of cyclin B1 and securin during meiosis I. a, Representative cdc20-GFP fluorescence recordings in oocytes microinjected with cdh1MO (n=16); 5MM-cdh1MO (n=23), cdh1MO and cdh1 cRNA (n=14); or no further injections (n=22). Note that cdh1 knockdown prevented cdc20-GFP degradation between GVB and PB. (b and c), Western blot and densitometric analysis of endogenous cdc20, cyclin B1 and securin levels (50 per lane) at the times indicated relative to GVB (0 h is GV arrest). In (b) oocytes were uninjected; note the early loss in cdc20 at 4h, ahead of both cyclin B1 or securin. In (c) oocytes were microinjected with cdh1MO and cdh1 cRNA as indicated; note that cdh1 knockdown abolished cdc20 loss and brought forward cyclin B1 and securin degradation. Cdc20 degradation at 4 h was recovered by cdh1 cRNA rescue. Actin was used as a loading control. Blots are representative of 2 further replicates. d, Cyclin B1-YFP and cdc20-CeFP fluorescence levels recorded simultaneously during maturation, with or without microinjection of cdh1MO as indicated. Representative traces (top, n=40; bottom n=44) of cyclin B1-YFP and cdc20-CeFP fluorescence during maturation. In (a) and (d) the timing of GVB and PB extrusion are marked by arrowheads. (b) and (c) Uncropped Westerns for cdc20, securin and cyclin B1 are displayed in the Supplementary Information, Fig S4.
To demonstrate again that the degradation of cdc20 occurs ahead of cyclin B1, cyclin B1-YFP and cdc20-Cerulean Fluorescent Protein (CeFP) cRNA were expressed in the same oocyte and their expression monitored during oocyte maturation. Cdc20 degradation preceded cyclin B1, in a period soon after GVB (Fig 3d). There was no observable loss in cyclin B1 at this time; its degradation began as the period of cdc20 degradation ended (Fig 3d and Supplementary Fig S3c). All these findings taken together show that during MI, APCcdh1 has substrate preference for cdc20 over cyclin B1 and securin; and this holds for both endogenous protein and proteins expressed from cRNA. This may be explained by the manner in which these substrates are degraded, processive or distributive ubiquitination, which underlies the temporal ordering of degradation for mitotic APCcdh1 substrates14. In addition the binding of accessory factors to APCcdh1 during prometaphase I may either enhance the degradation of cdc20, or decrease that of cyclin B1 and securin at this time; a phenomenon previously observed for a number of APCcdh1 substrates15-17.
When cdh1 knockdown oocytes were co-expressing cyclin B1-YFP and cdc20-CeFP cRNA no degradation of cdc20 was seen (Fig 3d), consistent with prometaphase APCcdh1 activity leading to loss of cdc20. However, cyclin B1-YFP degradation proceeded at a normal rate, confirming its degradation during MI is APCcdh1 independent. Cdh1 knockdown however brought forward the time of cyclin B1 degradation to the point at which cdc20 would normally have been degraded (Supplementary Fig S3c). In fact regardless of the actual time of onset for cdc20 degradation, cyclin B1 degradation followed 2 h later (Supplementary Fig 3d).
Cdc20 is essential for MI in lower eukaryotes but dispensable in frogs18-22. Therefore, we wanted to establish the cdc20 dependency of MI in mouse. During maturation, cdc20 quickly re-accumulated after its loss, making it possible that nascent cdc20 was necessary for APCcdc20 mediated cyclin B1 and securin loss. Protein synthesis was needed for MI completion since cycloheximide added to oocytes at 4 h post GVB, when cdc20 levels were low, blocked PB extrusion (control, 91% PB extrusion rates, n=34; with cycloheximide 30%, n=37). We used a cdc20 morpholino (cdc20MO; hereafter ‘cdc20 knockdown’) to block cdc20 re-accumulation during MI (Fig 4a) and therefore examine directly the role played by cdc20 protein synthesis in MI. Cdc20 knockdown specifically blocked PB extrusion (Fig 4b), since it was not observed with a 5-base mismatch morpholino (5MM-cdc20MO) and high rates of PB extrusion could be restored by cdc20 cRNA microinjection (Fig 4a, b; hereafter ‘cdc20 rescue’). These data suggested that cdc20 re-accumulation during MI was needed for securin and cyclin B1 degradation. To examine this directly, we measured the rate of degradation of both cyclin B1 (Fig 4c) and securin (Supplementary Fig S2e) and found them to be substantially reduced following cdc20 knockdown. This was not due to the microinjection procedure because normal rates of cyclin B1 degradation were observed following 5MM-cdc20MO microinjection (Fig 4c). These data show that re-accumulation of cdc20 was needed for completion on MI and that in an unperturbed MI, APCcdc20 contributed to cyclin B1 and securin degradation. In cdh1 knockdown oocytes cdc20 was also responsible for the cyclin B1 and securin degradation. We could not knockdown cdc20 in a cdh1 background because cdc20 loss was dependent on APCcdh1, however large injections of cdc20-CeFP (4-fold greater pipette concentration than used elsewhere), acted as a dominant-negative construct to block polar body extrusion and inhibit cyclin B1 degradation in cdh1 knockdown oocytes (Fig 4d). These data, along with the observed ability of mad2 to block degradation (Fig 2d), strongly suggests that APCcdc20 is responsible for cyclin B1 and securin degradation in cdh1 knockdown oocytes.
Although CDK1 can phosphorylate and so inhibit cdh1 binding to the APC23, 24, its rise in oocytes during prometaphase I was found to be sufficiently slow to allow APCcdh1 to function. Δ90cyclin B1 cRNA, which accelerated CDK1 activity (Fig 5a) and increased phosphorylated cdh1 (Fig 5b, phosphorylated cdh1 migrates more slowly23), reduced by a half the period of APCcdh1 mediated cdc20 degradation (Fig 5d,e). During oocyte maturation we also observed a loss of endogenous cdh1 in a period between 6 to 7 h after GVB (Fig 5c). This loss in cdh1, which can be degraded through both APC- and SCF-dependent mechanisms25, 26, suggested that the switch from APCcdh1 to APCcdc20 activity could be regulated by both CDK1 activity and cdh1 degradation. In fact they appeared to act redundantly in switching off APCcdh1, because it was only when both cdh1 cRNA was overexpressed and CDK1 inhibited, that the period over which APCcdh1 was active increased (Fig 5e).
Figure 5.

CDK1 regulation of prometaphase APCcdh1. a, H1 kinase assays of maturing oocytes at the times indicated, normalised with respect to time 0 h (GV arrest). Some GV oocytes were microinjected with Δ90cyclin B1 cRNA (Δ90cycB1) before maturation (n=5). Δ90cyclin B1 increased the rate of rise of H1 kinase activity following GVB. (b and c) cdh1 expression in oocytes at the times indicated after GVB. In (b), cdh1MO, or cRNA for Δ90cyclin B1 and cdh1 were microinjected as indicated. d, cdc20-GFP fluorescence changes in a control (black) and Δ90cyclin B1 cRNA injected oocytes (grey), plotted with respect to time as a percentage of the maximum recorded fluorescence before any cdc20 loss. Time t is the time taken to reach the minimum recorded fluorescence from the start of cdc20 degradation and is plotted in (e). e, The period of cdc20-GFP degradation (t), calculated from experiments performed as in part (d), in oocytes injected with cRNA for Δ90cyclin B1 and cdh1 or treated with roscovitine (ROS). Numbers examined in parentheses with oocytes pooled for each condition from at least 2 independent experiments. In (a) and (e) error bars are s.d. and asterisks mark timepoints that are significantly different from: time 0 (a) or no additions (e) (p<0.05, ANOVA).
In summary we have shown that in prometaphase, APCcdh1 is active and has a role to play in oocyte maturation, slowing passage through MI and allowing time for homologues to congress. APCcdh1 activity preferentially targeted cdc20 for degradation over cyclin B1 and securin, whose later degradation was mediated by cdc20. Cdc20 resynthesis was therefore essential for completion of MI. Cdh1 knockdown moved forward the period of APCcdc20 activity despite a lack of homologue congression, suggesting that the various SAC components in oocytes9, 27, 28, do not act to monitor bivalent orientation; a finding suggested previously from studies in mice carrying a univalent X chromosome29. The present work therefore describes a mechanism controlling the timing of mammalian meiosis which has not been observed for other cell divisions.
METHODS
Oocyte collection and culture
GV oocytes were collected from four to six week old MFI mice (Harlan, Bicester, UK) mice 44-52 h after intraperitoneal injection of 7.5 IU pregnant mares' serum gonadotrophin (Calbiochem, Nottingham, UK). For bench handling, microinjections, and imaging experiments, oocytes were cultured in medium M2 following mechanical removal of granulosa cells. For long-term incubation, oocytes were cultured in MEM (22571-020; Gibco, Paisley, UK) with 20% foetal calf serum in a 5% CO2 humidified incubator at 37°C. Milrinone (1 μM: Sigma-Aldrich, Poole, UK) was used to arrest GV oocytes. Chromosome spreads and H1 kinase assays on oocytes were performed as described previously7,30.
cRNA and morpholinos
Cdc20, cdh1, cyclin B1, Δ90cyclin B1 and mad2 cRNA were made as described previously7,31, using a modified pRN3 vector designed to produce protein C-terminally coupled to CFP (Cyan), CeFP (Cerulean), GFP (Green), Venus (Yellow), YFP (Yellow), or unlabelled. cRNA was synthesised using T3 mMESSAGE mMACHINE (Ambion, Huntingdon, UK), and dissolved in nuclease-free water to a concentration of approximately 1 μg μl−1 before microinjection. The following morpholinos (Gene Tools LLC, OR, USA) were used at a micropipette concentration of 1mM: cdh1MO, 5′-CCTTCGCTCATAGTCCTGGTCCATG-3′; 5MM-cdh1MO, 5′-GTACCTGGTCCTGATACTCGCTTCC-3′; cdc20MO , 5′-CGCTCTCGAACACGAACTGCGCCAT-3′; and 5MM-cdc20MO, 5′-CGGTCTCAAACACCAAGTGCGGCAT -3′. For all experiments involving cdh1MO and 5MM-cdh1MO, oocytes were injected with morpholino for 24 h prior to release into milrinone-free medium. This allows for >90% knockdown in cdh1, with around 70% of oocytes remaining GV arrested7. For all experiments involving cdc20MO or 5MM-cdc20MO, oocytes were microinjected in milrinone-arresting medium, then washed free of milrinone following completion of microinjection.
Microinjections and imaging
All microinjections into oocytes were made on the heated stage of a Nikon TE300 inverted microscope as described previously7. Briefly, micropipettes were inserted into cells using the negative capacitance overcompensation facility on an electrophysiological amplifier. A single 0.1-0.3% volume injection was achieved using a timed injection on a Pneumatic PicoPump (World Precision Instruments, Stevenage, UK). Brightfield and epi-fluorescence images were recorded using a Sony Interline MicroMax CCD camera. Confocal sections were performed on a LeicaSP2. Metamorph and Metafluor software (Universal Imaging Corp., Downington, PA) were used for image capture and data analysis.
Immunoblotting
Western blotting was performed as described previously7 using antibodies against cdc20 (sc-8358, Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 1:200, cdh1 (ab3242; Abcam, Cambridge, UK) at 1:200, securin (ab3305; Abcam) at 1:1000, cyclin B1 (ab72; Abcam) at 1:400, and actin (ab3280; Abcam) at 1:400. Blots were incubated in primary antibodies overnight at 4°C, using nonfat milk (5%) as a blocking agent. Anti-rabbit IgG (for polyclonals; P0448; Dako, Ely, UK), anti-mouse IgG (for monoclonals; P0447; Dako) and ECL Plus (RPN2132; GE Healthcare; Little Chalfont, UK) were used as secondary detection reagents according to the manufacturer's instructions.
Immunofluorescence microscopy
Oocytes were fixed with 3.7% paraformaldehyde in phosphate-buffered saline containing 1% polyvinylpyrrolidine (PBS+PVP) and then transferred to the same containing 2% Triton-X100. Fixing and permeabilising were for 30 mins each at room temperature. Oocytes were then washed extensively in PBS+PVP before incubation with 1:400 anti-tubulin (ab6160; Abcam). Following further washes oocytes were incubated with 1:20 TRITC-labelled rabbit anti-mouse IgG (Dako) and Hoechst 33258 to stain chromatin. These incubations were at 37°C in PBS+PVP.
Statistical analysis
All 1-way ANOVA statistical analysis was performed using Minitab release 14, with a 95% confidence level and Fisher's post-hoc analysis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the technical support of Olaf Stemmann (Max Planck Institute of Biochemistry, Munich, Germany) in performing the H1 kinase assays. This work was supported by a grant from the Wellcome Trust to K.T.J.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2001;2:280–91. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 2.Acquaviva C, Pines J. The anaphase-promoting complex/cyclosome: APC/C. J. Cell. Sci. 2006;119:2401–4. doi: 10.1242/jcs.02937. [DOI] [PubMed] [Google Scholar]
- 3.Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006;7:644–56. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 4.Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCFSkp2-Cks1 ubiquitin ligase by the APC/CCdh1 ubiquitin ligase. Nature. 2004;428:190–3. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- 5.Wei W, et al. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–8. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- 6.Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16:55–63. doi: 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Reis A, Chang HY, Levasseur M, Jones KT. APCcdh1 activity in mouse oocytes prevents entry into the first meiotic division. Nat. Cell Biol. 2006;8:539–40. doi: 10.1038/ncb1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marangos P, Verschuren EW, Chen R, Jackson PK, Carroll J. Prophase I arrest and progression to metaphase I in mouse oocytes are controlled by Emi1-dependent regulation of APCCdh1. J. Cell Biol. 2007;176:65–75. doi: 10.1083/jcb.200607070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homer HA, et al. Mad2 prevents aneuploidy and premature proteolysis of cyclin B and securin during meiosis I in mouse oocytes. Genes Dev. 2005;19:202–7. doi: 10.1101/gad.328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagting A, et al. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J. Cell Biol. 2002;157:1125–37. doi: 10.1083/jcb.200111001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbert M, et al. Homologue disjunction in mouse oocytes requires proteolysis of securin and cyclin B1. Nat. Cell Biol. 2003;5:1023–5. doi: 10.1038/ncb1062. [DOI] [PubMed] [Google Scholar]
- 12.Hampl A, Eppig JJ. Analysis of the mechanism(s) of metaphase I arrest in maturing mouse oocytes. Development. 1995;121:925–33. doi: 10.1242/dev.121.4.925. [DOI] [PubMed] [Google Scholar]
- 13.Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, Peters JM. TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr. Biol . 2003;13:1459–68. doi: 10.1016/s0960-9822(03)00581-5. [DOI] [PubMed] [Google Scholar]
- 14.Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 2006;124:89–103. doi: 10.1016/j.cell.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Binne UK, et al. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 2007;9:225–32. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- 16.Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-beta signaling by targeting SnoN for destruction. Mol. Cell. 2001;8:1027–39. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- 17.Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438:1036–9. doi: 10.1038/nature04221. [DOI] [PubMed] [Google Scholar]
- 18.Salah SM, Nasmyth K. Destruction of the securin Pds1p occurs at the onset of anaphase during both meiotic divisions in yeast. Chromosoma. 2000;109:27–34. doi: 10.1007/s004120050409. [DOI] [PubMed] [Google Scholar]
- 19.Taieb FE, Gross SD, Lewellyn AL, Maller JL. Activation of the anaphase-promoting complex and degradation of cyclin B is not required for progression from Meiosis I to II in Xenopus oocytes. Curr. Biol. 2001;11:508–13. doi: 10.1016/s0960-9822(01)00145-2. [DOI] [PubMed] [Google Scholar]
- 20.Peter M, et al. The APC is dispensable for first meiotic anaphase in Xenopus oocytes. Nat. Cell Biol. 2001;3:83–7. doi: 10.1038/35050607. [DOI] [PubMed] [Google Scholar]
- 21.Lee BH, Amon A. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science. 2003;300:482–6. doi: 10.1126/science.1081846. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa R, Law E, Tang L, Rose AM. The Cdc20 homolog, FZY-1, and its interacting protein, IFY-1, are required for proper chromosome segregation in Caenorhabditis elegans. Curr. Biol. 2002;12:2118–23. doi: 10.1016/s0960-9822(02)01392-1. [DOI] [PubMed] [Google Scholar]
- 23.Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–4. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- 24.Listovsky T, Zor A, Laronne A, Brandeis M. Cdk1 is essential for mammalian cyclosome/APC regulation. Exp. Cell Res. 2000;255:184–91. doi: 10.1006/excr.1999.4788. [DOI] [PubMed] [Google Scholar]
- 25.Listovsky T, et al. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 2004;23:1619–26. doi: 10.1038/sj.emboj.7600149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benmaamar R, Pagano M. Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle. 2005;4:1230–2. doi: 10.4161/cc.4.9.2048. [DOI] [PubMed] [Google Scholar]
- 27.Brunet S, Pahlavan G, Taylor S, Maro B. Functionality of the spindle checkpoint during the first meiotic division of mammalian oocytes. Reproduction. 2003;126:443–50. doi: 10.1530/rep.0.1260443. [DOI] [PubMed] [Google Scholar]
- 28.Wassmann K, Niault T, Maro B. Metaphase I arrest upon activation of the Mad2-dependent spindle checkpoint in mouse oocytes. Curr. Biol. 2003;13:1596–608. doi: 10.1016/j.cub.2003.08.052. [DOI] [PubMed] [Google Scholar]
- 29.LeMaire-Adkins R, Radke K, Hunt PA. Lack of checkpoint control at the metaphase/anaphase transition: a mechanism of meiotic nondisjunction in mammalian females. J. Cell Biol . 1997;139:1611–9. doi: 10.1083/jcb.139.7.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorr IH, et al. Essential CDK1-inhibitory role for separase during meiosis I in vertebrate oocytes. Nat. Cell Biol. 2006;8:1035–7. doi: 10.1038/ncb1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madgwick S, Hansen DV, Levasseur M, Jackson PK, Jones KT. Mouse Emi2 is required to enter meiosis II by reestablishing cyclin B1 during interkinesis. J. Cell Biol. 2006;174:791–801. doi: 10.1083/jcb.200604140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.